The Role of p53-Mediated Signaling in the Therapeutic Response of Colorectal Cancer to 9F, a Spermine-Modified Naphthalene Diimide Derivative

 ,
,
Abstract
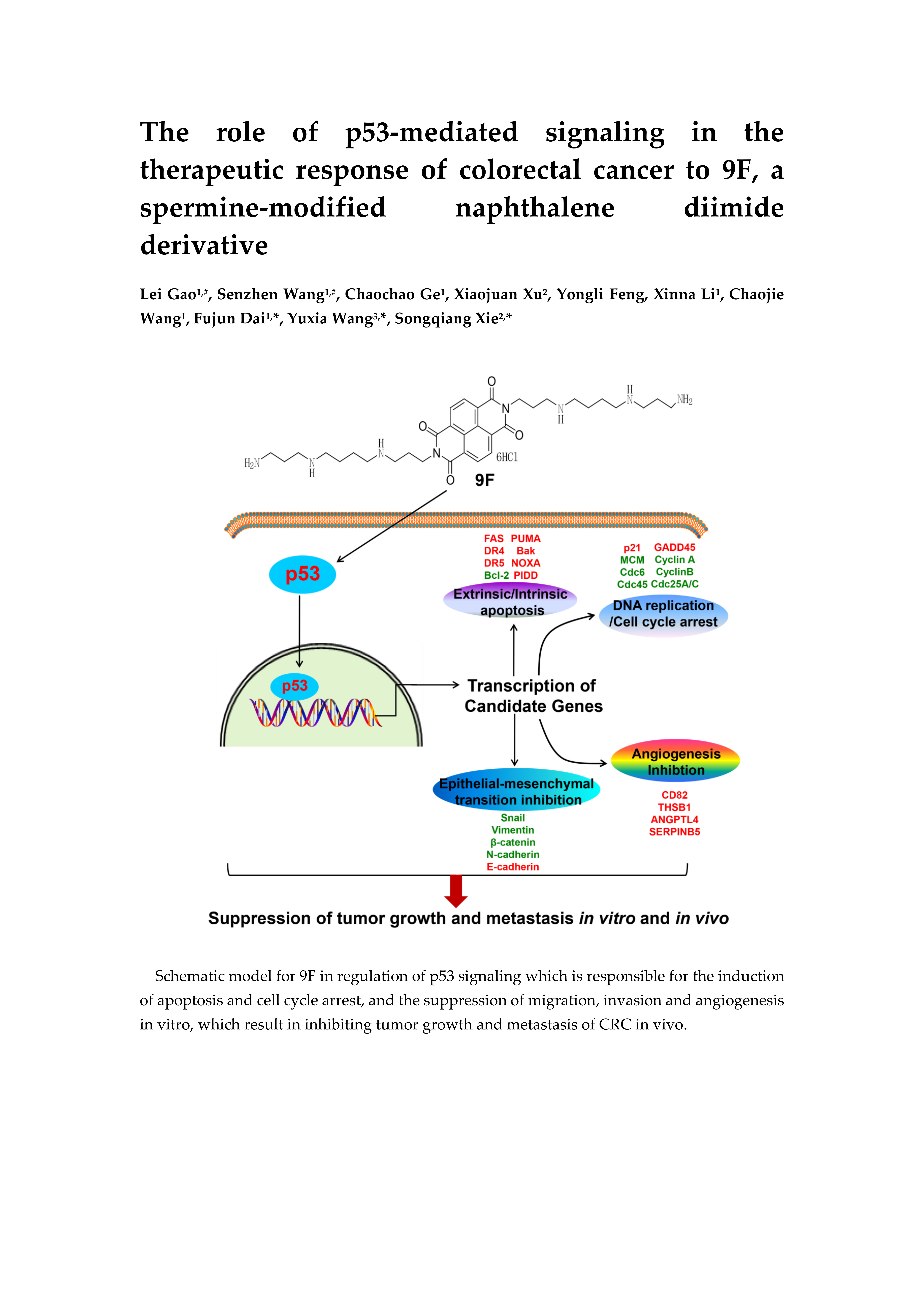
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. 9F Inhibits Cell Growth and Induces Cell Deathof CRC
2.2. Identification of p53-Mediated Pathway in 9F-Treated CRC Cells
2.3. 9F Induces Cell Cycle Arrest and Disrupts DNA Replication of CRCCells
2.4. Both the Mitochondria-Mediated Pathway and the Death Receptor-Induced Pathway Play an Important Role in 9F-Induced Apoptosis
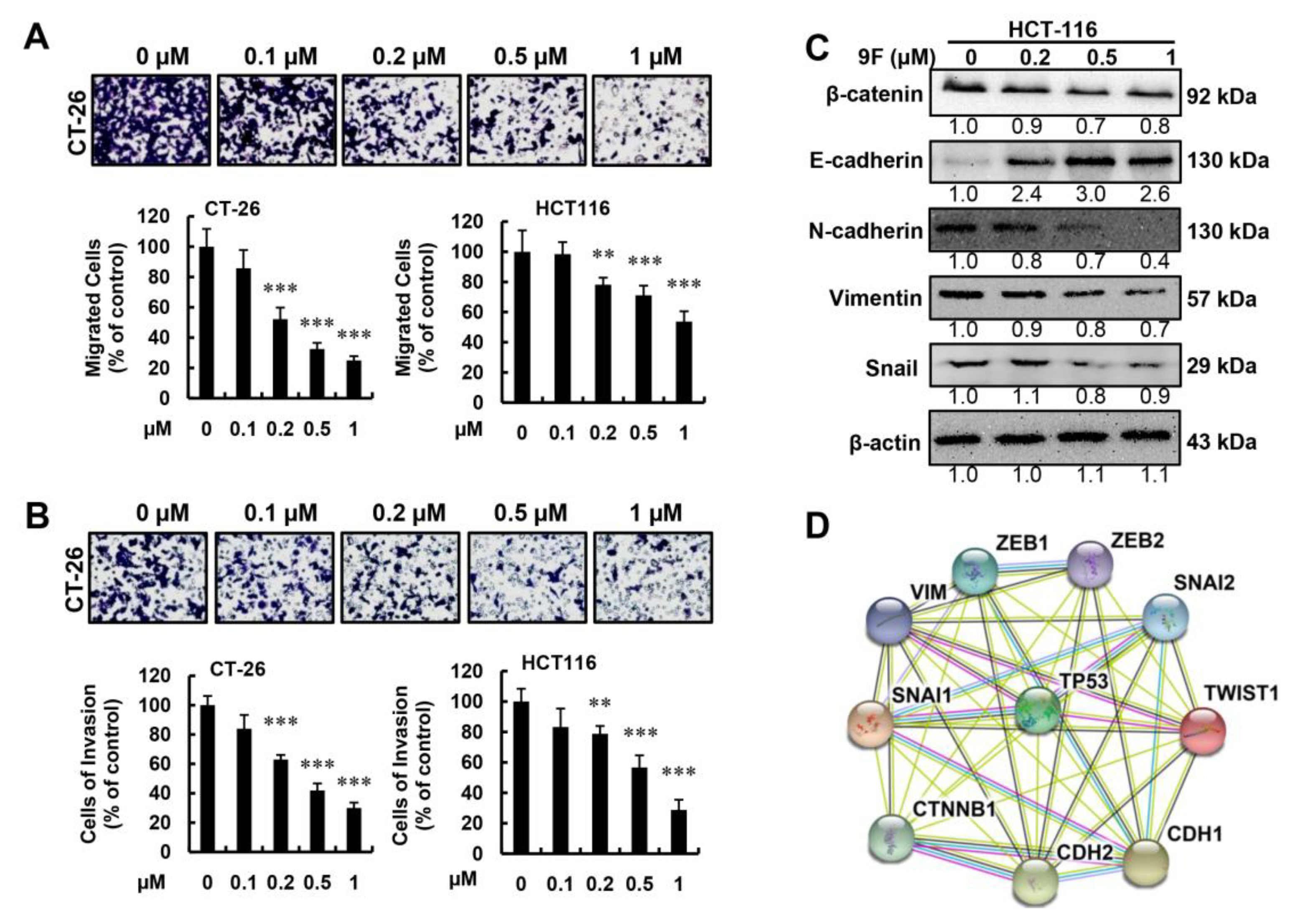
2.5. 9F Inhibits Migration and Invasion of CRC Cells
2.6. 9F Suppresses Proliferation, Migration and Tube Formation of Endothelial Cells
2.7. 9F Suppresses Tumor Growth and Lung Metastasis of CRC In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. MTT Assay
4.3. 2D Colony Formation Assay
4.4. Apoptosis Detection
4.5. Cell Cycle Assay
4.6. Immunofluorescence Assay
4.7. Western Bloting Assay
4.8. Boyden Chamber Migration and Invasion Assay
4.9. Angiogenesis Assay
4.10. Mice, Tumor Model and Treatment
4.11. Hematoxylin and Eosin Staining
4.12. RNA-Sequencing and Bioinformatics Analysis
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fang, Z.; Gong, C.; Yu, S.; Zhou, W.; Hassan, W.; Li, H.; Wang, X.; Hu, Y.; Gu, K.; Chen, X.; et al. NFYB-induced high expression of E2F1 contributes to oxaliplatin resistance in colorectal cancer via the enhancement of CHK1 signaling. Cancer Lett. 2018, 415, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.T.; Jiang, J.K.; Chang, S.C.; Yang, S.H.; Lin, C.C.; Lin, H.H.; Wang, H.S.; Chen, W.S.; Lin, T.C.; Lin, J.K. Improved outcomes of colorectal cancer patients with liver metastases in the era of the multidisciplinary teams. Int. J. Colorectal Dis. 2016, 31, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Ambrosi, G.; Wandmacher, A.M.; Rauscher, B.; Betge, J.; Rindtorff, N.; Haussler, R.S.; Hinsenkamp, I.; Bamberg, L.; Hessling, B.; et al. MEK inhibitors activate Wnt signalling and induce stem cell plasticity in colorectal cancer. Nat. Commun. 2019, 10, 2197. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Wei, L.; Yu, J.; Zhang, L. Regorafenib Inhibits Colorectal Tumor Growth through PUMA-Mediated Apoptosis. Clin. Cancer Res. 2014, 20, 3472–3484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.Y.; Kana, W.N.; Meyerhardt, J.A.; Ogino, S.; Wang, M.L.; Fuchs, C.S.; Giovannucci, E.L.; Chan, A.T. Fiber Intake and Survival After Colorectal Cancer Diagnosis. JAMA Oncol. 2018, 4, 71–79. [Google Scholar] [CrossRef]
