Novel Quinoline Compounds Active in Cancer Cells through Coupled DNA Methyltransferase Inhibition and Degradation

 ,
,  , ,
, ,  , , , , , , ,
, , , , , , ,  ,
,  ,
,  , and
, and
Abstract
:
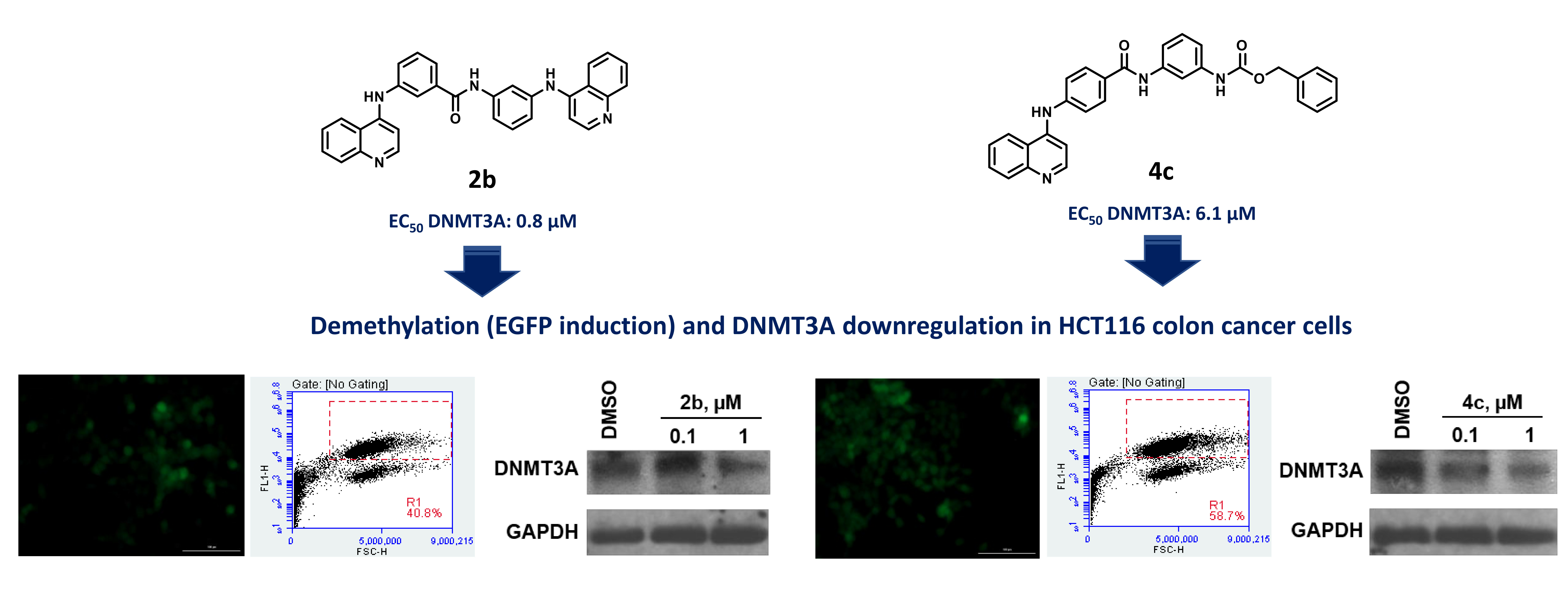
1. Introduction
2. Results
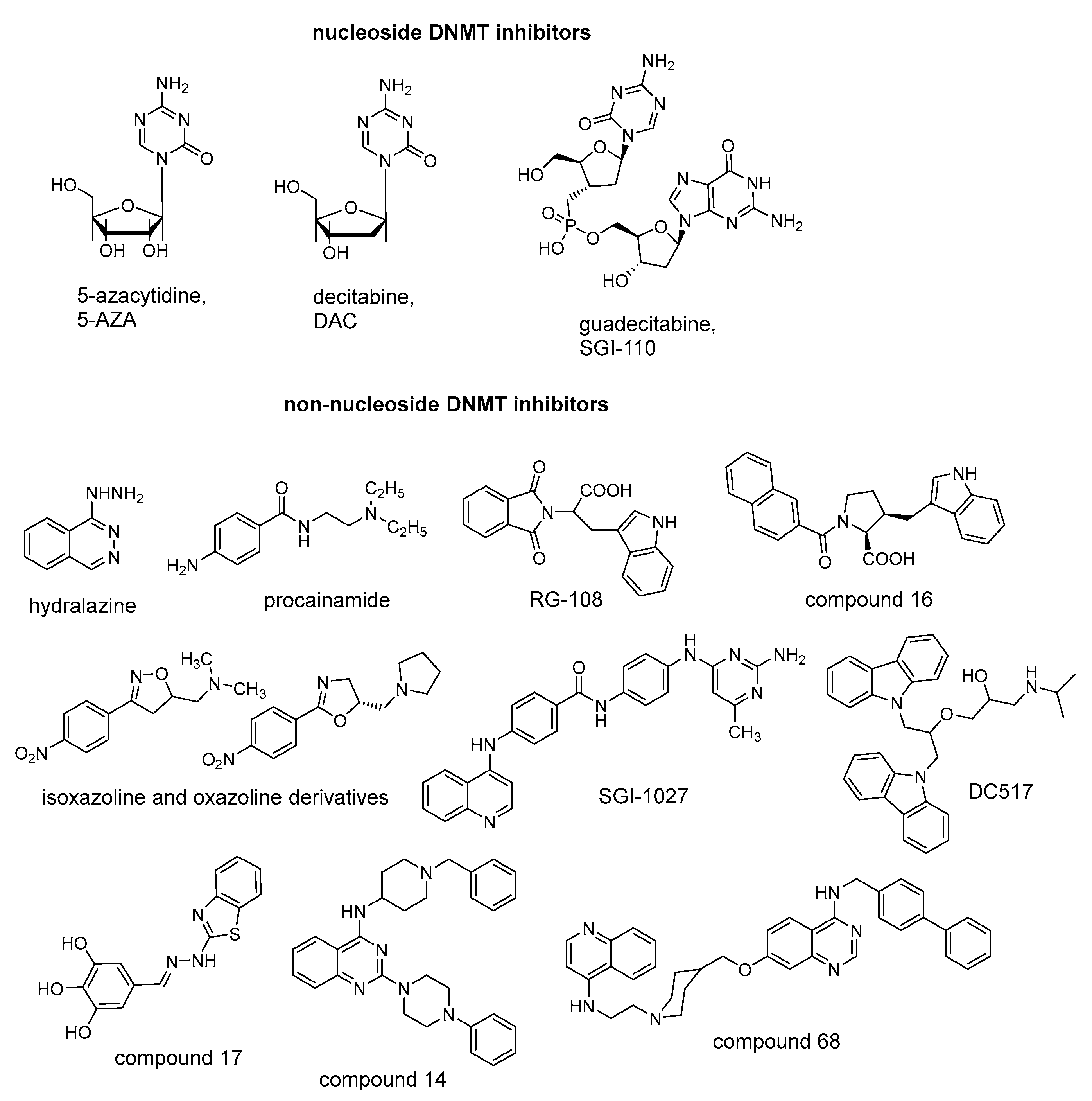
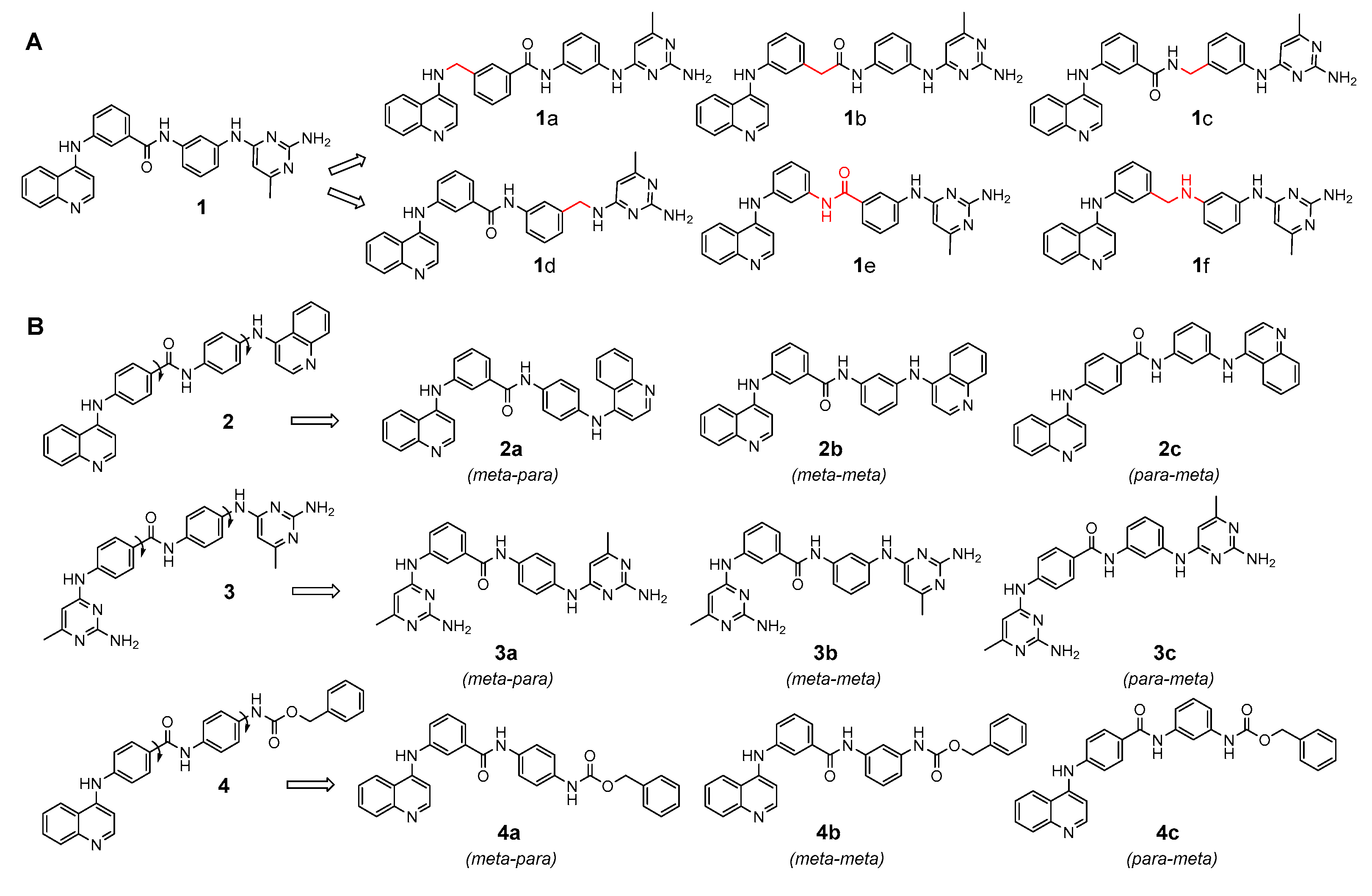
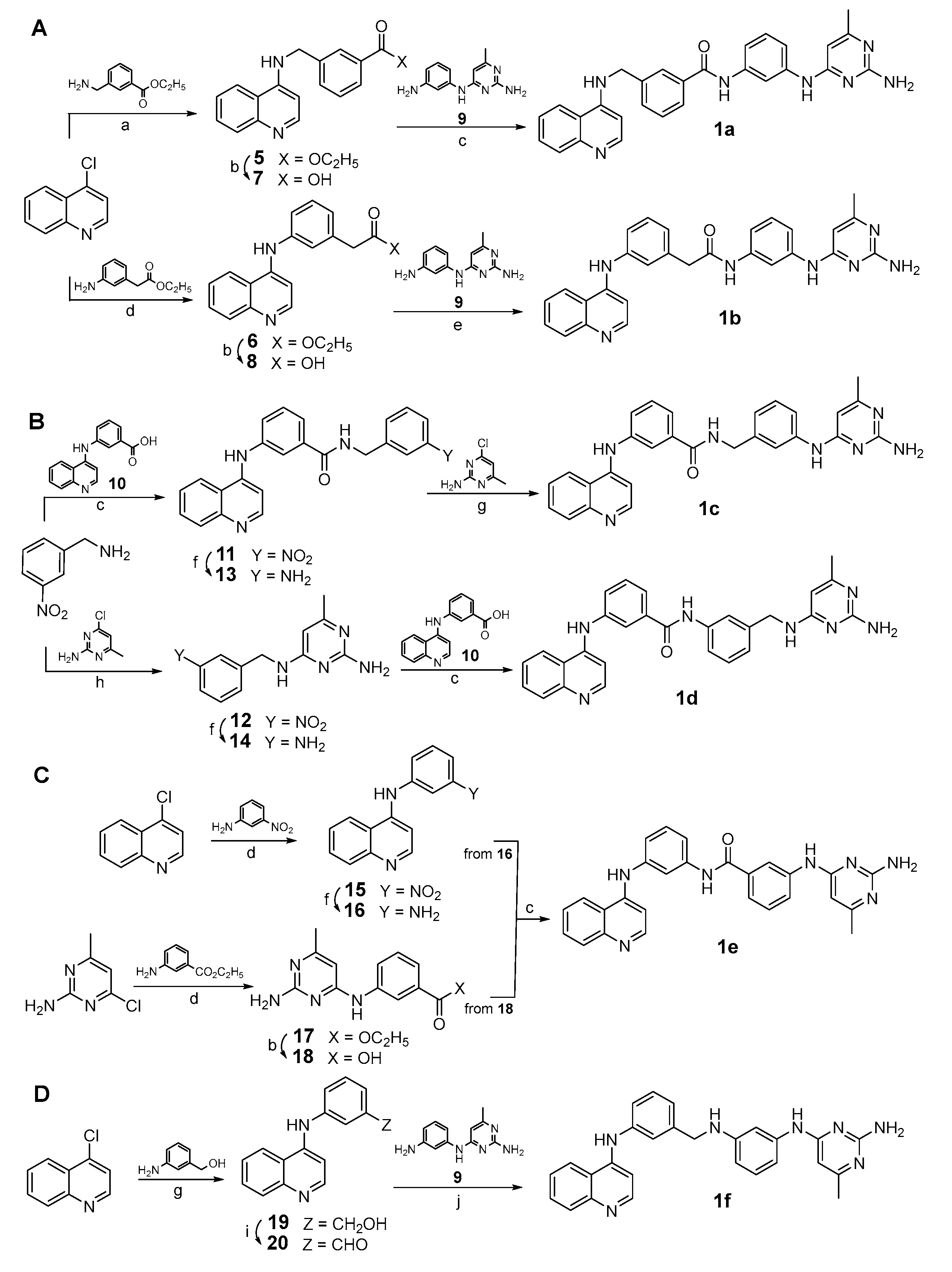
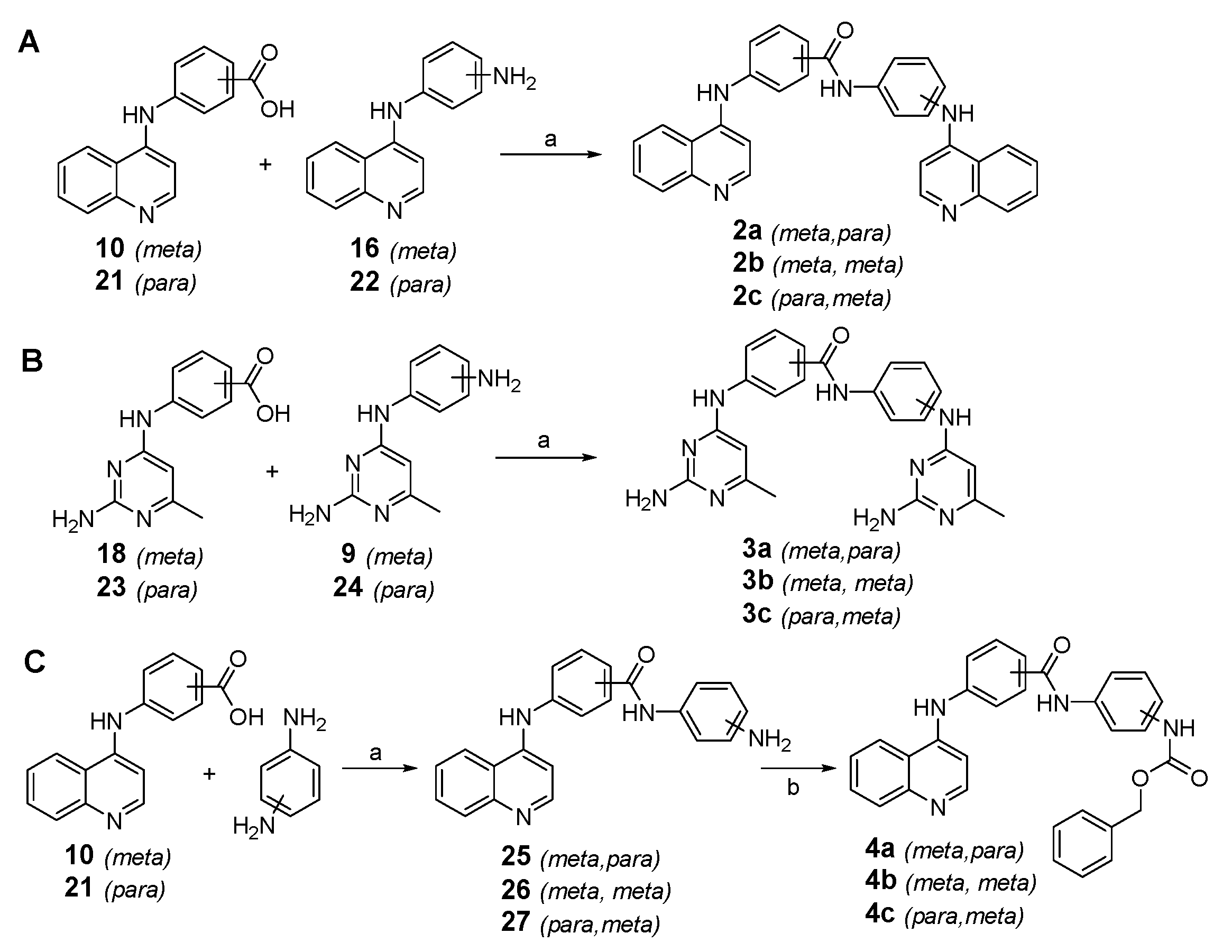
2.1. Design and Synthesis of Quinoline-Based DNMTi
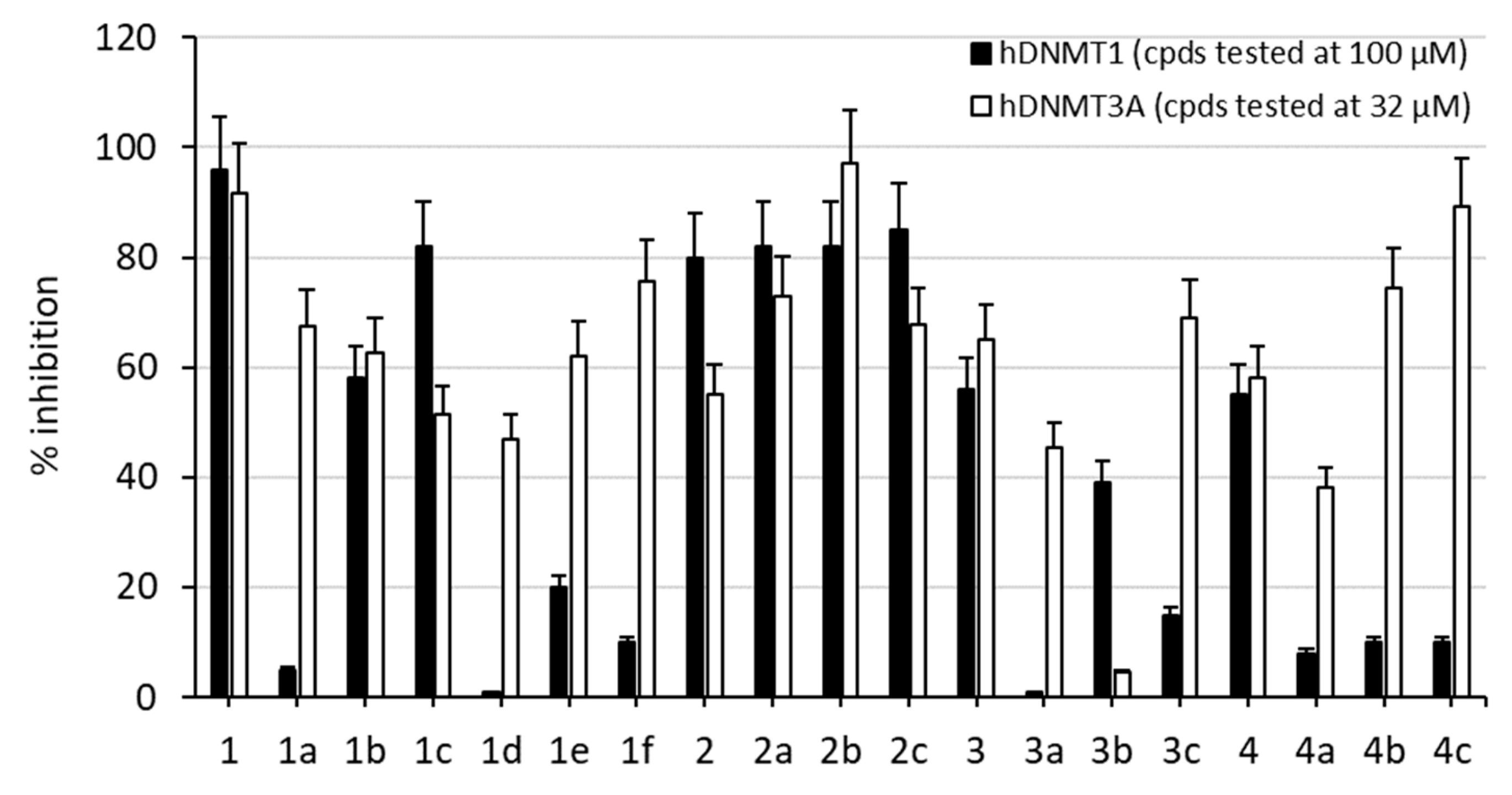
2.2. Enzyme Assays and Antiproliferative Activities Against U937 AML and HL60 APL Cells
2.3. Target Modulation by 2a, 4a, and 4c in KG-1 Leukemia Cells
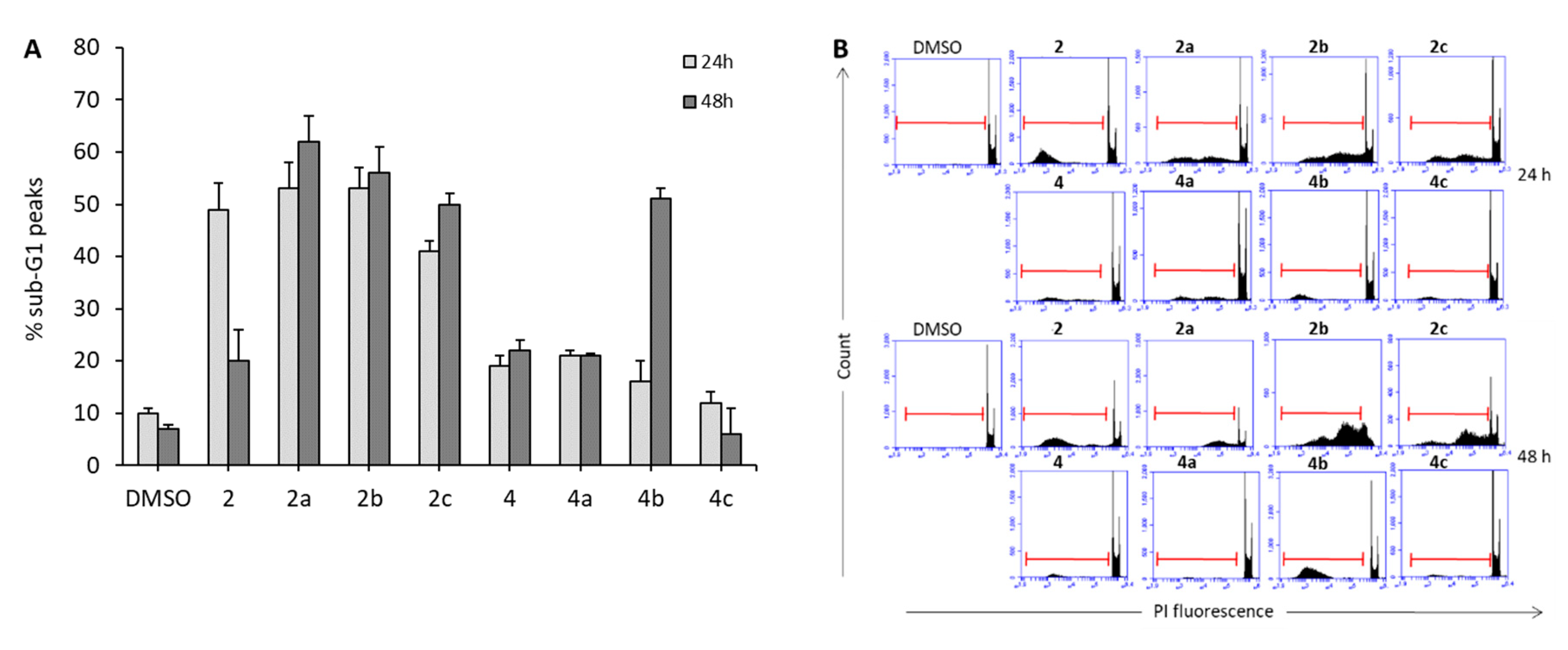
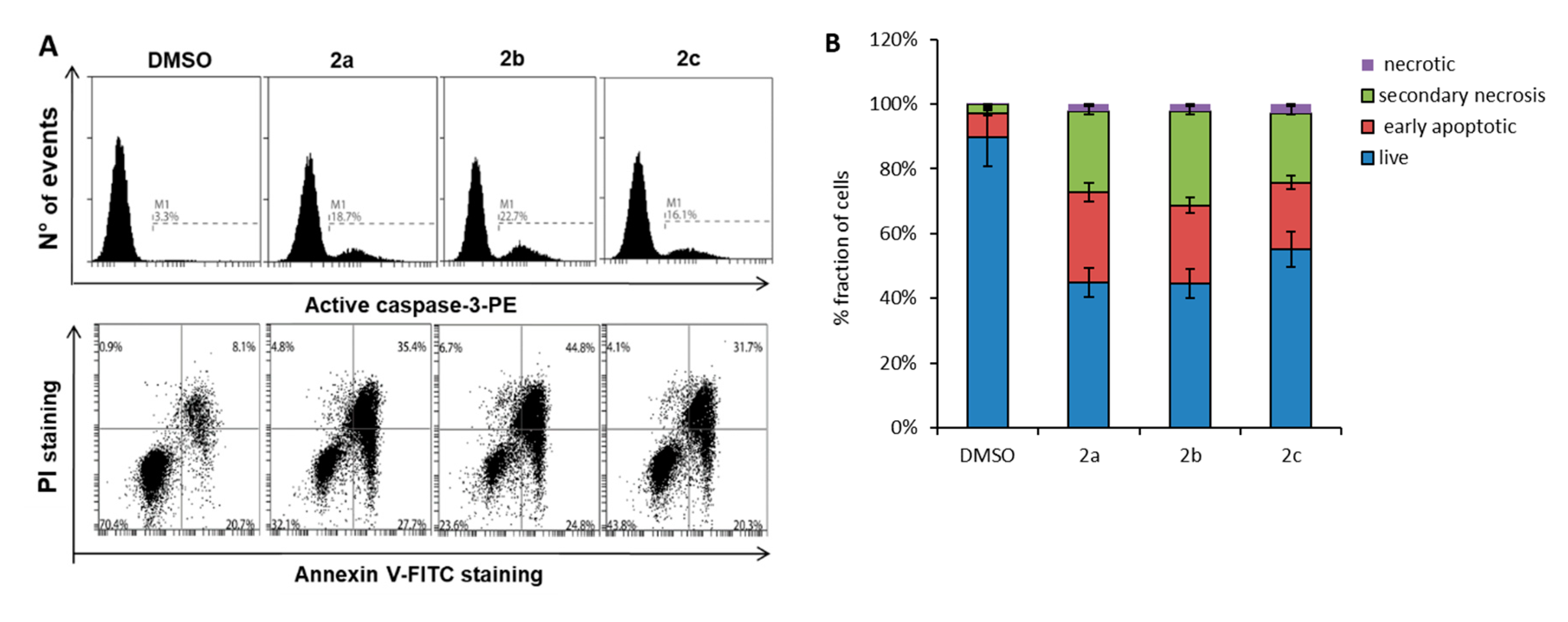
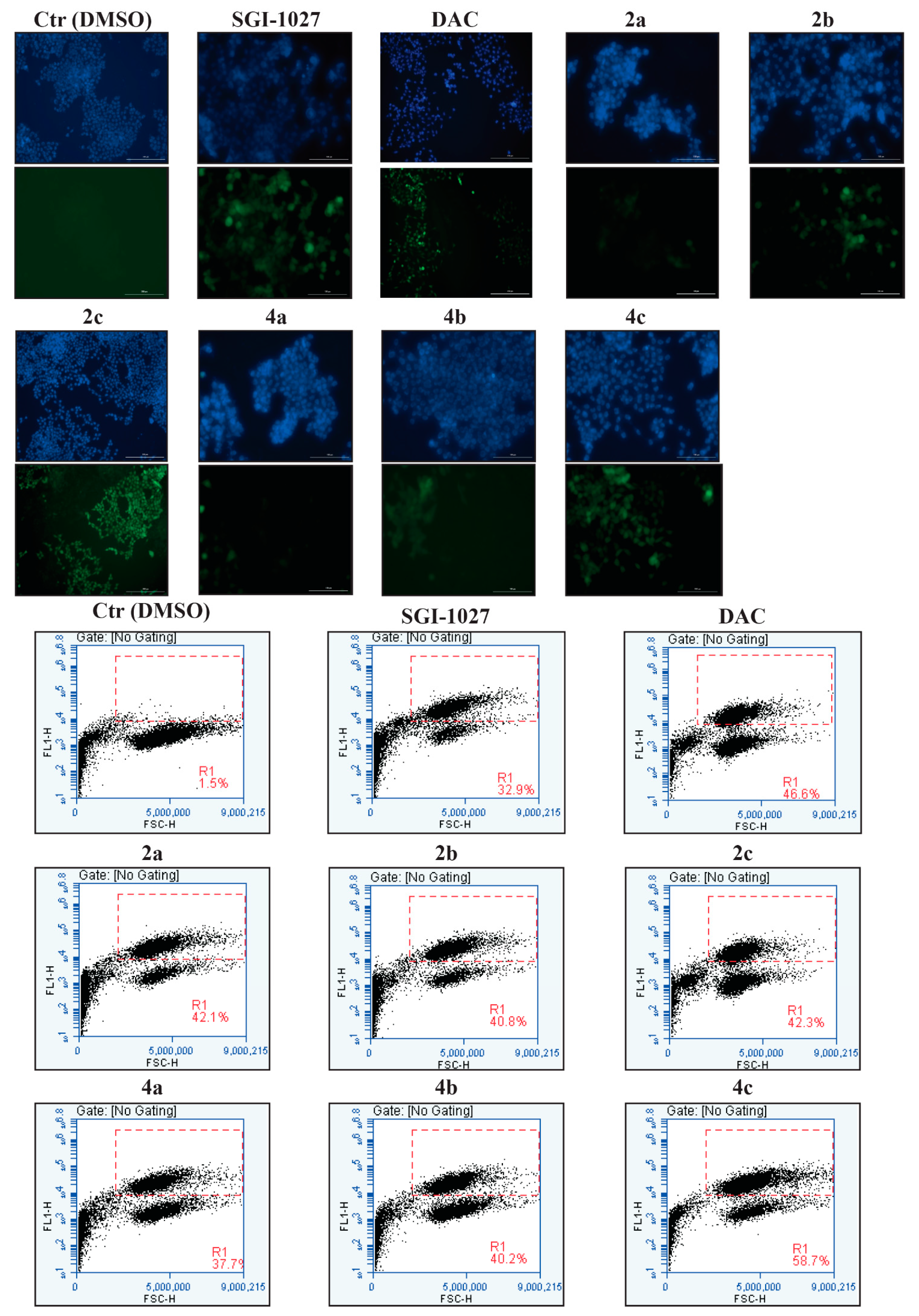
2.4. Compounds 2a–c and 4b Induce Apoptosis in U937 Leukemia Cells
2.5. Effects of 2a–c and 4a–c on Solid Cancer Cell Proliferation
2.6. Target Modulation by 2a–c and 4a–c in HCT116 Cells
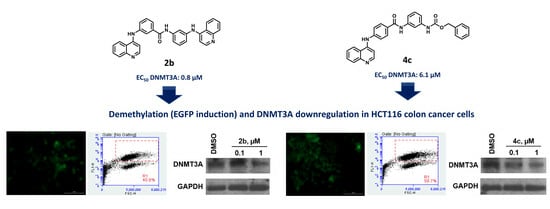
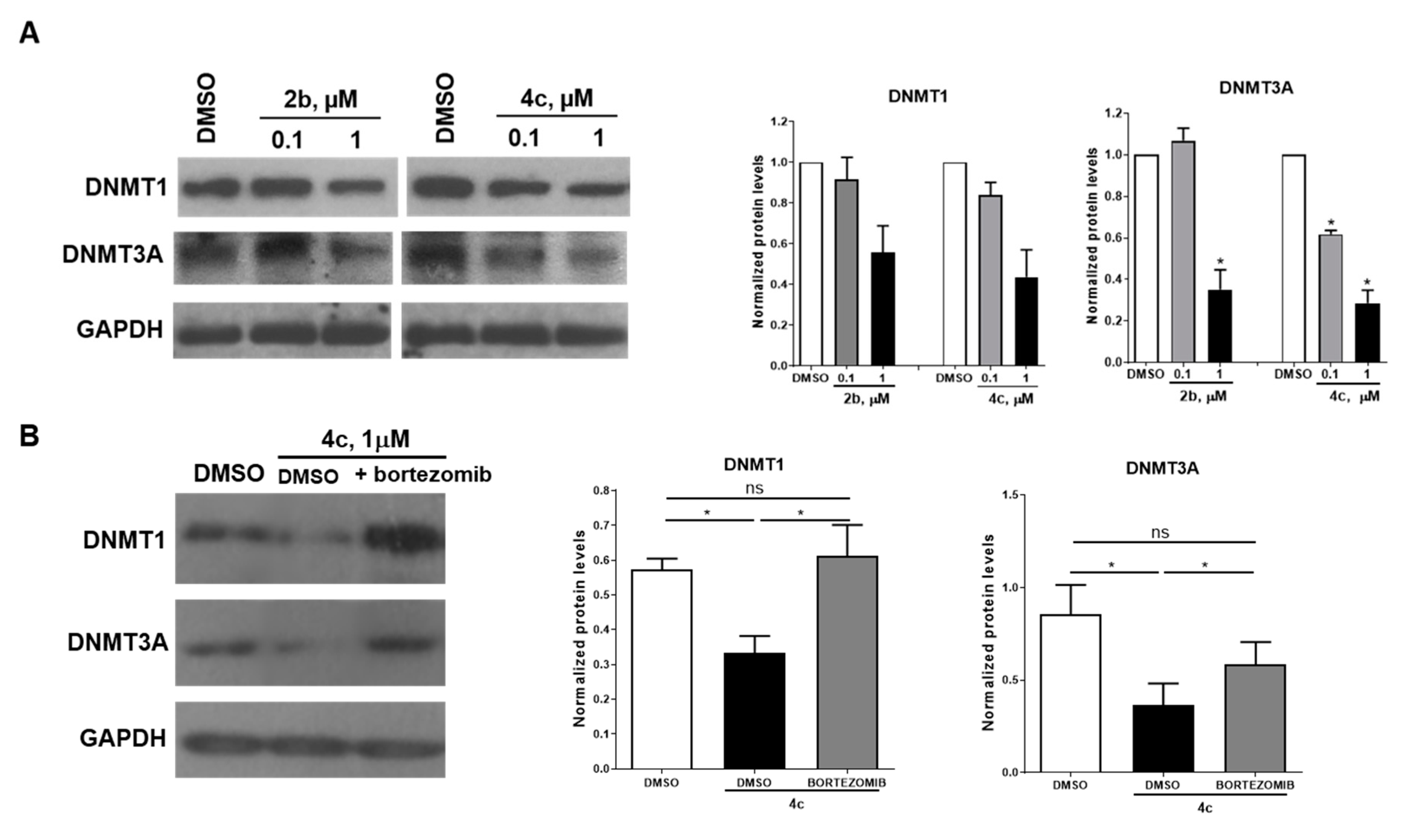
2.7. Selected Compounds 2b and 4c Induce a Downregulation of DNMT Proteins
2.8. Off-Target Effects: Potential DNA G-Quadruplex (G4) Stabilization and Kinase Inhibition
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.2. DNA Methyltransferase Assays
4.2.1. DNMT1 Assay
4.2.2. DNMT3A Assay
4.3. CMV-luc Assay in KG-1 Cells
4.4. UCHL1 Promoter Demethylation Assay (Luciferase Activity Induction) in HCT116 Colon Cancer Cells
4.5. Antiproliferative Assays
4.5.1. Cell Lines and Culture Conditions
4.5.2. Cell Proliferation Experiments
4.5.3. IC50 Values Determination and Statistical Analyses
4.6. Flow Cytometric Analysis of Cell Cycle and Apoptosis
4.7. Western Blot Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liu, G.; Zhou, F.; Su, B.; Li, Y. DNA methylation profiles in cancer diagnosis and therapeutics. Clin. Exp. Med. 2018, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation, methyltransferases, and cancer. Oncogene 2001, 20, 3139–3155. [Google Scholar] [CrossRef]
