Pancreatic Ductal Adenocarcinoma (PDAC) Organoids: The Shining Light at the End of the Tunnel for Drug Response Prediction and Personalized Medicine


Abstract
:Simple Summary
Abstract
1. PDAC Development and Classification
2. The Challenges of Pancreatic Ductal Adenocarcinoma (PDAC)
3. PDAC Organoids
3.1. What Is an Organoid?
3.2. Establishment of Mouse and Human PDAC Organoids
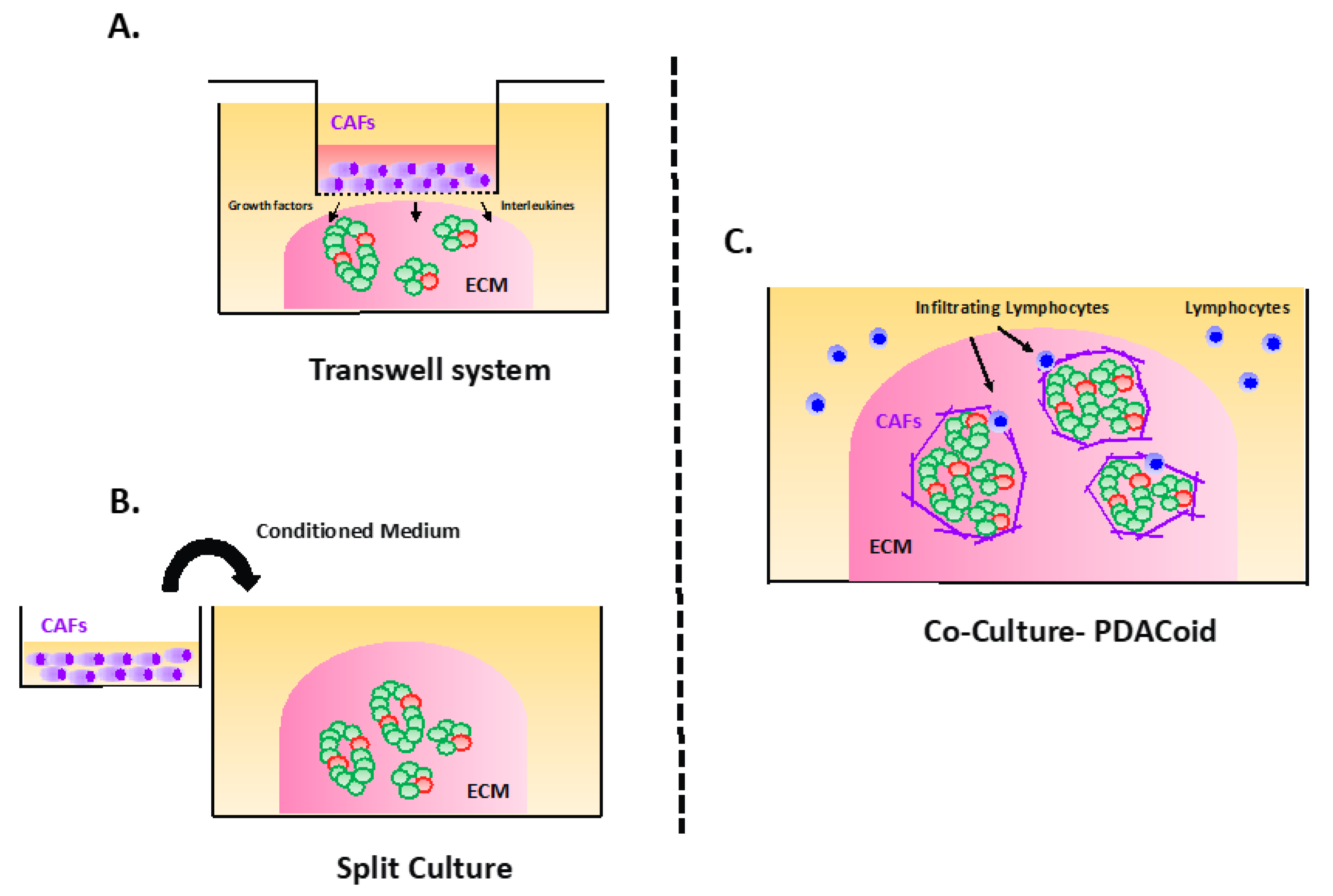
3.3. The “PDACoids”
3.4. Towards a Chemically Defined PDAC Organoid Culture
4. PDAC Organoids and Personalized Medicine
4.1. Sources of PDAC Organoids
4.2. CTC-Derived Organoids
4.3. Drug Guidance Based on Patient-Derived Organoids (PDO)
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Cronin, K.A.; Lake, A.J.; Scott, S.; Sherman, R.L.; Noone, A.M.; Howlader, N.; Henley, S.J.; Anderson, R.N.; Firth, A.U.; Ma, J.; et al. Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer 2018, 124, 2785–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpelan-Holmstrom, M.; Nordling, S.; Pukkala, E.; Sankila, R.; Luttges, J.; Kloppel, G.; Haglund, C. Does anyone survive pancreatic ductal adenocarcinoma? A nationwide study re-evaluating the data of the Finnish Cancer Registry. Gut 2005, 54, 385–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, A.W.; Schwerdel, D.; Costa, I.G.; Hackert, T.; Strobel, O.; Lam, S.; Barth, T.F.; Schroppel, B.; Meining, A.; Buchler, M.W.; et al. Detection of Hot-Spot Mutations in Circulating Cell-Free DNA From Patients With Intraductal Papillary Mucinous Neoplasms of the Pancreas. Gastroenterology 2016, 151, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Kuboki, Y.; Tanji, E.; Yoshida, S.; Hatori, T.; Yamamoto, M.; Shibata, N.; Shimizu, K.; Kamatani, N.; Shiratori, K. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci. Rep. 2011, 1, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoda, W.; Sasaki, E.; Murakami, Y.; Yamao, K.; Shimizu, Y.; Yatabe, Y. GNAS mutation is a frequent event in pancreatic intraductal papillary mucinous neoplasms and associated adenocarcinomas. Virchows. Arch. 2015, 466, 665–674. [Google Scholar] [CrossRef]
- Fischer, C.G.; Wood, L.D. From somatic mutation to early detection: Insights from molecular characterization of pancreatic cancer precursor lesions. J. Pathol. 2018, 246, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.C.; Bardeesy, N.; Mizukami, Y. Diversity of Precursor Lesions For Pancreatic Cancer: The Genetics and Biology of Intraductal Papillary Mucinous Neoplasm. Clin. Transl. Gastroenterol. 2017, 8, e86. [Google Scholar] [CrossRef]
- Sethi, V.; Giri, B.; Saluja, A.; Dudeja, V. Insights into the Pathogenesis of Pancreatic Cystic Neoplasms. Dig. Dis. Sci. 2017, 62, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Liu, X.; Suriawinata, A.A. Pancreatic Ductal Adenocarcinoma and Its Precursor Lesions: Histopathology, Cytopathology, and Molecular Pathology. Am. J. Pathol. 2019, 189, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Distler, M.; Aust, D.; Weitz, J.; Pilarsky, C.; Grutzmann, R. Precursor lesions for sporadic pancreatic cancer: PanIN, IPMN, and MCN. Biomed. Res. Int. 2014, 2014, 474905. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e3. [Google Scholar] [CrossRef] [Green Version]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Lomberk, G.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.; Yan, H.; Elarouci, N.; et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun. 2018, 9, 1978. [Google Scholar] [CrossRef] [PubMed]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Chiaravalli, M.; Reni, M.; O’Reilly, E.M. Pancreatic ductal adenocarcinoma: State-of-the-art 2017 and new therapeutic strategies. Cancer Treat. Rev. 2017, 60, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Bilimoria, K.Y.; Bentrem, D.J.; Ko, C.Y.; Stewart, A.K.; Winchester, D.P.; Talamonti, M.S. National failure to operate on early stage pancreatic cancer. Ann. Surg. 2007, 246, 173–180. [Google Scholar] [CrossRef]