- Tandon, R.; Luxami, V.; Kaur, H.; Tandon, N.; Paul, K. 1,8-Naphthalimide: A Potent DNA Intercalator and Target for Cancer Therapy. Chem. Rec. 2017, 17, 956–993. [Google Scholar] [CrossRef]
- Zhang, G.; An, Y.; Lu, X.; Zhong, H.; Zhu, Y.; Wu, Y.; Ma, F.; Yang, J.; Liu, Y.; Zhou, Z.; et al. A Novel Naphthalimide Compound Restores p53 Function in Non-small Cell Lung Cancer by Reorganizing the Bak.Bcl-xl Complex and Triggering Transcriptional Regulation. J. Biol. Chem. 2016, 291, 4211–4225. [Google Scholar] [CrossRef] [Green Version]
- Tomczyk, M.D.; Walczak, K.Z. l,8-Naphthalimide based DNA intercalators and anticancer agents. A systematic review from 2007 to 2017. Eur. J. Med. Chem. 2018, 159, 393–422. [Google Scholar] [CrossRef]
- Mijatovic, T.; Mahieu, T.; De Neve, N.; Dewelle, J.; Simon, G.; Dehoux, M.J.M.; van der Aar, E.; Haibe-Kains, B.; Bontempi, G.; Deacestecker, C.; et al. UNBS5162, a novel naphthalimide that decreases CXCL chemokine expression in experimental prostate cancers. Neoplasia 2008, 10, 573–586. [Google Scholar] [CrossRef]
- Zhu, H.; Huang, M.; Yang, F.; Chen, Y.; Miao, Z.H.; Qian, X.H.; Xu, Y.F.; Qin, Y.X.; Luo, H.B.; Shen, X.; et al. R16, a novel amonafide analogue, induces apoptosis and G2-M arrest via poisoning topoisomerase II. Mol. Cancer Ther. 2007, 6, 484–495. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Sun, L.; Zhang, H.; Xu, Y.; Qian, X.; Lu, Y.; Li, Q.; Ni, L.; Liu, J. A ROS-mediated lysosomal-mitochondrial pathway is induced by a novel Amonafide analogue, 7c, in human Hela cervix carcinoma cells. Cancer Lett. 2013, 333, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Dabiri, Y.; Schmid, A.; Theobald, J.; Blagojevic, B.; Streciwilk, W.; Ott, I.; Wolfl, S.; Cheng, X. A Ruthenium(II) N-Heterocyclic Carbene (NHC) Complex with Naphthalimide Ligand Triggers Apoptosis in Colorectal Cancer Cells via Activating the ROS-p38 MAPK Pathway. Int. J. Mol. Sci. 2018, 19, 3964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brider, T.; Redko, B.; Grynszpan, F.; Gellerman, G. Three overlooked chemical approaches toward 3-naphthalimide amonafide N-derivatives. Tetrahedron Lett. 2014, 55, 6675–6679. [Google Scholar] [CrossRef]
- Seliga, R.; Pilatova, M.; Sarissky, M.; Viglasky, V.; Walko, M.; Mojzis, J. Novel naphthalimide polyamine derivatives as potential antitumor agents. Mol. Biol. Rep. 2013, 40, 4129–4137. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Veale, E.B.; Phelan, C.M.; Murphy, S.A.; Tocci, G.M.; Gillespie, L.J.; Frimannsson, D.O.; Kelly, J.M.; Gunnlaugsson, T. Recent advances in the development of 1,8-naphthalimide based DNA targeting binders, anticancer and fluorescent cellular imaging agents. Chem. Soc. Rev. 2013, 42, 1601–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malviya, V.K.; Liu, P.Y.; Otoole, R.; Alberts, D.S.; Surwit, E.; Rosenoff, S.; Ward, J.H.; Yu, A.; Osullivan, J.; Craig, J.B. Phase-Ii Trial of Amonafide in Patients with Advanced Metastatic or Recurrent Endometrial Adenocarcinoma—A Southwest-Oncology-Group Study. Am. J. Clin. Oncol.-Cancer 1994, 17, 37–40. [Google Scholar] [CrossRef]
- Kornek, G.; Raderer, M.; Depisch, D.; Haider, K.; Fazeny, B.; Dittrich, C.; Scheithauer, W. Amonafide as First-Line Chemotherapy for Metastatic Breast-Cancer. Eur. J. Cancer 1994, 30A, 398–400. [Google Scholar] [CrossRef]
- Stone, R.M.; Mazzola, E.; Neuberg, D.; Allen, S.L.; Pigneux, A.; Stuart, R.K.; Wetzler, M.; Rizzieri, D.; Erba, H.P.; Damon, L.; et al. Phase III Open-Label Randomized Study of Cytarabine in Combination with Amonafide L-Malate or Daunorubicin as Induction Therapy for Patients with Secondary Acute Myeloid Leukemia. J. Clin. Oncol. 2015, 33, 1252. [Google Scholar] [CrossRef]
- Lizzi, F.; Veronesi, G.; Belluti, F.; Bergamini, C.; Lopez-Sanchez, A.; Kaiser, M.; Brun, R.; Krauth-Siegel, R.L.; Hall, D.G.; Rivas, L.; et al. Conjugation of quinones with natural polyamines: Toward an expanded antitrypanosomatid profile. J. Med. Chem. 2012, 55, 10490–10500. [Google Scholar] [CrossRef]
- Simoni, E.; Bergamini, C.; Fato, R.; Tarozzi, A.; Bains, S.; Motterlini, R.; Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Hrelia, P.; et al. Polyamine conjugation of curcumin analogues toward the discovery of mitochondria-directed neuroprotective agents. J. Med. Chem. 2010, 53, 7264–7268. [Google Scholar] [CrossRef]
- Bombarde, O.; Larminat, F.; Gomez, D.; Frit, P.; Racca, C.; Gomes, B.; Guilbaud, N.; Calsou, P. The DNA-Binding Polyamine Moiety in the Vectorized DNA Topoisomerase II Inhibitor F14512 Alters Reparability of the Consequent Enzyme-Linked DNA Double-Strand Breaks. Mol. Cancer Ther. 2017, 16, 2166–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oviatt, A.A.; Kuriappan, J.A.; Minniti, E.; Vann, K.R.; Onuorah, P.; Minarini, A.; De Vivo, M.; Osheroff, N. Polyamine-containing etoposide derivatives as poisons of human type II topoisomerases: Differential effects on topoisomerase II alpha and II beta. Bioorg. Med. Chem. Lett. 2018, 28, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Kruczynski, A.; Pillon, A.; Creancier, L.; Vandenberghe, I.; Gomes, B.; Brel, V.; Fournier, E.; Annereau, J.P.; Currie, E.; Guminski, Y.; et al. F14512, a polyamine-vectorized anti-cancer drug, currently in clinical trials exhibits a marked preclinical anti-leukemic activity. Leukemia 2013, 27, 2139–2148. [Google Scholar] [CrossRef] [PubMed]