- Zwergel, C.; Valente, S.; Mai, A. DNA Methyltransferases Inhibitors from Natural Sources. Curr. Top. Med. Chem. 2016, 16, 680–696. [Google Scholar] [CrossRef]
- Gros, C.; Fahy, J.; Halby, L.; Dufau, I.; Erdmann, A.; Gregoire, J.M.; Ausseil, F.; Vispe, S.; Arimondo, P.B. DNA methylation inhibitors in cancer: Recent and future approaches. Biochimie 2012, 94, 2280–2296. [Google Scholar] [CrossRef]
- Hervouet, E.; Vallette, F.M.; Cartron, P.F. Dnmt1/Transcription factor interactions: An alternative mechanism of DNA methylation inheritance. Genes. Cancer 2010, 1, 434–443. [Google Scholar] [CrossRef]
- Wang, J.; Hevi, S.; Kurash, J.K.; Lei, H.; Gay, F.; Bajko, J.; Su, H.; Sun, W.; Chang, H.; Xu, G.; et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2009, 41, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Daniel, F.I.; Cherubini, K.; Yurgel, L.S.; de Figueiredo, M.A.; Salum, F.G. The role of epigenetic transcription repression and DNA methyltransferases in cancer. Cancer 2011, 117, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Pechalrieu, D.; Etievant, C.; Arimondo, P.B. DNA methyltransferase inhibitors in cancer: From pharmacology to translational studies. Biochem. Pharmacol. 2017, 129, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.B.; Jeong, S.; Egger, G.; Liang, G.; Phiasivongsa, P.; Tang, C.; Redkar, S.; Jones, P.A. Delivery of 5-aza-2’-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007, 67, 6400–6408. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Search Term: DNA Methyltransferase AND Inhibition. Available online: www.clinicaltrials.gov (accessed on 26 June 2019).
- Castillo-Aguilera, O.; Depreux, P.; Halby, L.; Arimondo, P.B.; Goossens, L. DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge. Biomolecules 2017, 7, 3. [Google Scholar] [CrossRef]
- Brueckner, B.; Garcia Boy, R.; Siedlecki, P.; Musch, T.; Kliem, H.C.; Zielenkiewicz, P.; Suhai, S.; Wiessler, M.; Lyko, F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005, 65, 6305–6311. [Google Scholar] [CrossRef]