- Buanes, T.A. Role of surgery in pancreatic cancer. World J. Gastroenterol. 2017, 23, 3765–3770. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.M.; Cameron, J.L.; Campbell, K.A.; Arnold, M.A.; Chang, D.C.; Coleman, J.; Hodgin, M.B.; Sauter, P.K.; Hruban, R.H.; Riall, T.S.; et al. 1423 pancreaticoduodenectomies for pancreatic cancer: A single-institution experience. J. Gastrointest. Surg. 2006, 10, 1199–1210. [Google Scholar] [CrossRef]
- Moletta, L.; Serafini, S.; Valmasoni, M.; Pierobon, E.S.; Ponzoni, A.; Sperti, C. Surgery for Recurrent Pancreatic Cancer: Is It Effective? Cancers 2019, 11, 991. [Google Scholar] [CrossRef] [Green Version]
- Srinivasa, S.; Parks, R. Emerging concepts in the management of pancreatic ductal adenocarcinoma. Laparosc. Endosc. Robot. Surg. 2019, 2, 83–88. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, D.; El-Maraghi, R.H.; Hammel, P.; Heinemann, V.; Kunzmann, V.; Sastre, J.; Scheithauer, W.; Siena, S.; Tabernero, J.; Teixeira, L.; et al. nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: Long-term survival from a phase III trial. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Gunturu, K.S.; Yao, X.; Cong, X.; Thumar, J.R.; Hochster, H.S.; Stein, S.M.; Lacy, J. FOLFIRINOX for locally advanced and metastatic pancreatic cancer: Single institution retrospective review of efficacy and toxicity. Med. Oncol. 2013, 30, 361. [Google Scholar] [CrossRef]
- Kapalczynska, M.; Kolenda, T.; Przybyla, W.; Zajaczkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Blizniak, R.; Luczewski, L.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Brait, M.; Izumchenko, E.; Kagohara, L.T.; Long, S.; Wysocki, P.T.; Faherty, B.; Fertig, E.J.; Khor, T.O.; Bruckheimer, E.; Baia, G.; et al. Comparative mutational landscape analysis of patient-derived tumour xenografts. Br. J. Cancer 2017, 116, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Coleman, O.; Henry, M.; O’Neill, F.; Roche, S.; Swan, N.; Boyle, L.; Murphy, J.; Meiller, J.; Conlon, N.T.; Geoghegan, J.; et al. A Comparative Quantitative LC-MS/MS Profiling Analysis of Human Pancreatic Adenocarcinoma, Adjacent-Normal Tissue, and Patient-Derived Tumour Xenografts. Proteomes 2018, 6, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinska, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [Green Version]
- Izumchenko, E.; Paz, K.; Ciznadija, D.; Sloma, I.; Katz, A.; Vasquez-Dunddel, D.; Ben-Zvi, I.; Stebbing, J.; McGuire, W.; Harris, W.; et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol. 2017, 28, 2595–2605. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Viqueira, B.; Jimeno, A.; Cusatis, G.; Zhang, X.; Iacobuzio-Donahue, C.; Karikari, C.; Shi, C.; Danenberg, K.; Danenberg, P.V.; Kuramochi, H.; et al. An in vivo platform for translational drug development in pancreatic cancer. Clin. Cancer Res. 2006, 12, 4652–4661. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, X.W.; Zhao, Z.Y.; Dong, B.; Guan, X.Y.; Tian, X.Y.; Qian, H.G.; Hao, C.Y. Establishment of pancreatic cancer patient-derived xenograft models and comparison of the differences among the generations. Am. J. Transl. Res. 2019, 11, 3128–3139. [Google Scholar]
- Choi, S.I.; Jeon, A.R.; Kim, M.K.; Lee, Y.S.; Im, J.E.; Koh, J.W.; Han, S.S.; Kong, S.Y.; Yoon, K.A.; Koh, Y.H.; et al. Development of Patient-Derived Preclinical Platform for Metastatic Pancreatic Cancer: PDOX and a Subsequent Organoid Model System Using Percutaneous Biopsy Samples. Front. Oncol. 2019, 9, 875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenberg, K.R.; Gao, J.; Oppel, F.; Frank, S.; Kang, N.; Dieter, S.M.; Herbst, F.; Mohrmann, L.; Dubash, T.D.; Schulz, E.R.; et al. Systematic Generation of Patient-Derived Tumor Models in Pancreatic Cancer. Cells 2019, 8, 142. [Google Scholar] [CrossRef] [Green Version]