- Tierny, D.; Serres, F.; Segaoula, Z.; Bemelmans, I.; Bouchaert, E.; Petain, A.; Brel, V.; Couffin, S.; Marchal, T.; Nguyen, L.; et al. Phase I Clinical Pharmacology Study of F14512, a New Polyamine-Vectorized Anticancer Drug, in Naturally Occurring Canine Lymphoma. Clin. Cancer Res. 2015, 21, 5314–5323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leary, A.; Le Tourneau, C.; Varga, A.; Sablin, M.P.; Gomez-Roca, C.; Guilbaud, N.; Petain, A.; Pavlyuk, M.; Delord, J.P. Phase I dose-escalation study of F14512, a polyamine-vectorized topoisomerase II inhibitor, in patients with platinum-refractory or resistant ovarian cancer. Investig. New Drugs 2019, 37, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Meek, D.W. Regulation of the p53 response and its relationship to cancer. Biochem. J. 2015, 469, 325–346. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. TIG 2012, 28, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Tsai, A.C.; Pan, S.L.; Sun, H.L.; Wang, C.Y.; Peng, C.Y.; Wang, S.W.; Chang, Y.L.; Kuo, S.C.; Lee, K.H.; Teng, C.M. CHM-1, a new vascular targeting agent, induces apoptosis of human umbilical vein endothelial cells via p53-mediated death receptor 5 up-regulation. J. Biol. Chem. 2010, 285, 5497–5506. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.F.; Yang, J.S.; Tsai, F.J.; Chiang, N.N.; Lu, C.C.; Huang, Y.S.; Chen, C.; Chen, F.A. Kaempferol induces ATM/p53-mediated death receptor and mitochondrial apoptosis in human umbilical vein endothelial cells. Int. J. Oncol. 2016, 48, 2007–2014. [Google Scholar] [CrossRef] [Green Version]
- Lourenco, A.R.; Ban, Y.; Crowley, M.J.; Lee, S.B.; Ramchandani, D.; Du, W.; Elemento, O.; George, J.T.; Jolly, M.K.; Levine, H.; et al. Differential contributions of pre- and post-EMT tumor cells in breast cancer metastasis. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Li, Y.; Li, B.; Zhang, M.; Liu, Y.; Yao, Y.; Li, D. NMIIA promotes tumor growth and metastasis by activating the Wnt/beta-catenin signaling pathway and EMT in pancreatic cancer. Oncogene 2019, 38, 5500–5515. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Jackstadt, R.; Siemens, H.; Li, H.; Kirchner, T.; Hermeking, H. p53-induced miR-15a/16-1 and AP4 form a double-negative feedback loop to regulate epithelial-mesenchymal transition and metastasis in colorectal cancer. Cancer Res. 2014, 74, 532–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Jones, R.; Liu, J.C.; Deng, T.; Robinson, T.; Chung, P.E.D.; Wang, S.; Herschkowitz, J.I.; Egan, S.E.; Perou, C.M.; et al. RB1 and p53 at the crossroad of EMT and triple negative breast cancer. Cell Cycle 2011, 10, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Kim, H.S.; Li, X.Y.; Lee, I.; Choi, H.S.