An All-In-One Transcriptome-Based Assay to Identify Therapy-Guiding Genomic Aberrations in Nonsmall Cell Lung Cancer Patients

 ,
,  , ,
, , 

Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Sequencing Results
2.2. Detection of SNVs and INDELs
2.3. Fusion Gene Detection
2.4. RNA Input Limit
2.5. Quality Criteria for Successful Mutation Detection
3. Discussion
4. Materials and Methods
4.1. Sample Information
4.2. RNA Isolation
4.3. Design of All-In-One Lung Cancer Assay
4.4. Library Preparation
4.5. NGS Data Analysis
4.6. Detection of Fusion Gene Transcripts by NanoString
4.7. Variant Detection by Droplet Digital (dd) PCR
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Petersen, I.; Warth, A. Lung cancer: Developments, concepts, and specific aspects of the new WHO classification. J. Cancer Res. Clin. Oncol. 2016, 142, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Califano, R.; Abidin, A.; Tariq, N.U.; Economopoulou, P.; Metro, P.; Giannis Mountzios, G. Beyond EGFR and ALK inhibition: Unravelling and exploiting novel genetic alterations in advanced non small-cell lung cancer. Cancer Treat. Rev. 2015, 41, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Ellison, G.; Zhu, G.; Moulis, A.; Dearden, S.; Speake, G.; Rose McCormack, R. EGFR mutation testing in lung cancer: A review of available methods and their use for analysis of tumour tissue and cytology samples. J. Clin. Pathol. 2013, 66, 79–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.-I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Vakiani, E.; Solit, D.B. KRAS and BRAF: Drug targets and predictive biomarkers. J. Pathol. 2011, 223, 219–229. [Google Scholar] [CrossRef]
- Shea, M.; Costa, D.B.; Rangachari, D. Management of advanced non-small cell lung cancers with known mutations or rearrangements: Latest evidence and treatment approaches. Ther. Adv. Respir. Dis. 2016, 10, 113–129. [Google Scholar] [CrossRef] [Green Version]
- Shim, H.S.; Choi, Y.-L.; Kim, L.; Chang, S.; Kim, W.-S.; Roh, M.S.; Kim, T.-J.; Ha, S.Y.; Chung, J.-H.; Jang, S.J.; et al. Molecular Testing of Lung Cancers. J. Pathol. Trans. Med. 2017, 51, 242–254. [Google Scholar] [CrossRef] [Green Version]
- Hiley, C.T.; Le Quesne, J.; Santis, G.; Sharpe, R.; de Castro, D.G.; Middleton, G.; Swanton, C. Challenges in molecular testing in non-small-cell lung cancer patients with advanced disease. Lancet 2016, 388, 1002–1011. [Google Scholar] [CrossRef] [Green Version]
- Han, J.-Y.; Kim, S.H.; Lee, Y.-S.; Lee, S.-Y.; Hwang, J.-A.; Kim, J.-Y.; Yoon, S.J.; Lee, G.K. Comparison of targeted next-generation sequencing with conventional sequencing for predicting the responsiveness to epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI) therapy in never-smokers with lung adenocarcinoma. Lung Cancer 2014, 85, 161–167. [Google Scholar] [CrossRef]