- Mead, B.E.; Karp, J.M. All models are wrong, but some organoids may be useful. Genome Biol. 2019, 20, 66. [Google Scholar] [CrossRef] [PubMed]
- Gendoo, D.M.A.; Denroche, R.E.; Zhang, A.; Radulovich, N.; Jang, G.H.; Lemire, M.; Fischer, S.; Chadwick, D.; Lungu, I.M.; Ibrahimov, E.; et al. Whole genomes define concordance of matched primary, xenograft, and organoid models of pancreas cancer. PLoS Comput. Biol. 2019, 15, e1006596. [Google Scholar] [CrossRef] [Green Version]
- Romero-Calvo, I.; Weber, C.R.; Ray, M.; Brown, M.; Kirby, K.; Nandi, R.K.; Long, T.M.; Sparrow, S.M.; Ugolkov, A.; Qiang, W.; et al. Human Organoids Share Structural and Genetic Features with Primary Pancreatic Adenocarcinoma Tumors. Mol. Cancer Res. 2019, 17, 70–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharick, J.T.; Jeffery, J.J.; Karim, M.R.; Walsh, C.M.; Esbona, K.; Cook, R.S.; Skala, M.C. Cellular Metabolic Heterogeneity In Vivo Is Recapitulated in Tumor Organoids. Neoplasia 2019, 21, 615–626. [Google Scholar] [CrossRef]
- Smith, E.; Cochrane, W.J. Cystic organoid teratoma; report of a case. Can. Med. Assoc. J. 1946, 55, 151. [Google Scholar]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Weeber, F.; Ooft, S.N.; Dijkstra, K.K.; Voest, E.E. Tumor Organoids as a Pre-clinical Cancer Model for Drug Discovery. Cell Chem. Biol. 2017, 24, 1092–1100. [Google Scholar] [CrossRef]
- Weiswald, L.B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kunz-Schughart, L.A.; Kreutz, M.; Knuechel, R. Multicellular spheroids: A three-dimensional in vitro culture system to study tumour biology. Int. J. Exp. Pathol. 1998, 79, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, M.; Pignatta, S.; Arienti, C.; Bonafe, M.; Tesei, A. Anticancer drug discovery using multicellular tumor spheroid models. Expert Opin. Drug Discov. 2019, 14, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Simian, M.; Bissell, M.J. Organoids: A historical perspective of thinking in three dimensions. J. Cell Biol. 2017, 216, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Huch, M. Disease modelling in human organoids. Dis. Model Mech. 2019, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huch, M.; Bonfanti, P.; Boj, S.F.; Sato, T.; Loomans, C.J.M.; Van De Wetering, M.; Sojoodi, M.; Li, V.S.W.; Schuijers, J.; Gracanin, A.; et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 2013, 32, 2708–2721. [Google Scholar] [CrossRef] [Green Version]
- Reichert, M.; Takano, S.; Heeg, S.; Bakir, B.; Botta, G.P.; Rustgi, A.K. Isolation, culture and genetic manipulation of mouse pancreatic ductal cells. Nat. Protoc. 2013, 8, 1354–1365. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [Green Version]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Engle, D.D.; Tuveson, D.A.; Clevers, H. Model organoids provide new research opportunities for ductal pancreatic cancer. Mol. Cell. Oncol. 2016, 3, 9–11. [Google Scholar] [CrossRef] [Green Version]
- Broutier, L.; Andersson-Rolf, A.; Hindley, C.J.; Boj, S.F.; Clevers, H.; Koo, B.K.; Huch, M. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat. Protoc. 2016, 11, 1724–1743. [Google Scholar] [CrossRef]
- Huang, L.; Holtzinger, A.; Jagan, I.; Begora, M.; Lohse, I.; Ngai, N.; Nostro, C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 1–13. [Google Scholar] [CrossRef]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018, 22, 454–467.e456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, A.J.; Castellanos, J.A.; Nagathihalli, N.S.; Merchant, N.B.; Skala, M.C. Optical Imaging of Drug-Induced Metabolism Changes in Murine and Human Pancreatic Cancer Organoids Reveals Heterogeneous Drug Response. Pancreas 2016, 45, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Snyder, E.R.; Liu, Y.; Gu, X.; Wang, J.; Flowers, B.M.; Kim, Y.J.; Park, S.; Szot, G.L.; Hruban, R.H.; et al. Reconstituting development of pancreatic intraepithelial neoplasia from primary human pancreas duct cells. Nat. Commun. 2017, 8, 14686. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.; et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 2014, 20, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Ootani, A.; Kuo, C. An Air-Liquid Interface Culture System for 3D Organoid Culture of Diverse Primary Gastrointestinal Tissues. Methods Mol. Biol. 2016, 1422, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.A.; Tiriac, H.; Tuveson, D.A. Generation and Culture of Human Pancreatic Ductal Adenocarcinoma Organoids from Resected Tumor Specimens. Methods Mol. Biol. 2019, 1882, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.A.; Tuveson, D.A. Generation and Culture of Tumor and Metastatic Organoids from Murine Models of Pancreatic Ductal Adenocarcinoma. Methods Mol. Biol. 2019, 1882, 117–133. [Google Scholar] [CrossRef]
- Hou, S.; Tiriac, H.; Sridharan, B.P.; Scampavia, L.; Madoux, F.; Seldin, J.; Souza, G.R.; Watson, D.; Tuveson, D.; Spicer, T.P. Advanced Development of Primary Pancreatic Organoid Tumor Models for High-Throughput Phenotypic Drug Screening. SLAS Discov. 2018, 23, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Willert, K.H. Isolation and application of bioactive Wnt proteins. Methods Mol. Biol. 2008, 468, 17–29. [Google Scholar] [CrossRef]
- Molenaar, M.; van de Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destree, O.; Clevers, H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Veeman, M.T.; Slusarski, D.C.; Kaykas, A.; Louie, S.H.; Moon, R.T. Zebrafish Prickle, a Modulator of Noncanonical Wnt/Fz Signaling, Regulates Gastrulation Movements. Curr. Biol. 2003, 13, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Fuerer, C.; Nusse, R. Lentiviral vectors to probe and manipulate the Wnt signaling pathway. PLoS ONE 2010, 5, e9370. [Google Scholar] [CrossRef]
- Barolo, S. Transgenic Wnt/TCF pathway reporters: All you need is Lef? Oncogene 2006, 25, 7505–7511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Noort, M.; Meeldijk, J.; van der Zee, R.; Destree, O.; Clevers, H. Wnt signaling controls the phosphorylation status of beta-catenin. J. Biol. Chem. 2002, 277, 17901–17905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamai, K.; Zeng, X.; Liu, C.; Zhang, X.; Harada, Y.; Chang, Z.; He, X. A mechanism for Wnt coreceptor activation. Mol. Cell 2004, 13, 149–156. [Google Scholar] [CrossRef]
- Urbischek, M.; Rannikmae, H.; Foets, T.; Ravn, K.; Hyvonen, M.; de la Roche, M. Organoid culture media formulated with growth factors of defined cellular activity. Sci. Rep. 2019, 9, 6193. [Google Scholar] [CrossRef] [Green Version]
- Driehuis, E.; van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad Sci. USA 2019. [Google Scholar] [CrossRef]
- Willert, K.; Nusse, R. Wnt proteins. Cold Spring Harb. Perspect. Biol. 2012, 4, a007864. [Google Scholar] [CrossRef]
- Mihara, E.; Hirai, H.; Yamamoto, H.; Tamura-Kawakami, K.; Matano, M.; Kikuchi, A.; Sato, T.; Takagi, J. Active and water-soluble form of lipidated Wnt protein is maintained by a serum glycoprotein afamin/alpha-albumin. Elife 2016, 5. [Google Scholar] [CrossRef]
- Tuysuz, N.; van Bloois, L.; van den Brink, S.; Begthel, H.; Verstegen, M.M.; Cruz, L.J.; Hui, L.; van der Laan, L.J.; de Jonge, J.; Vries, R.; et al. Lipid-mediated Wnt protein stabilization enables serum-free culture of human organ stem cells. Nat. Commun. 2017, 8, 14578. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, Y.; Cui, K.; Guo, Y.; Zhang, X.; Qin, J. Advances in Hydrogels in Organoids and Organs-on-a-Chip. Adv. Mater. 2019, 31, e1902042. [Google Scholar] [CrossRef] [PubMed]
- Brassard, J.A.; Lutolf, M.P. Engineering Stem Cell Self-organization to Build Better Organoids. Cell Stem Cell 2019, 24, 860–876. [Google Scholar] [CrossRef] [PubMed]