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Yoon, D.; Fearon, E.R.; et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J. Cell. Biol. 2011, 195, 417–433. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Chen, H.C.; Zhang, D.Z.; Fu, J.J. Twist: A molecular target in cancer therapeutics. Tumor Biol. 2013, 34, 2497–2506. [Google Scholar] [CrossRef]
- Fenouille, N.; Tichet, M.; Dufies, M.; Pottier, A.; Mogha, A.; Soo, J.K.; Rocchi, S.; Mallavialle, A.; Galibert, M.D.; Khammari, A.; et al. The Epithelial-Mesenchymal Transition (EMT) Regulatory Factor SLUG (SNAI2) Is a Downstream Target of SPARC and AKT in Promoting Melanoma Cell Invasion. PLoS ONE 2012, 7, e40378. [Google Scholar] [CrossRef] [Green Version]
- Pinho, A.V.; Rooman, I.; Real, F.X. p53-dependent regulation of growth, epithelial-mesenchymal transition and stemness in normal pancreatic epithelial cells. Cell Cycle 2011, 10, 1312–1321. [Google Scholar] [CrossRef]
- Powell, E.; Piwnica-Worms, D.; Piwnica-Worms, H. Contribution of p53 to metastasis. Cancer Discov. 2014, 4, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Dai, F.J.; Li, Q.; Wang, Y.X.; Ge, C.C.; Feng, C.Y.; Xie, S.Q.; He, H.Y.; Xu, X.J.; Wang, C.J. Design, Synthesis, and Biological Evaluation of Mitochondria Targeted Flavone-Naphthalimide-Polyamine Conjugates with Antimetastatic Activity. J. Med. Chem. 2017, 60, 2071–2083. [Google Scholar] [CrossRef]
- Wang, Y.X.; Zhang, X.B.; Zhao, J.; Xie, S.Q.; Wang, C.J. Nonhematotoxic Naphthalene Diimide Modified by Polyamine: Synthesis and Biological Evaluation. J. Med. Chem. 2012, 55, 3502–3512. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.J.; He, H.Y.; Xu, X.J.; Chen, S.; Wang, C.J.; Feng, C.Y.; Tian, Z.Y.; Dong, H.Y.; Xie, S.Q. Synthesis and biological evaluation of naphthalimide-polyamine conjugates modified by alkylation as anticancer agents through p53 pathway. Bioorg. Chem. 2018, 77, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, D.; Northfelt, D.W.; Chalasani, P.; Rensvold, D.; Kurtin, S.; Von Hoff, D.D.; Borad, M.J.; Tibes, R. Phase I trial of UNBS5162, a novel naphthalimide in patients with advanced solid tumors or lymphoma. Int. J. Clin. Oncol. 2013, 18, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Gellerman, G. Recent Developments in the Synthesis and Applications of Anticancer Amonafide Derivatives. A Mini Review. Lett. Drug Des. Discov. 2016, 13, 47–63. [Google Scholar] [CrossRef]