- Wong, S.Q.; Fellowes, A.; Doig, K.; Ellul, J.; Bosma, T.J.; Irwin, D.; Vedururu, R.; Tan, A.Y.-C.; Weiss, J.; Chan, K.S.; et al. Assessing the clinical value of targeted massively parallel sequencing in a longitudinal, prospective population-based study of cancer patients. Br. J. Cancer 2015, 112, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Tsoulos, N.; Papadopoulou, E.; Metaxa-Mariatou, V.; Tsaousis, G.; Efstathiadou, C.; Tounta, G.; Scapeti, A.; Bourkoula, E.; Zarogoulidis, P.; Pentheroudakis, G.; et al. Tumor molecular profiling of NSCLC patients using next generation sequencing. Oncol. Rep. 2017, 38, 3419–3429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, M.; Sakai, K.; Terashima, M.; Kaneda, H.; Hayashi, H.; Tanaka, K.; Okamoto, K.; Takahama, T.; Yoshida, T.; Iwasa, T.; et al. Clinical application of amplicon-based next-generation sequencing to therapeutic decision making in lung cancer. Ann. Oncol. 2015, 26, 2477–2482. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, K.E.; Karlsson, A.; Leveen, P.; Brunnström, H.; Reuterswärd, C.; Holm, K.; Jönsson, M.; Annersten, K.; Rosengren, F.; Jirström, K.; et al. Clinical framework for next generation sequencing based analysis of treatment predictive mutations and multiplexed gene fusion detection in non-small cell lung cancer. Oncotarget 2017, 8, 34796–34810. [Google Scholar] [CrossRef] [Green Version]
- Scolnick, J.A.; Dimon, M.; Wang, I.C.; Huelga, S.C.; Amorese, D.A. An Efficient Method for Identifying Gene Fusions by Targeted RNA Sequencing from Fresh Frozen and FFPE Samples. PLoS ONE 2015, 10, e0128916. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.R.; Patel, K.P.; Routbort, M.J.; Reddy, N.G.; Barkoh, B.A.; Handal, B.; Kanagal-Shamanna, R.; Greaves, W.O.; Medeiros, L.J.; Aldape, K.D.; et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J. Mol. Diagn. 2013, 15, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Guseva, N.V.; Jaber, O.; Stence, A.A.; Sompallae, K.; Bashir, A.; Sompallae, R.; Bossler, A.D.; Jensen, C.S.; Ma, D. Simultaneous detection of single-nucleotide variant, deletion/insertion, and fusion in lung and thyroid carcinoma using cytology specimen and an RNA-based next-generation sequencing assay. Cancer Cytopathol. 2018, 126, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, J.N.; Bloom, R.; Forys, J.T.; Hiken, J.; Armstrong, J.R.; Branson, J.; McNulty, S.; Velu, P.D.; Pepin, K.; Abel, H.; et al. Genomic heterogeneity of ALK fusion breakpoints in non-small-cell lung cancer. Mod. Pathol. 2018, 31, 791. [Google Scholar] [CrossRef]
- Shah, N.; Teso, K.; Zilli, K.; Menon, S. Novel fusion protein ALK-MPRIP exhibits ALK activation and sensitivity to crizotinib. J. Thorac. Oncol. 2015, 10, S691–S692. [Google Scholar]