- Magin, C.M.; Alge, D.L.; Anseth, K.S. Bio-inspired 3D microenvironments: A new dimension in tissue engineering. Biomed. Mater. 2016, 11, 022001. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Joisher, H.; Ganguly, A. Polymeric Scaffolds for Pancreatic Tissue Engineering: A Review. Rev. Diabet. Stud. 2018, 14, 334–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salg, G.A.; Giese, N.A.; Schenk, M.; Huttner, F.J.; Felix, K.; Probst, P.; Diener, M.K.; Hackert, T.; Kenngott, H.G. The emerging field of pancreatic tissue engineering: A systematic review and evidence map of scaffold materials and scaffolding techniques for insulin-secreting cells. J. Tissue Eng. 2019, 10, 2041731419884708. [Google Scholar] [CrossRef]
- Greggio, C.; De Franceschi, F.; Figueiredo-Larsen, M.; Gobaa, S.; Ranga, A.; Semb, H.; Lutolf, M.; Grapin-Botton, A. Artificial three-dimensional niches deconstruct pancreas development in vitro. Development 2013, 140, 4452–4462. [Google Scholar] [CrossRef] [Green Version]
- Sackett, S.D.; Tremmel, D.M.; Ma, F.; Feeney, A.K.; Maguire, R.M.; Brown, M.E.; Zhou, Y.; Li, X.; O’Brien, C.; Li, L.; et al. Extracellular matrix scaffold and hydrogel derived from decellularized and delipidized human pancreas. Sci. Rep. 2018, 8, 10452. [Google Scholar] [CrossRef]
- Broguiere, N.; Isenmann, L.; Hirt, C.; Ringel, T.; Placzek, S.; Cavalli, E.; Ringnalda, F.; Villiger, L.; Zullig, R.; Lehmann, R.; et al. Growth of Epithelial Organoids in a Defined Hydrogel. Adv. Mater. 2018, 30, e1801621. [Google Scholar] [CrossRef]
- Tiriac, H.; Bucobo, J.C.; Tzimas, D.; Grewel, S.; Lacomb, J.F.; Rowehl, L.M.; Nagula, S.; Wu, M.; Kim, J.; Sasson, A.; et al. Successful creation of pancreatic cancer organoids by means of EUS-guided fine-needle biopsy sampling for personalized cancer treatment. Gastrointest. Endosc. 2018, 87, 1474–1480. [Google Scholar] [CrossRef]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschênes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidard, F.C.; Huguet, F.; Louvet, C.; Mineur, L.; Bouche, O.; Chibaudel, B.; Artru, P.; Desseigne, F.; Bachet, J.B.; Mathiot, C.; et al. Circulating tumor cells in locally advanced pancreatic adenocarcinoma: The ancillary CirCe 07 study to the LAP 07 trial. Ann. Oncol. 2013, 24, 2057–2061. [Google Scholar] [CrossRef]
- Ankeny, J.S.; Court, C.M.; Hou, S.; Li, Q.; Song, M.; Wu, D.; Chen, J.F.; Lee, T.; Lin, M.; Sho, S.; et al. Circulating tumour cells as a biomarker for diagnosis and staging in pancreatic cancer. Br. J. Cancer 2016, 114, 1367–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buscail, E.; Alix-Panabieres, C.; Quincy, P.; Cauvin, T.; Chauvet, A.; Degrandi, O.; Caumont, C.; Verdon, S.; Lamrissi, I.; Moranvillier, I.; et al. High Clinical Value of Liquid Biopsy to Detect Circulating Tumor Cells and Tumor Exosomes in Pancreatic Ductal Adenocarcinoma Patients Eligible for Up-Front Surgery. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amantini, C.; Morelli, M.B.; Nabissi, M.; Piva, F.; Marinelli, O.; Maggi, F.; Bianchi, F.; Bittoni, A.; Berardi, R.; Giampieri, R.; et al. Expression Profiling of Circulating Tumor Cells in Pancreatic Ductal Adenocarcinoma Patients: Biomarkers Predicting Overall Survival. Front Oncol. 2019, 9, 874. [Google Scholar] [CrossRef]
- Rosenbaum, M.W.; Cauley, C.E.; Kulemann, B.; Liss, A.S.; Castillo, C.F.; Warshaw, A.L.; Lillemoe, K.D.; Thayer, S.P.; Pitman, M.B. Cytologic characteristics of circulating epithelioid cells in pancreatic disease. Cancer Cytopathol. 2017, 125, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Kulemann, B.; Pitman, M.B.; Liss, A.S.; Valsangkar, N.; Fernandez-Del Castillo, C.; Lillemoe, K.D.; Hoeppner, J.; Mino-Kenudson, M.; Warshaw, A.L.; Thayer, S.P. Circulating tumor cells found in patients with localized and advanced pancreatic cancer. Pancreas 2015, 44, 547–550. [Google Scholar] [CrossRef] [Green Version]
- Kulemann, B.; Rosch, S.; Seifert, S.; Timme, S.; Bronsert, P.; Seifert, G.; Martini, V.; Kuvendjiska, J.; Glatz, T.; Hussung, S.; et al. Pancreatic cancer: Circulating Tumor Cells and Primary Tumors show Heterogeneous KRAS Mutations. Sci. Rep. 2017, 7, 4510. [Google Scholar] [CrossRef] [Green Version]