- Gao, H.; Jin, S.; Song, Y.; Fu, M.; Wang, M.; Liu, Z.; Wu, M.; Zhan, Q. B23 regulates GADD45a nuclear translocation and contributes to GADD45a-induced cell cycle G2-M arrest. J. Biol. Chem. 2005, 280, 10988–10996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Farnebo, M.; Bykov, V.J.N.; Wiman, K.G. The p53 tumor suppressor: A master regulator of diverse cellular processes and therapeutic target in cancer. Biochem. Biophys. Res. Commun. 2010, 396, 85–89. [Google Scholar] [CrossRef]
- Cazzalini, O.; Scovassi, A.I.; Savio, M.; Stivala, L.A.; Prosperi, E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat. Res. 2010, 704, 12–20. [Google Scholar] [CrossRef]
- Ren, B.; Cam, H.; Takahashi, Y.; Volkert, T.; Terragni, J.; Young, R.A.; Dynlacht, B.D. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Gene Dev. 2002, 16, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Sheng, C.B.; Mendler, I.H.; Rieke, S.; Snyder, P.; Jentsch, M.; Friedrich, D.; Drossel, B.; Loewer, A. PCNA-Mediated Degradation of p21 Coordinates the DNA Damage Response and Cell Cycle Regulation in Individual Cells. Cell Rep. 2019, 27, 48. [Google Scholar] [CrossRef] [Green Version]
- Lei, M. The MCM complex: Its role in DNA replication and implications for cancer therapy. Curr. Cancer Drug Targets 2005, 5, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Hesketh, E.L.; Knight, J.R.; Wilson, R.H.; Chong, J.P.; Coverley, D. Transient association of MCM complex proteins with the nuclear matrix during initiation of mammalian DNA replication. Cell Cycle 2015, 14, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Dai, Q.; Park, D.; Deng, X. Targeting DNA Replication Stress for Cancer Therapy. Genes 2016, 7, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.M.; Li, J.; Zhao, H.; Tan, L.F. Ruthenium(II) polypyridyl complexes with 1,8-naphthalimide group as DNA binder, photonuclease, and dual inhibitors of topoisomerases I and II alpha. J. Inorg. Biochem. 2016, 163, 88–94. [Google Scholar] [CrossRef]
- Ji, L.Y.; Yang, S.M.; Li, S.S.; Liu, S.; Tang, S.N.; Liu, Z.Q.; Meng, X.B.; Yu, S.W. A novel triazolonaphthalimide induces apoptosis and inhibits tumor growth by targeting DNA and DNA-associated processes. Oncotarget 2017, 8, 37394–37408. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [Green Version]