- Asgatay, S.; Champion, C.; Marloie, G.; Drujon, T.; Senamaud-Beaufort, C.; Ceccaldi, A.; Erdmann, A.; Rajavelu, A.; Schambel, P.; Jeltsch, A.; et al. Synthesis and evaluation of analogues of N-phthaloyl-l-tryptophan (RG108) as inhibitors of DNA methyltransferase 1. J. Med. Chem 2014, 57, 421–434. [Google Scholar] [CrossRef]
- Castellano, S.; Kuck, D.; Viviano, M.; Yoo, J.; Lopez-Vallejo, F.; Conti, P.; Tamborini, L.; Pinto, A.; Medina-Franco, J.L.; Sbardella, G. Synthesis and biochemical evaluation of delta(2)-isoxazoline derivatives as DNA methyltransferase 1 inhibitors. J. Med. Chem. 2011, 54, 7663–7677. [Google Scholar] [CrossRef]
- Castellano, S.; Kuck, D.; Sala, M.; Novellino, E.; Lyko, F.; Sbardella, G. Constrained analogues of procaine as novel small molecule inhibitors of DNA methyltransferase-1. J. Med. Chem. 2008, 51, 2321–2325. [Google Scholar] [CrossRef]
- Datta, J.; Ghoshal, K.; Denny, W.A.; Gamage, S.A.; Brooke, D.G.; Phiasivongsa, P.; Redkar, S.; Jacob, S.T. A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res. 2009, 69, 4277–4285. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Y.; Zhou, W.; Li, S.; Peng, J.; Shi, Z.; Hu, J.; Liu, Y.C.; Ding, H.; Lin, Y.; et al. Identifying novel selective non-nucleoside DNA methyltransferase 1 inhibitors through docking-based virtual screening. J. Med. Chem. 2014, 57, 9028–9041. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, A.; Menon, Y.; Gros, C.; Masson, V.; Aussagues, Y.; Ausseil, F.; Novosad, N.; Schambel, P.; Baltas, M.; Arimondo, P.B. Identification and optimization of hydrazone-gallate derivatives as specific inhibitors of DNA methyltransferase 3A. Future Med. Chem. 2016, 8, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Rotili, D.; Tarantino, D.; Marrocco, B.; Gros, C.; Masson, V.; Poughon, V.; Ausseil, F.; Chang, Y.; Labella, D.; Cosconati, S.; et al. Properly substituted analogues of BIX-01294 lose inhibition of G9a histone methyltransferase and gain selective anti-DNA methyltransferase 3A activity. PLoS ONE 2014, 9, e96941. [Google Scholar] [CrossRef] [PubMed]