- Effenberger, K.E.; Schroeder, C.; Hanssen, A.; Wolter, S.; Eulenburg, C.; Tachezy, M.; Gebauer, F.; Izbicki, J.R.; Pantel, K.; Bockhorn, M. Improved Risk Stratification by Circulating Tumor Cell Counts in Pancreatic Cancer. Clin. Cancer Res. 2018, 24, 2844–2850. [Google Scholar] [CrossRef] [Green Version]
- Gemenetzis, G.; Groot, V.P.; Yu, J.; Ding, D.; Teinor, J.A.; Javed, A.A.; Wood, L.D.; Burkhart, R.A.; Cameron, J.L.; Makary, M.A.; et al. Circulating Tumor Cells Dynamics in Pancreatic Adenocarcinoma Correlate With Disease Status: Results of the Prospective CLUSTER Study. Ann. Surg. 2018, 268, 408–420. [Google Scholar] [CrossRef]
- Varillas, J.I.; Zhang, J.; Chen, K.; Barnes, I.I.; Liu, C.; George, T.J.; Fan, Z.H. Microfluidic Isolation of Circulating Tumor Cells and Cancer Stem-Like Cells from Patients with Pancreatic Ductal Adenocarcinoma. Theranostics 2019, 9, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Lapin, M.; Tjensvoll, K.; Oltedal, S.; Javle, M.; Smaaland, R.; Gilje, B.; Nordgard, O. Single-cell mRNA profiling reveals transcriptional heterogeneity among pancreatic circulating tumour cells. BMC Cancer 2017, 17, 390. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wu, G.; Cheng, K.S.; Chen, A.; Neoh, K.H.; Chen, S.; Tang, Z.; Lee, P.F.; Dai, M.; Han, R.P.S. CTC phenotyping for a preoperative assessment of tumor metastasis and overall survival of pancreatic ductal adenocarcinoma patients. EBioMedicine 2019, 46, 133–149. [Google Scholar] [CrossRef]
- Stott, S.L.; Hsu, C.H.; Tsukrov, D.I.; Yu, M.; Miyamoto, D.T.; Waltman, B.A.; Rothenberg, S.M.; Shah, A.M.; Smas, M.E.; Korir, G.K.; et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad Sci. USA 2010, 107, 18392–18397. [Google Scholar] [CrossRef] [Green Version]
- Arnoletti, J.P.; Fanaian, N.; Reza, J.; Sause, R.; Almodovar, A.J.; Srivastava, M.; Patel, S.; Veldhuis, P.P.; Griffith, E.; Shao, Y.P.; et al. Pancreatic and bile duct cancer circulating tumor cells (CTC) form immune-resistant multi-cell type clusters in the portal venous circulation. Cancer Biol. Ther. 2018, 19, 887–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cauley, C.E.; Pitman, M.B.; Zhou, J.; Perkins, J.; Kuleman, B.; Liss, A.S.; Fernandez-Del Castillo, C.; Warshaw, A.L.; Lillemoe, K.D.; Thayer, S.P. Circulating Epithelial Cells in Patients with Pancreatic Lesions: Clinical and Pathologic Findings. J. Am. Coll. Surg. 2015, 221, 699–707. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Thege, F.I.; Santana, S.M.; Lannin, T.B.; Saha, T.N.; Tsai, S.; Maggs, L.R.; Kochman, M.L.; Ginsberg, G.G.; Lieb, J.G.; et al. Detection of circulating pancreas epithelial cells in patients with pancreatic cystic lesions. Gastroenterology 2014, 146, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Poruk, K.E.; Valero, V., 3rd; He, J.; Ahuja, N.; Cameron, J.L.; Weiss, M.J.; Lennon, A.M.; Goggins, M.; Wood, L.D.; Wolfgang, C.L. Circulating Epithelial Cells in Intraductal Papillary Mucinous Neoplasms and Cystic Pancreatic Lesions. Pancreas 2017, 46, 943–947. [Google Scholar] [CrossRef]
- Praharaj, P.P.; Bhutia, S.K.; Nagrath, S.; Bitting, R.L.; Deep, G. Circulating tumor cell-derived organoids: Current challenges and promises in medical research and precision medicine. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shiratsuchi, H.; Lin, J.; Chen, G.; Reddy, R.M.; Azizi, E.; Fouladdel, S.; Chang, A.C.; Lin, L.; Jiang, H.; et al. Expansion of CTCs from early stage lung cancer patients using a microfluidic co-culture model. Oncotarget 2014, 5, 12383–12397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernandez-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frappart, P.-O.; Walter, K.; Gout, J.; Beutel, A.K.; Morawe, M.; Arnold, F.; Breunig, M.; Barth, T.F.E.; Marienfeld, R.; Schulte, L.; et al. Pancreatic cancer-derived organoids—A disease modeling tool to predict drug response. United Eur. Gastroenterol. J. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seppala, T.T.; Zimmerman, J.W.; Sereni, E.; Plenker, D.; Suri, R.; Rozich, N.; Blair, A.; Thomas, D.L., 2nd; Teinor, J.; Javed, A.; et al. Patient-derived Organoid Pharmacotyping is a Clinically Tractable Strategy for Precision Medicine in Pancreatic Cancer. Ann. Surg. 2020, 272, 427–435. [Google Scholar] [CrossRef]
