Targeted Dual Intervention-Oriented Drug-Encapsulated (DIODE) Nanoformulations for Improved Treatment of Pancreatic Cancer

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
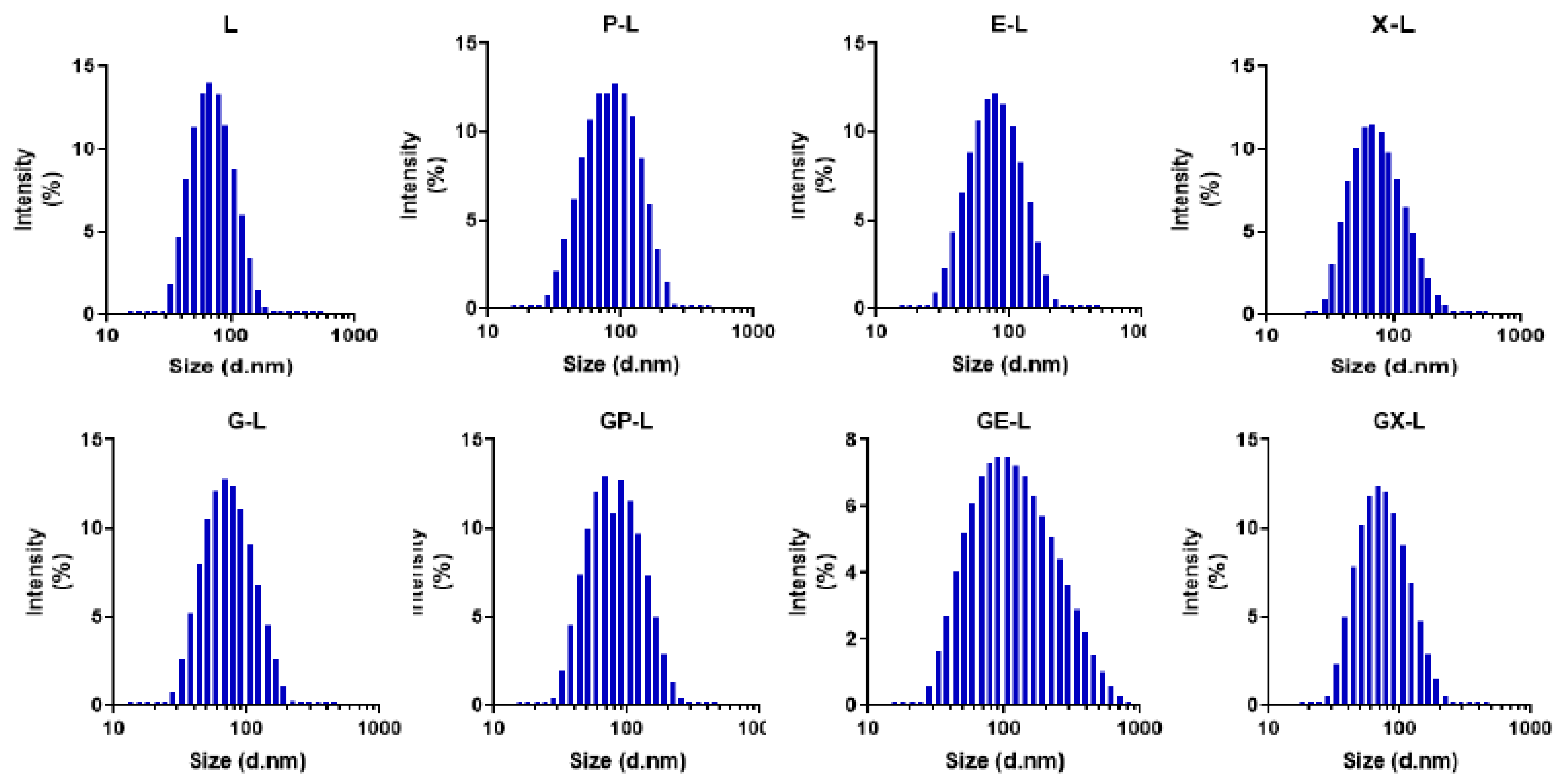