- Anai, S.; Takeshita, M.; Ando, N.; Ikematsu, Y.; Mishima, S.; Ishida, K.; Inoue, K. A case of lung adenocarcinoma resistant to crizotinib harboring a novel EML4-ALK variant, exon 6 of EML4 fused to exon 18 of ALK. J. Thorac. Oncol. 2016, 11, e126–e128. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-X.; Cartegni, L.; Zhang, M.Q.; Krainer, A.R. A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat. Genet. 2001, 27, 55. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Zhang, D.; Chen, K.; Lu, J.; Wu, J.; Cao, X.; Ying, L.; Jin, Q.; Ye, Y.; Xie, Z.; et al. High performance of targeted next generation sequencing on variance detection in clinical tumor specimens in comparison with current conventional methods. J. Exp. Clin. Cancer Res. 2017, 36, 017–0591. [Google Scholar] [CrossRef] [PubMed]
- Pfarr, N.; Kirchner, M.; Lehmann, U.; Leichsenring, J.; Merkelbach-Bruse, S.; Glade, J.; Hummel, M.; Stögbauer, F.; Lehmann, A.; Trautmann, M.; et al. Testing NTRK testing: Wet-lab and in silico comparison of RNA-based targeted sequencing assays. Genes Chromosomes Cancer 2020, 59, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Krypuy, M.; Newnham, G.M.; Thomas, D.M.; Conron, M.; Dobrovic, A. High resolution melting analysis for the rapid and sensitive detection of mutations in clinical samples: KRAS codon 12 and 13 mutations in non-small cell lung cancer. BMC Cancer 2006, 6, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimamura, T.; Lowell, A.M.; Engelman, J.A.; Shapiro, G.I. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res. 2005, 65, 6401–6408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumagalli, D.; Gavin, P.G.; Taniyama, Y.; Kim, S.I.; Choi, H.J.; Paik, S.; Pogue-Geile, K.L. A rapid, sensitive, reproducible and cost-effective method for mutation profiling of colon cancer and metastatic lymph nodes. BMC Cancer 2010, 10, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [Green Version]
- Das, A.K.; Sato, M.; Story, M.D.; Peyton, M.; Graves, R.; Redpath, S.; Girard, L.; Gazdar, A.F.; Shay, J.W.; Minna, J.D.; et al. Non–small cell lung cancers with kinase domain mutations in the epidermal growth factor receptor are sensitive to ionizing radiation. Cancer Res. 2006, 66, 9601–9608. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S.; Lauriault, V.; Kolakowski, G.R.; Brandhuber, B.J.; et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discov. 2017, 7, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Klampfer, L.; Huang, J.; Corner, G.; Mariadason, J.; Arango, D.; Sasazuki, T.; Shirasawa, S.; Augenlicht, L. Oncogenic Ki-ras inhibits the expression of interferon-responsive genes through inhibition of STAT1 and STAT2 expression. J. Biol. Chem. 2003, 278, 46278–46287. [Google Scholar] [CrossRef] [Green Version]