- Raimondi, G.; Mato-Berciano, A.; Pascual-Sabater, S.; Rovira-Rigau, M.; Cuatrecasas, M.; Fondevila, C.; Sanchez-Cabus, S.; Begthel, H.; Boj, S.F.; Clevers, H.; et al. Patient-derived pancreatic tumour organoids identify therapeutic responses to oncolytic adenoviruses. EBioMedicine 2020, 56, 102786. [Google Scholar] [CrossRef]
- Miyabayashi, K.; Baker, L.A.; Deschenes, A.; Traub, B.; Caligiuri, G.; Plenker, D.; Alagesan, B.; Belleau, P.; Li, S.; Kendall, J.; et al. Intraductal transplantation models of human pancreatic ductal adenocarcinoma reveal progressive transition of molecular subtypes. Cancer Discov. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ponz-Sarvise, M.; Corbo, V.; Tiriac, H.; Engle, D.D.; Frese, K.K.; Oni, T.E.; Hwang, C.I.; Ohlund, D.; Chio, I.I.C.; Baker, L.A.; et al. Identification of Resistance Pathways Specific to Malignancy Using Organoid Models of Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 6742–6755. [Google Scholar] [CrossRef] [Green Version]
- Bode, K.J.; Mueller, S.; Schweinlin, M.; Metzger, M.; Brunner, T. A fast and simple fluorometric method to detect cell death in 3D intestinal organoids. Biotechniques 2019, 67, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Phan, N.; Hong, J.J.; Tofig, B.; Mapua, M.; Elashoff, D.; Moatamed, N.A.; Huang, J.; Memarzadeh, S.; Damoiseaux, R.; Soragni, A. A simple high-throughput approach identifies actionable drug sensitivities in patient-derived tumor organoids. Commun. Biol. 2019, 2, 78. [Google Scholar] [CrossRef]
- Huang, S.; Pang, L. Comparing statistical methods for quantifying drug sensitivity based on in vitro dose-response assays. Assay. Drug. Dev. Technol. 2012, 10, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Lemire, M.; Zhang, A.; Chan-Seng-Yue, M.; Wilson, G.; Grant, R.C.; Merico, D.; Lungu, I.; et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 2019, 35, 267–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohwieler, M.; Illing, A.; Hermann, P.C.; Mayer, T.; Stockmann, M.; Perkhofer, L.; Eiseler, T.; Antony, J.S.; Muller, M.; Renz, S.; et al. Human pluripotent stem cell-derived acinar/ductal organoids generate human pancreas upon orthotopic transplantation and allow disease modelling. Gut 2017, 66, 473–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korytnikov, R.; Nostro, M.C. Generation of polyhormonal and multipotent pancreatic progenitor lineages from human pluripotent stem cells. Methods 2016, 101, 56–64. [Google Scholar] [CrossRef]
- Russ, H.A.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015, 34, 1759–1772. [Google Scholar] [CrossRef] [Green Version]
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133. [Google Scholar] [CrossRef]
- Pagliuca, F.W.; Millman, J.R.; Gurtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic beta cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Medium | ECM/Matrix | Tissues |
|---|---|---|---|
| Huch et al., 2013 [55] | AdvDMEM/F12, R-spondin1-conditioned medium (10%), Noggin-conditioned medium (10%), B27 (1×), N-acetylcysteine (1.25 mM), EGF (50 ng/mL), gastrin I (10 mM), FGF10 (100 ng/mL), nicotinamide (10 mM), Y-27632 (10 µM) | Matrigel 100% (Dome) | Primary mouse tissue Mouse PDAC |
| Reichert et al., 2013 [56] | AdvDMEM/F12, D-glucose (4.7 mg/mL), nicotinamide (1.22 mg/mL), soybean trypsin inhibitor (0.1 mg/mL), Primocin (0.2%), Nu-Serum IV (5%), ITS + premix (0.5%), bovine pituitary extract (25 µg/mL), mEGF (20 ng/mL), cholera toxin (100 ng/mL), T3 (5 nM), Dexamethasone (5 nM) | Collagen | Primary mouse tissue Mouse PDAC |