- Weinmann, L.; Wischhusen, J.; Demma, M.J.; Naumann, U.; Roth, P.; Dasmahapatra, B.; Weller, M. A novel p53 rescue compound induces p53-dependent growth arrest and sensitises glioma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 2008, 15, 718–729. [Google Scholar] [CrossRef]
- Sheikh, M.S.; Burns, T.F.; Huang, Y.; Wu, G.S.; Amundson, S.; Brooks, K.S.; Fornace, A.J.; El-Deiry, W.S. p53-dependent and -independent regulation of the death receptor KILLER/DR5 gene expression in response to genotoxic stress and tumor necrosis factor alpha. Cancer Res. 1998, 58, 1593–1598. [Google Scholar]
- Guan, B.X.; Yue, P.; Clayman, G.L.; Sun, S.Y. Evidence that the death receptor DR4 is a DNA damage-inducible, p53-regulated gene. J. Cell. Physiol. 2001, 188, 98–105. [Google Scholar] [CrossRef]
- Liu, X.G.; Yue, P.; Khuri, F.R.; Sun, S.Y. p53 upregulates death receptor 4 expression through an intronic p53 binding site. Cancer Res. 2004, 64, 5078–5083. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.Q.; Li, Q.A.; Zhang, Y.H.; Wang, J.H.; Mei, Z.H.; Zhao, J.; Wang, C.J. NPC-16, a novel naphthalimide-polyamine conjugate, induced apoptosis and autophagy in human hepatoma HepG2 cells and Bel-7402 cells. Apoptosis 2011, 16, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Valentini, A.M.; Pirrelli, M.; Renna, L.; Armentano, R.; Caruso, M.L. P53 and beta-catenin in colorectal cancer progression. Curr. Pharm. Design 2003, 9, 1932–1936. [Google Scholar] [CrossRef] [PubMed]
- Sadot, E.; Geiger, B.; Oren, M.; Ben-Ze’ev, A. Down-regulation of beta-catenin by activated p53. Mol. Cell. Biol. 2001, 21, 6768–6781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, S.; Yonekawa, A.; Harada, C.; Hamada, M.; Katagiri, W.; Nakazawa, M.; Yura, Y. Involvement of the Wnt-beta-catenin pathway in invasion and migration of oral squamous carcinoma cells. Int. J. Oncol. 2010, 37, 1095–1103. [Google Scholar] [CrossRef] [Green Version]
- Tanigawa, N.; Amaya, H.; Matsumura, M.; Lu, C.D.; Kitaoka, A.; Matsuyama, K.; Muraoka, R. Tumor angiogenesis and mode of metastasis in patients with colorectal cancer. Cancer Res. 1997, 57, 1043–1046. [Google Scholar]
- Ellis, L.M. Angiogenesis and its role in colorectal tumor and metastasis formation. Semin. Oncol. 2004, 31, 3–9. [Google Scholar] [CrossRef]
- Mousa, L.; Salem, M.E.; Mikhail, S. Biomarkers of Angiogenesis in Colorectal Cancer. Biomark. Cancer 2015, 7, 13–19. [Google Scholar] [CrossRef]