- Halby, L.; Menon, Y.; Rilova, E.; Pechalrieu, D.; Masson, V.; Faux, C.; Bouhlel, M.A.; David-Cordonnier, M.H.; Novosad, N.; Aussagues, Y.; et al. Rational Design of Bisubstrate-Type Analogues as Inhibitors of DNA Methyltransferases in Cancer Cells. J. Med. Chem. 2017, 60, 4665–4679. [Google Scholar] [CrossRef] [PubMed]
- Valente, S.; Liu, Y.; Schnekenburger, M.; Zwergel, C.; Cosconati, S.; Gros, C.; Tardugno, M.; Labella, D.; Florean, C.; Minden, S.; et al. Selective non-nucleoside inhibitors of human DNA methyltransferases active in cancer including in cancer stem cells. J. Med. Chem. 2014, 57, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Gros, C.; Fleury, L.; Nahoum, V.; Faux, C.; Valente, S.; Labella, D.; Cantagrel, F.; Rilova, E.; Bouhlel, M.A.; David-Cordonnier, M.H.; et al. New insights on the mechanism of quinoline-based DNA Methyltransferase inhibitors. J. Biol. Chem. 2015, 290, 6293–6302. [Google Scholar] [CrossRef]
- Manara, M.C.; Valente, S.; Cristalli, C.; Nicoletti, G.; Landuzzi, L.; Zwergel, C.; Mazzone, R.; Stazi, G.; Arimondo, P.B.; Pasello, M.; et al. A Quinoline-Based DNA Methyltransferase Inhibitor as a Possible Adjuvant in Osteosarcoma Therapy. Mol. Cancer Ther. 2018, 17, 1881–1892. [Google Scholar] [CrossRef]
- Zwergel, C.; Schnekenburger, M.; Sarno, F.; Battistelli, C.; Manara, M.C.; Stazi, G.; Mazzone, R.; Fioravanti, R.; Gros, C.; Ausseil, F.; et al. Identification of a novel quinoline-based DNA demethylating compound highly potent in cancer cells. Clin. Epigenetics 2019, 11, 68. [Google Scholar] [CrossRef]
- Okochi-Takada, E.; Hattori, N.; Ito, A.; Niwa, T.; Wakabayashi, M.; Kimura, K.; Yoshida, M.; Ushijima, T. Establishment of a high-throughput detection system for DNA demethylating agents. Epigenetics 2018, 13, 147–155. [Google Scholar] [CrossRef]
- Shirude, P.S.; Gillies, E.R.; Ladame, S.; Godde, F.; Shin-Ya, K.; Huc, I.; Balasubramanian, S. Macrocyclic and helical oligoamides as a new class of G-quadruplex ligands. J. Am. Chem. Soc. 2007, 129, 11890–11891. [Google Scholar] [CrossRef]