| Li et al., 2014 [65] | Ham’s F12, FCS (20%) | 3D collagen gel/double dish culture system/air–liquid interface | Primary mouse tissue Mouse PDAC |
| Boj et al., 2015 [57] | AdvDMEM/F12, HEPES (1×), WNT-conditioned medium (50%), R-spondin1-conditioned medium (10%), Noggin-conditioned medium (10%), GlutaMAX (1×), B27 (1×), N-acetylcysteine (1mM), EGF (50 ng/ mL), gastrin I (10 mM), FGF10 (100 ng/mL), nicotinamide (10 mM), A83-01 (0.5 µM), PGE2 (1 µM), Primocin (1 mg/mL) | Matrigel 100% (Dome) | Primary human tissue Human PDAC |
| Huang et al., 2015 [60] | AdvDMEM/F12, B27 (1×), ascorbic acid (50 µg/mL), insulin (20 µg/mL), hydroxycortisone (0.25 µg/mL), FGF2 (100 ng/mL), all-trans retinoic acid (100 nM), Y-27632 (10 µM) | Coated Matrigel 100% + Medium 5% Matrigel | Human PDAC |
| Walsh et al., 2016 [63] | RPMI, FCS (10%), EGF (10 ng/mL) | Matrigel 50% | Human PDAC Mouse PDAC |
| Broutier et al., 2016 [59] | AdvDMEM/F12, HEPES (10 mM), R-spondin1-conditioned medium (5%), GlutaMAX (1×), B27 (1×), N-acetylcysteine (1 mM), EGF (50 ng/mL), gastrin I (10 mM), FGF10 (100 ng/mL), nicotinamide (10 mM), Noggin (25 ng/mL) | BME2 100% (Dome) | Primary mouse tissue |
| Broutier et al., 2016 [59] | AdvDMEM/F12, HEPES (10 mM), WNT-conditioned medium (50%), R-spondin1-conditioned medium (10%), GlutaMAX (1×), B27 (1×), N-acetylcysteine (1 mM), EGF (50 ng/mL), gastrin I (10 mM), FGF10 (100 ng/mL), nicotinamide (10 mM), A83-01 (5 µM), N2 (1%), PGE2 (3 µM), Noggin (25 ng/mL) | BME2 100% (Dome) | Primary human tissue |
| Lee et al., 2017 [64] | AdvDMEM/F12, hEGF (50 ng/mL), hR-spondin-1 (500 ng/mL), hFGF10 (50 ng/mL), mNoggin (100 ng/mL), nicotinamide (10 mM) | GFR-Matrigel 100% (Dome) | Primary human pancreas tissue |
| Seino et al., 2018 [62] | AdvDMEM/F12, HEPES (10 mM), GlutaMAX (2 mM), B27 (1×), gastrin I (10 nM), N-acetylcysteine (1 mM), mEGF (50ng/mL), Noggin (100 ng/mL), R-spondin1-conditioned medium (10%), Afamin-Wnt-3A-conditioned medium (25%), A83-01 (500 nM), SB202190 (10 µM) | GFR-Matrigel 100% (Dome) | Human PDAC |
| Tsai et al. 2018 [61] | IntestiCult™ (mouse), B27 (1×), gastrin I (10 nM), hEGF (100 ng/mL), hFGF10 (100 ng/mL), Nicotinamide (10 mM), A83-01 (500 nM), N-acetylcysteine (1.5 mM), Primocin (1 mg/mL), Y-27632 (10.5 µM) | GFR-Matrigel 100% (Dome) | Human PDAC Primary human pancreas tissue |
| Romero-Calvo et al., 2018 [45] | IntestiCult™ (mouse), B27 (1×), gastrin I (10 nM), FGF10 (100 ng/mL), nicotinamide (10 mM), A83-01 (500 nM), N-acetylcysteine (10 mM), Primocin (1 mg/mL), Y-27632 (10.5 µM) | GFR-Matrigel 100% (Dome) | Human PDAC-PDX and primary tumors |
| Choi et al., 2019 [41] | AdvDMEM/F12, B27, N-acetylcysteine, EGF, FGF10, Rspondin-1, Noggin * | GFR-Matrigel 100% (Dome) | Human PDAC-PDX from metastatic PDAC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frappart, P.-O.; Hofmann, T.G. Pancreatic Ductal Adenocarcinoma (PDAC) Organoids: The Shining Light at the End of the Tunnel for Drug Response Prediction and Personalized Medicine. Cancers 2020, 12, 2750. https://doi.org/10.3390/cancers12102750
Frappart P-O, Hofmann TG. Pancreatic Ductal Adenocarcinoma (PDAC) Organoids: The Shining Light at the End of the Tunnel for Drug Response Prediction and Personalized Medicine. Cancers. 2020; 12(10):2750. https://doi.org/10.3390/cancers12102750
Chicago/Turabian StyleFrappart, Pierre-Olivier, and Thomas G. Hofmann. 2020. "Pancreatic Ductal Adenocarcinoma (PDAC) Organoids: The Shining Light at the End of the Tunnel for Drug Response Prediction and Personalized Medicine" Cancers 12, no. 10: 2750. https://doi.org/10.3390/cancers12102750
APA StyleFrappart, P. -O., & Hofmann, T. G. (2020). Pancreatic Ductal Adenocarcinoma (PDAC) Organoids: The Shining Light at the End of the Tunnel for Drug Response Prediction and Personalized Medicine. Cancers, 12(10), 2750. https://doi.org/10.3390/cancers12102750