- Samuels, Y.; Diaz, L.A.; Schmidt-Kittler, O.; Cummins, J.M.; Delong, L.; Cheong, I.; Rago, C.; Huso, D.L.; Lengauer, C.; Kinzler, K.W.; et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 2005, 7, 561–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.J.; Kannu, P.; Sharma, S.; Neyt, C.; Zhang, D.; Paria, N.; Daniel, P.B.; Whetstone, H.; Sprenger, H.G.; Hammerschmidt, P.; et al. Mutations preventing regulated exon skipping in MET cause osteofibrous dysplasia. Am. J. Hum. Genet. 2015, 97, 837–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006, 66, 283–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, H.; Shigematsu, H.; Nomura, M.; Lockwood, W.W.; Sato, M.; Okumura, N.; Soh, J.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 2008, 68, 6913–6921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driessen, E.M.C.; van Roon, E.H.J.; Spijkers-Hagelstein, J.A.P.; Schneider, P.; de Lorenzo, P.; Valsecchi, M.G.; Pieters, R.; Stam, R.W. Frequencies and prognostic impact of RAS mutations in MLL-rearranged acute lymph, oblastic leukemia in infants. Haematologica 2013, 98, 937–944. [Google Scholar] [CrossRef]
- Lee, G.D.; Lee, S.E.; Oh, D.-Y.; Yu, D.B.; Jeong, H.M.; Kim, J.; Hong, S.; Jung, H.S.; Oh, E.; Song, J.Y.; et al. MET Exon 14 Skipping mutations in lung adenocarcinoma: Clinicopathologic implications and prognostic values. J. Thorac. Oncol. 2017, 12, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Barchi, L.; Acquadro, A.; Alonso, D.; Aprea, G.; Bassolino, L.; Demurtas, O.; Ferrante, P.; Gramazio, P.; Mini, P.; Portis, E.; et al. Single primer enrichment technology (spet) for high-throughput genotyping in tomato and eggplant germplasm. Front. Plant Sci. 2019, 10, 1005. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650. [Google Scholar] [CrossRef]
- Riedel, R.F.; Crawford, J. Small-cell lung cancer: A review of clinical trials. Semin. J. Thorac. Cardiovasc. Surg. 2003, 15, 448–456. [Google Scholar] [CrossRef]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef]
- Rudin, C.M.; Durinck, S.; Stawiski, E.W.; Poirier, J.T.; Modrusan, Z.; Shames, D.S.; Bergbower, E.A.; Guan, Y.; Shin, J.; Guillory, J.; et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 2012, 44, 1111–1116. [Google Scholar] [CrossRef]
- Pelosi, G.; Pasini, F.; Sonzogni, A.; Maffini, F.; Maisonneuve, P.; Iannucci, A.; Terzi, A.; De Manzoni, G.; Bresaola, E.; Viale, G. Prognostic implications of neuroendocrine differentiation and hormone production in patients with Stage I non-small cell lung carcinoma. Cancer 2003, 97, 2487–2497. [Google Scholar] [CrossRef]
- Byelas, H.; Kanterakis, A.; Swertz, M.A. Towards a Molgenis-based computational framework. In Proceedings of the IEEE 19th EUROMICRO International Conference on Parallel, Distributed and Network-Based Computing, Belfast, UK, 27 February–1 March 2013; Kilpatrick, P., Milligan, P., Stotzka, R., Eds.; CPS: Belfast, UK, 2013; pp. 331–339. [Google Scholar]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310. [Google Scholar] [CrossRef] [Green Version]