- Brito, H.; Martins, A.C.; Lavrado, J.; Mendes, E.; Francisco, A.P.; Santos, S.A.; Ohnmacht, S.A.; Kim, N.S.; Rodrigues, C.M.; Moreira, R.; et al. Targeting KRAS Oncogene in Colon Cancer Cells with 7-Carboxylate Indolo [3,2-b]quinoline Tri-Alkylamine Derivatives. PLoS ONE 2015, 10, e0126891. [Google Scholar] [CrossRef] [PubMed]
- Lavrado, J.; Brito, H.; Borralho, P.M.; Ohnmacht, S.A.; Kim, N.S.; Leitao, C.; Pisco, S.; Gunaratnam, M.; Rodrigues, C.M.; Moreira, R.; et al. KRAS oncogene repression in colon cancer cell lines by G-quadruplex binding indolo[3,2-c]quinolines. Sci. Rep. 2015, 5, 9696. [Google Scholar] [CrossRef]
- Zeng, D.Y.; Kuang, G.T.; Wang, S.K.; Peng, W.; Lin, S.L.; Zhang, Q.; Su, X.X.; Hu, M.H.; Wang, H.; Tan, J.H.; et al. Discovery of Novel 11-Triazole Substituted Benzofuro[3,2-b]quinolone Derivatives as c-myc G-Quadruplex Specific Stabilizers via Click Chemistry. J. Med. Chem. 2017, 60, 5407–5423. [Google Scholar] [CrossRef]
- Cogoi, S.; Xodo, L.E. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006, 34, 2536–2549. [Google Scholar] [CrossRef] [PubMed]
- Musiol, R. An overview of quinoline as a privileged scaffold in cancer drug discovery. Expert Opin. Drug Discov. 2017, 12, 583–597. [Google Scholar] [CrossRef]
- Hsieh, M.Y.; Van Etten, R.A. IKK-dependent activation of NF-kappaB contributes to myeloid and lymphoid leukemogenesis by BCR-ABL1. Blood 2014, 123, 2401–2411. [Google Scholar] [CrossRef] [PubMed]
- Saiya-Cork, K.; Collins, R.; Parkin, B.; Ouillette, P.; Kuizon, E.; Kujawski, L.; Erba, H.; Campagnaro, E.; Shedden, K.; Kaminski, M.; et al. A pathobiological role of the insulin receptor in chronic lymphocytic leukemia. Clin. Cancer Res. 2011, 17, 2679–2692. [Google Scholar] [CrossRef]