- Watanabe, K.; Hasegawa, Y.; Yamashita, H.; Shimizu, K.; Ding, Y.Y.; Abe, M.; Ohta, H.; Imagawa, K.; Hojo, K.; Maki, H.; et al. Vasohibin as an endothelium-derived negative feedback regulator of angiogenesis. J. Clin. Investig. 2004, 114, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Hiraki, Y.; Inoue, H.; Iyama, K.; Kamizono, A.; Ochiai, M.; Shukunami, C.; Iijima, S.; Suzuki, F.; Kondo, J. Identification of chondromodulin I as a novel endothelial cell growth inhibitor—Purification and its localization in the avascular zone of epiphysical cartilage. J. Biol. Chem. 1997, 272, 32419–32426. [Google Scholar] [CrossRef] [Green Version]
- Lawler, J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J. Cell. Mol. Med. 2002, 6, 1–12. [Google Scholar] [CrossRef]
- Teodoro, J.G.; Evans, S.K.; Green, M.R. Inhibition of tumor angiogenesis by p53: A new role for the guardian of the genome. J. Mol. Med. 2007, 85, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.B.; Li, D.Y.; Zhang, H.F.; Xue, H.Z.; Pan, C.E.; Zhao, S.H.; Wang, L. Resveratrol Inhibits Invasion and Metastasis of Hepatocellular Carcinoma Cells. J. Anim. Vet. Adv. 2010, 9, 3117–3124. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.S.; Li, M.S.; Du, J.; Jiang, Q.Y.; Wang, L.; Yan, S.Y.; Yu, D.M.; Deng, J.B. Neuronal apoptosis in the developing cerebellum. Anat. Histol. Ebryol. 2011, 40, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.J.; Gao, L.; Zhao, Y.; Wang, C.J.; Xie, S.Q. Farrerol inhibited angiogenesis through Akt/mTOR, Erk and Jak2/Stat3 signal pathway. Phytomedicine 2016, 23, 686–693. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, L.; Ge, C.; Wang, S.; Xu, X.; Feng, Y.; Li, X.; Wang, C.; Wang, Y.; Dai, F.; Xie, S. The Role of p53-Mediated Signaling in the Therapeutic Response of Colorectal Cancer to 9F, a Spermine-Modified Naphthalene Diimide Derivative. Cancers 2020, 12, 528. https://doi.org/10.3390/cancers12030528
Gao L, Ge C, Wang S, Xu X, Feng Y, Li X, Wang C, Wang Y, Dai F, Xie S. The Role of p53-Mediated Signaling in the Therapeutic Response of Colorectal Cancer to 9F, a Spermine-Modified Naphthalene Diimide Derivative. Cancers. 2020; 12(3):528. https://doi.org/10.3390/cancers12030528
Chicago/Turabian StyleGao, Lei, Chaochao Ge, Senzhen Wang, Xiaojuan Xu, Yongli Feng, Xinna Li, Chaojie Wang, Yuxia Wang, Fujun Dai, and Songqiang Xie. 2020. "The Role of p53-Mediated Signaling in the Therapeutic Response of Colorectal Cancer to 9F, a Spermine-Modified Naphthalene Diimide Derivative" Cancers 12, no. 3: 528. https://doi.org/10.3390/cancers12030528
APA StyleGao, L., Ge, C., Wang, S., Xu, X., Feng, Y., Li, X., Wang, C., Wang, Y., Dai, F., & Xie, S. (2020). The Role of p53-Mediated Signaling in the Therapeutic Response of Colorectal Cancer to 9F, a Spermine-Modified Naphthalene Diimide Derivative. Cancers, 12(3), 528. https://doi.org/10.3390/cancers12030528