- Nicorici, D.; Şatalan, M.; Edgren, H.; Kangaspeska, S.; Murumägi, A.; Kallioniemi, O.; Virtanen, S.; Kilkku, O. FusionCatcher—A tool for finding somatic fusion genes in paired-end RNA-sequencing data. BioRxiv 2014, 011650. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT—The BLAST-Like Alignment Tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, E.; Totoki, Y.; Nakamura, H.; Morizane, C.; Nara, S.; Hama, N.; Suzuki, M.; Furukawa, E.; Kato, M.; Hayashi, H.; et al. Clinical utility of circulating tumor DNA for molecular assessment in pancreatic cancer. Sci. Rep. 2015, 5, 18425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sample ID | Origin | DV200 | Known Variants Detected at DNA Level | Results of All-In-One Transcriptome-Based Assay a | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Amino Acid Change | MD Test or Reference | Tool | Mutant Reads | Total Reads | VAF c | Status | |||
| Variants Known at DNA Level | ||||||||||
| P35 | PE | 81 | AKT1 | p.E17K | NGS | IGV | 81 | 292 | 28% | confirmed |
| P13 | PE | 89 | ALK | p.G1269A | NGS | IGV | 44 | 240 | 18% | confirmed |
| P13 | PE | 89 | ALK | p.I1171N | NGS | IGV and Pipeline | 331 | 336 | 99% | confirmed |
| P07 | FFPE | 65 | ALK | p.L1196M | NGS | IGV | 1 | 3 | 33% | not confirmed |
| P35 | PE | 81 | BRAF | p.V600E | NGS | IGV and Pipeline | 31 | 60 | 52% | confirmed |
| P25 | FFPE | 71 | BRAF | p.V600E | NGS | IGV and Pipeline | 33 | 46 | 72% | confirmed |
| H1650 | cell line | nd | EGFR | p.E746_A750del | NGS | IGV and Pipeline | 46 | 76 | 61% | confirmed |
| H1975 | cell line | nd | EGFR | p.T790M | NGS | IGV and Pipeline | 347 | 425 | 82% | confirmed |
| H1975 | cell line | nd | EGFR | p.L858R | NGS | IGV and Pipeline | 564 | 684 | 82% | confirmed |
| H820 | cell line | 99 | EGFR | p.E746_A750del | NGS | IGV and Pipeline | 80 | 606 | 13% | confirmed |
| H820 | cell line | 99 | EGFR | p.T790M | NGS | IGV and Pipeline | 127 | 660 | 19% | confirmed |
| P04_S2 | PE | 88 | EGFR | p.L858R | NGS | IGV and Pipeline | 4661 | 4931 | 95% | confirmed |
| P05 | PE | 17 | EGFR | p.E746_A750del | 22 | IGV | 0 | 0 | not confirmed | |
| P04_S1 | FFPE | 26 | EGFR | p.L858R | 19,22 | IGV and Pipeline | 69 | 72 | 96% | confirmed |
| P06 | FFPE | 37 | EGFR | p.L747_P753delinsS | 22 | IGV and Pipeline | 8 | 17 | 47% | confirmed |
| P06 | FFPE | 37 | EGFR | p.T790M | 19,22 | IGV | 0 | 15 | 0% | not confirmed |
| P15 | FFPE | 40 | EGFR | p.E746_A750del | NGS | IGV and Pipeline | 51 | 76 | 67% | confirmed |
| P15 | FFPE | 40 | EGFR | p.T790M | NGS | IGV and Pipeline | 22 | 88 | 25% | confirmed |
| P17 | FFPE | 57 | EGFR | p.E746_A750del | NGS | IGV and Pipeline | 128 | 182 | 70% | confirmed |
| P17 | FFPE | 57 | EGFR | p.T790M | NGS | IGV and Pipeline | 62 | 127 | 49% | confirmed |
| P22 | FFPE | 69 | EGFR | p.E746_A750del | 19,22 | IGV | 13 | 38 | 34% | confirmed |
| P26 | FFPE | nd | EGFR | p.L858R | NGS | IGV | 0 | 6 | 0% | not confirmed |
| A549 | cell line | nd | KRAS | p.G12S | 18 | IGV and Pipeline | 512 | 513 | 100% | confirmed |