- Green, L.J.; Marder, P.; Ray, C.; Cook, C.A.; Jaken, S.; Musib, L.C.; Herbst, R.S.; Carducci, M.; Britten, C.D.; Basche, M.; et al. Development and validation of a drug activity biomarker that shows target inhibition in cancer patients receiving enzastaurin, a novel protein kinase C-beta inhibitor. Clin. Cancer Res. 2006, 12, 3408–3415. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi, F.; Zhang, J.; Harrison, J.S.; Wang, X.; Studzinski, G.P. Induction of differentiation of human leukemia cells by combinations of COX inhibitors and 1,25-dihydroxyvitamin D3 involves Raf1 but not Erk 1/2 signaling. Cell Cycle 2008, 7, 917–924. [Google Scholar] [CrossRef]
- Jensen, H.A.; Styskal, L.E.; Tasseff, R.; Bunaciu, R.P.; Congleton, J.; Varner, J.D.; Yen, A. The Src-family kinase inhibitor PP2 rescues inducible differentiation events in emergent retinoic acid-resistant myeloblastic leukemia cells. PLoS ONE 2013, 8, e58621. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, G.M.; Choi, Y.J.; Kim, H.J.; Kim, Y.J.; Jin, W. c-Src activation through a TrkA and c-Src interaction is essential for cell proliferation and hematological malignancies. Biochem. Biophys. Res. Commun. 2013, 441, 431–437. [Google Scholar] [CrossRef]
- Lee, B.H.; Yegnasubramanian, S.; Lin, X.; Nelson, W.G. Procainamide is a specific inhibitor of DNA methyltransferase 1. J. Biol. Chem. 2005, 280, 40749–40756. [Google Scholar] [CrossRef]
- Gros, C.; Chauvigne, L.; Poulet, A.; Menon, Y.; Ausseil, F.; Dufau, I.; Arimondo, P.B. Development of a universal radioactive DNA methyltransferase inhibition test for high-throughput screening and mechanistic studies. Nucleic Acids Res. 2013, 41, e185. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, A.; Rajavelu, A.; Champion, C.; Rampon, C.; Jurkowska, R.; Jankevicius, G.; Senamaud-Beaufort, C.; Ponger, L.; Gagey, N.; Ali, H.D.; et al. C5-DNA methyltransferase inhibitors: From screening to effects on zebrafish embryo development. Chembiochem 2011, 12, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Rilova, E.; Erdmann, A.; Gros, C.; Masson, V.; Aussagues, Y.; Poughon-Cassabois, V.; Rajavelu, A.; Jeltsch, A.; Menon, Y.; Novosad, N.; et al. Design, synthesis and biological evaluation of 4-amino-N- (4-aminophenyl)benzamide analogues of quinoline-based SGI-1027 as inhibitors of DNA methylation. ChemMedChem 2014, 9, 590–601. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | Enzyme Inhibition (EC50 ± SD, µM) 1 | Antiproliferative Activity (IC50 ± SD, µM) 2 | ||
|---|---|---|---|---|
| hDNMT1 | hDNMT3A | U937 | HL60 | |
| SGI-1027 3 | 10 ± 1 | 0.8 ± 0.1 | 0.70 ± 0.08 | 0.70 ± 0.16 |
| 13 | 5.7 ± 0.2 | 0.7 ± 0.5 | 6.90 ± 0.8 | 5.90 ± 0.7 |
| 1a | ND 4 | 21.0 ± 3.1 | 1.40 ± 0.2 | 18.1 ± 2.4 |
| 1b | ND | 19.0 ± 2.0 | 37.0 ± 5.9 | 40.0 ± 5.0 |
| 1c | 35 ± 7 | 31.0 ± 6.0 | 25.8 ± 3.2 | 36.0 ± 4.0 |
| 1d | ND | 51.0 ± 10.0 | 67.0 ± 8.8 | 67.0 ± 7.4 |
| 1e | ND | 21.0 ± 4.0 | 4.8 ± 0.6 | 9.9 ± 1.1 |
| 1f | ND | 12.0 ± 3.0 | 4.4 ± 0.7 | 1.9 ± 0.1 |
| 25 | 33 ± 1 | 27.0 ± 5.0 | 6.6 ± 0.9 | 65.0 ± 8.1 |
| 2a | 23 ± 4 | 6.0 ± 1.0 | 0.70 ± 0.09 | 0.20 ± 0.03 |
| 2b | 27 ± 5 | 0.8 ± 0.1 | 1.5 ± 0.1 | 1.6 ± 0.2 |
| 2c | 22 ± 3 | 5.6 ± 1.0 | 1.5 ± 0.1 | 1.9 ± 0.3 |
| 35 | 68 ± 4 | 18.0 ± 2.0 | 60.0 ± 8.2 | 119.0 ± 12.9 |
| 3a | ND | 41.0 ± 10.0 | 52.4 ± 7.6 | 83.0 ± 10.2 |
| 3b | ND | >100 | 123.0 ± 15.0 | 225.0 ± 28.5 |
| 3c | ND | 13.0 ± 0.9 | 66.0 ± 9.2 | 125.0 ± 16.2 |
| 46 | 67 ± 4 | 17.0 ± 3.0 | 2.3 ± 0.4 | 8.3 ± 1.0 |
| 4a7 | ND | >32 | 0.50 ± 0.08 | 0.30 ± 0.06 |
| 4b | ND | 13.0 ± 1.4 | 3.2 ± 0.6 | 1.9 ± 0.3 |
| 4c | ND | 6.1 ± 1.0 | 1.2 ± 0.2 | 0.30 ± 0.07 |
| Compd | Luciferase Expression (Fold ± SD 1) | ||||
|---|---|---|---|---|---|
| 0.5 µM | 1 µM | 5 µM | 10 µM | 25 µM | |
| 2a | 1.02 ± 0.04 | 1.05 ± 0.12 | 19.94 ± 4.26 | 4.66 ± 1.63 | 1.59 ± 0.59 |
| 4a | 1.05 ± 0.12 | 1.02 ± 0.07 | 1.04 ± 0.16 | 1.45 ± 0.39 | 1.60 ± 1.00 |
| 4c | 0.96 ± 0.01 | 0.87 ± 0.05 | 40.40 ± 12.75 | 28.51 ± 3.39 | 8.07 ± 1.96 |
| SGI-1027 | 1.07 ± 0.16 | 1.16 ± 0.20 | 25.54 ± 5.77 | 6.00 ± 2.57 | 3.24 ± 1.66 |
| DAC | 18.13 ± 5.52 | 19.80 ± 3.88 | 23.82 ± 5.44 | 20.96 ± 4.02 | ND 2 |
| Compd | Antiproliferative Activity (IC50 ± SD, µM) 1 | |||||
|---|---|---|---|---|---|---|
| H1299 | HCT116 | HeLa | M14 | HT1080 | MCF-7 | |
| 2 | 11.4 ± 1.8 | 13.3 ± 2.1 | 52.0 ± 5.7 | 80.0 ± 9.1 | 17.0 ± 2.1 | ND 2 |