| HCT116 | cell line | nd | KRAS | p.G13D | 24 | IGV and Pipeline | 223 | 456 | 49% | confirmed |
| KOPN-8 | cell line | 99 | KRAS | p.G12D | 29 | IGV and Pipeline | 99 | 177 | 56% | confirmed |
| P01 | PE | 90 | KRAS | p.G12D | NGS | IGV and Pipeline | 14 | 111 | 13% | confirmed |
| P03 | FFPE | nd | KRAS | p.G12A | NGS | IGV and Pipeline | 8 | 8 | 100% | confirmed |
| P23 | FFPE | 44 | KRAS | p.G12C | NGS | IGV | 0 | 1 | not confirmed | |
| P28 | FFPE | 38 | KRAS | p.G12A | NGS | IGV and Pipeline | 8 | 22 | 36% | confirmed |
| P31 | FFPE | 66 | KRAS | p.Q61H | NGS | IGV and Pipeline | 60 | 123 | 49% | confirmed |
| P39 | FFPE | 65 | KRAS | p.G12D | NGS | IGV and Pipeline | 8 | 12 | 67% | confirmed |
| P40 | FFPE | 68 | KRAS | p.G12F | NGS | IGV | 0 | 1 | not confirmed | |
| P32 | FFPE | 32 | KRAS | p.G12D | NGS | IGV | 0 | 2 | not confirmed | |
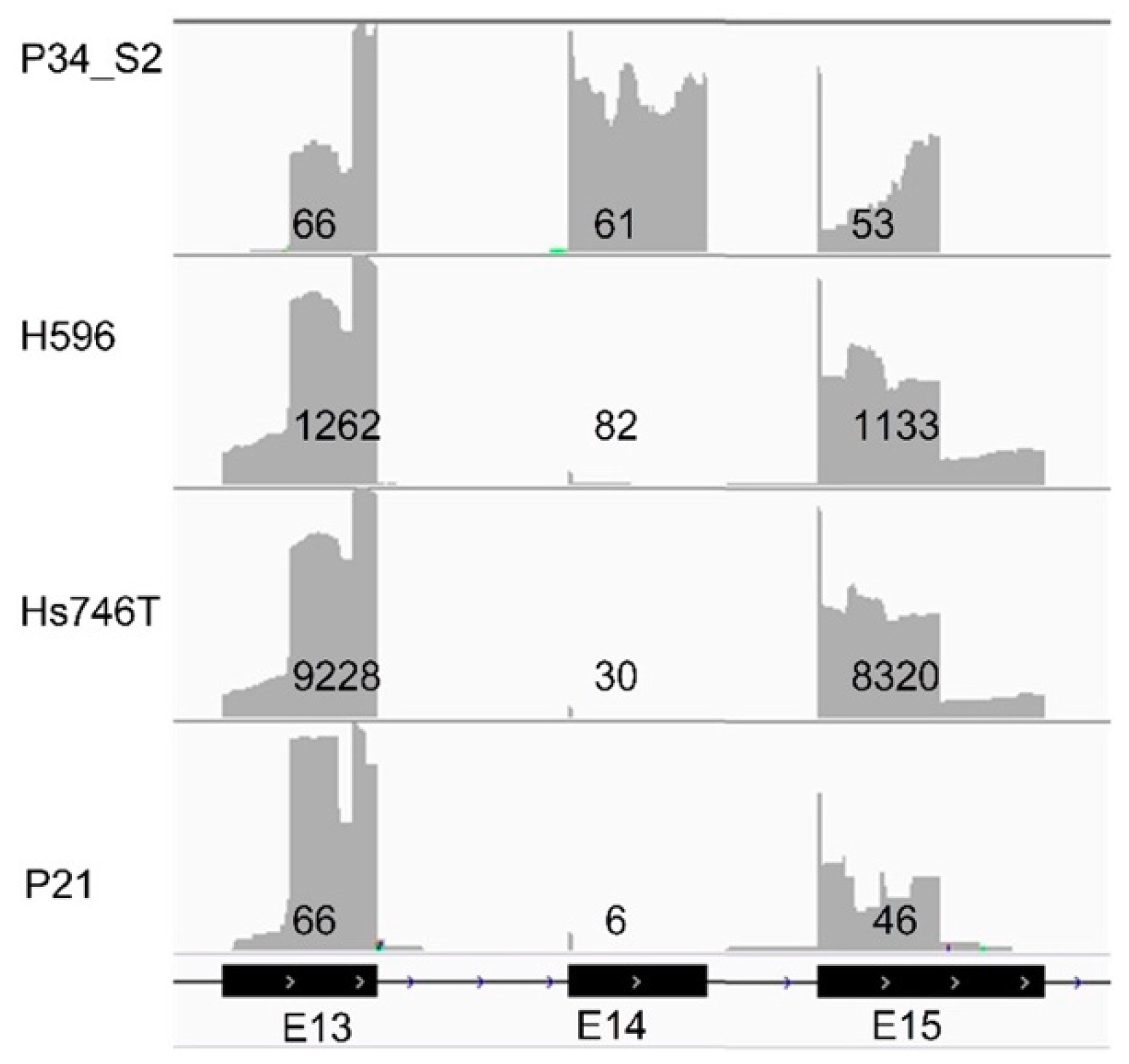
| H596 | cell line | nd | MET | Exon skipping mut. | 27 | IGV | 1116 | 1196 b | 93% | confirmed |
| Hs746T | cell line | 97 | MET | Exon skipping mut. | 30 | IGV | 8744 | 8774 b | 100% | confirmed |
| P21 | FFPE | 34 | MET | Exon skipping mut. | NGS | IGV | 50 | 56 b | 89% | confirmed |
| H1299 | cell line | nd | NRAS | p.Q61K | 20 | IGV and Pipeline | 1107 | 2549 | 43% | confirmed |
| H596 | cell line | nd | PIK3CA | p.E545K | 28 | IGV and Pipeline | 156 | 330 | 47% | confirmed |
| HCT116 | cell line | nd | PIK3CA | p.H1047R | 25 | IGV and Pipeline | 69 | 115 | 60% | confirmed |
| P02 | FFPE | 51 | PIK3CA | p.H1047L | NGS | IGV and Pipeline | 12 | 31 | 39% | confirmed |
| P26 | FFPE | nd | PIK3CA | p.E542K | NGS | IGV | 0 | 0 | not confirmed | |
| P37 | PE | 21 | ROS1 | p.D2033N | NGS | IGV and Pipeline | 3 | 3 | 100% | confirmed |
| Overview of Additional Variants that were Not Reported by MD | ||||||||||
| P03 | FFPE | nd | EGFR | p.V834L | NGS; FISH | Pipeline | 9 | 64 | 14% | na |
| P34_S1 | FFPE | 76 | KRAS | p.L56fs | NGS; FISH | Pipeline | 7 | 33 | 21% | na |
| P39 | FFPE | 65 | NRAS | p.G15fs | NGS; FISH | Pipeline | 5 | 22 | 23% | na |
| P40 | FFPE | 68 | BRAF | p.Y472fs | NGS; FISH | Pipeline | 6 | 13 | 46% | na |
| Sample ID | Origin | DV200 | MD Variant | Results of All-In-One Transcriptome-Based Assay | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | IHC | FISH | Fusion Transcript | IGV (Splitting Reads in Gene 1, Splitting Reads in Gene 2) | Fusion Catcher (Spanning, Splitting Reads) | Strand NGS (Splitting Reads) | Status | |||
| H2228 | cell line | nd | ALK | nd | nd | EML4_E6-ALK_E20 | 104, 59 | 41, 107 | 84 | confirmed |
| P07 | FFPE | 65 | ALK | + | + | not confirmed | ||||
| P08 | FFPE | 67 | ALK | + | + | KIF5B_E24-ALK_E20 | 5, 9 | confirmed | ||
| P13 | PE | 89 | ALK | + | nd | EML4_E6-ALK_E20 | 83, 238 | 58, 185 | 170 | confirmed |
| P14 | PE | 86 | ALK | + | nd | DCTN1_E26-ALK_E20 | 76, 21 | 20, 41 | 51 | confirmed |