| 2a | 14.7 ± 2.3 | 1.0 ± 0.2 | 0.14 ± 0.04 | 0.50 ± 0.08 | 0.50 ± 0.09 | 4.1 ± 0.7 |
| 2b | 5.3 ± 0.9 | 1.5 ± 0.3 | 2.0 ± 0.3 | 4.7 ± 0.6 | 2.7 ± 0.4 | 0.30 ± 0.06 |
| 2c | 14.0 ± 1.8 | 2.3 ± 0.5 | 4.2 ± 0.8 | 3.8 ± 0.7 | 2.6 ± 0.6 | 2.2 ± 0.4 |
| 4 | 5.9 ± 0.8 | 8.6 ± 1.0 | 3.2 ± 0.5 | 2.9 ± 0.4 | 1.7 ± 0.2 | ND |
| 4a | 45.0 ± 4.1 | 23.9 ± 2.6 | 77.0 ± 7.9 | 69.0 ± 8.1 | 19.5 ± 2.8 | ND |
| 4b | 40.0 ± 4.8 | 20.5 ± 2.8 | 65.0 ± 7.3 | 15.3 ± 2.0 | 13.1 ± 1.5 | ND |
| 4c | 6.9 ± 0.9 | 17.7 ± 2.4 | 7.4 ± 1.2 | 4.5 ± 0.8 | 3.9 ± 0.8 | ND |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zwergel, C.; Fioravanti, R.; Stazi, G.; Sarno, F.; Battistelli, C.; Romanelli, A.; Nebbioso, A.; Mendes, E.; Paulo, A.; Strippoli, R.; et al. Novel Quinoline Compounds Active in Cancer Cells through Coupled DNA Methyltransferase Inhibition and Degradation. Cancers 2020, 12, 447. https://doi.org/10.3390/cancers12020447
Zwergel C, Fioravanti R, Stazi G, Sarno F, Battistelli C, Romanelli A, Nebbioso A, Mendes E, Paulo A, Strippoli R, et al. Novel Quinoline Compounds Active in Cancer Cells through Coupled DNA Methyltransferase Inhibition and Degradation. Cancers. 2020; 12(2):447. https://doi.org/10.3390/cancers12020447
Chicago/Turabian StyleZwergel, Clemens, Rossella Fioravanti, Giulia Stazi, Federica Sarno, Cecilia Battistelli, Annalisa Romanelli, Angela Nebbioso, Eduarda Mendes, Alexandra Paulo, Raffaele Strippoli, and et al. 2020. "Novel Quinoline Compounds Active in Cancer Cells through Coupled DNA Methyltransferase Inhibition and Degradation" Cancers 12, no. 2: 447. https://doi.org/10.3390/cancers12020447
APA StyleZwergel, C., Fioravanti, R., Stazi, G., Sarno, F., Battistelli, C., Romanelli, A., Nebbioso, A., Mendes, E., Paulo, A., Strippoli, R., Tripodi, M., Pechalrieu, D., Arimondo, P. B., De Luca, T., Del Bufalo, D., Trisciuoglio, D., Altucci, L., Valente, S., & Mai, A. (2020). Novel Quinoline Compounds Active in Cancer Cells through Coupled DNA Methyltransferase Inhibition and Degradation. Cancers, 12(2), 447. https://doi.org/10.3390/cancers12020447