| P18 | Frozen | 86 | ALK | + | + | EML4_E6-ALK_E20 | 230, 143 | 74, 290 | 789 | confirmed |
| P33 | FFPE | 58 | ALK | + | + | EML4_E6-ALK_E20 | 6, 4 | confirmed | ||
| P34_S1 | Frozen | 82 | ALK | + | + | EML4_E6-ALK_E20 | 62, 41 | 44, 156 | 77 | confirmed |
| P34_S1 | FFPE | 76 | ALK | + | + | EML4_E6-ALK_E20 | 7, 3 | 2, 3 | 2 | confirmed |
| P34_S2 | FFPE | 70 | ALK | + | + | EML4_E6-ALK_E20 | 38, 17 | 10, 3 | 2 | confirmed |
| P36 | PE | 93 | ALK | + | + | EML4_E6-ALK_E18 | 49, 105 | 35, 156 | 86 | confirmed |
| P42 | PE | 53 | ALK | + | + | MPRIP_E21-ALK_E20 | 8, 20 | 5, 26 | 27 | confirmed |
| KM12 | cell line | 94 | NTRK1 | nd | nd | TPM3_E7-NTRK1_E9 | 188, 87 | 41, 153 | 340 | confirmed |
| P08 | FFPE | 67 | RET | nd | + | true negative | ||||
| P11 | FFPE | 81 | RET | nd | + | KIF5B_E15-RET_E12 | 2, 3 | confirmed | ||
| P37 | PE | 21 | ROS1 | nd | + | CD74_E6-ROS1_E34 | 0, 3 | 2, 3 | 1 | confirmed |
| P38 | FFPE | 55 | ROS1 | nd | + | EZR_E10-ROS1_E34 | 11, 2 | 4, 3 | confirmed | |
| P41 | FFPE | nd | ROS1 | nd | + | EZR_E10-ROS1_E34 | 19, 0 | 10, 9 | confirmed | |
| Variant Type | All Variants (Sensitivity) | Variants in Samples with DV200 >30 and Unique Read Count >50 K (Sensitivity) |
|---|---|---|
| SNVs/INDELs | 32/39 (82%) | 19/19 (100%) |
| MET exon skipping | 3/3 (100%) | 3/3 (100%) |
| Fusions | 17/18 (94%) | 13/13 (100%) |
| Overall | 52/60 (87%) | 35/35 (100%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, J.; Rybczynska, A.A.; Meng, P.; Terpstra, M.; Saber, A.; Sietzema, J.; Timens, W.; Schuuring, E.; Hiltermann, T.J.N.; Groen, H.J.M.; et al. An All-In-One Transcriptome-Based Assay to Identify Therapy-Guiding Genomic Aberrations in Nonsmall Cell Lung Cancer Patients. Cancers 2020, 12, 2843. https://doi.org/10.3390/cancers12102843
Wei J, Rybczynska AA, Meng P, Terpstra M, Saber A, Sietzema J, Timens W, Schuuring E, Hiltermann TJN, Groen HJM, et al. An All-In-One Transcriptome-Based Assay to Identify Therapy-Guiding Genomic Aberrations in Nonsmall Cell Lung Cancer Patients. Cancers. 2020; 12(10):2843. https://doi.org/10.3390/cancers12102843
Chicago/Turabian StyleWei, Jiacong, Anna A. Rybczynska, Pei Meng, Martijn Terpstra, Ali Saber, Jantine Sietzema, Wim Timens, Ed Schuuring, T. Jeroen N. Hiltermann, Harry. J.M. Groen, and et al. 2020. "An All-In-One Transcriptome-Based Assay to Identify Therapy-Guiding Genomic Aberrations in Nonsmall Cell Lung Cancer Patients" Cancers 12, no. 10: 2843. https://doi.org/10.3390/cancers12102843
APA StyleWei, J., Rybczynska, A. A., Meng, P., Terpstra, M., Saber, A., Sietzema, J., Timens, W., Schuuring, E., Hiltermann, T. J. N., Groen, H. J. M., van der Wekken, A. J., van den Berg, A., & Kok, K. (2020). An All-In-One Transcriptome-Based Assay to Identify Therapy-Guiding Genomic Aberrations in Nonsmall Cell Lung Cancer Patients. Cancers, 12(10), 2843. https://doi.org/10.3390/cancers12102843