2.1. Preparation and Characterization of Liposomes
2.2. Drug-Loading Efficiency and Encapsulation Efficiency
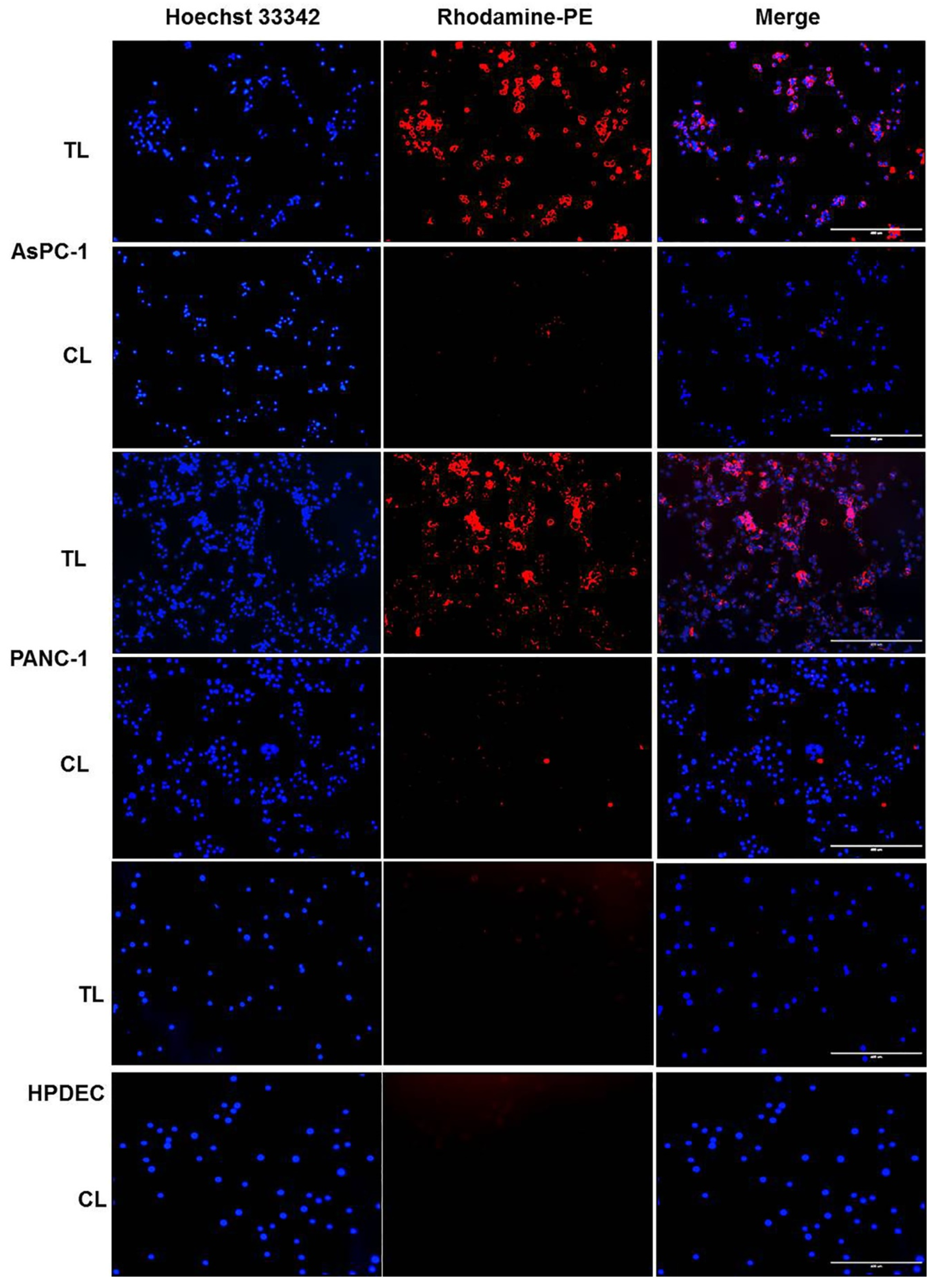
2.3. In Vitro Cellular Uptake of Rhodamine-PE-Labeled Liposomes in Pancreatic Cancer Cell Lines
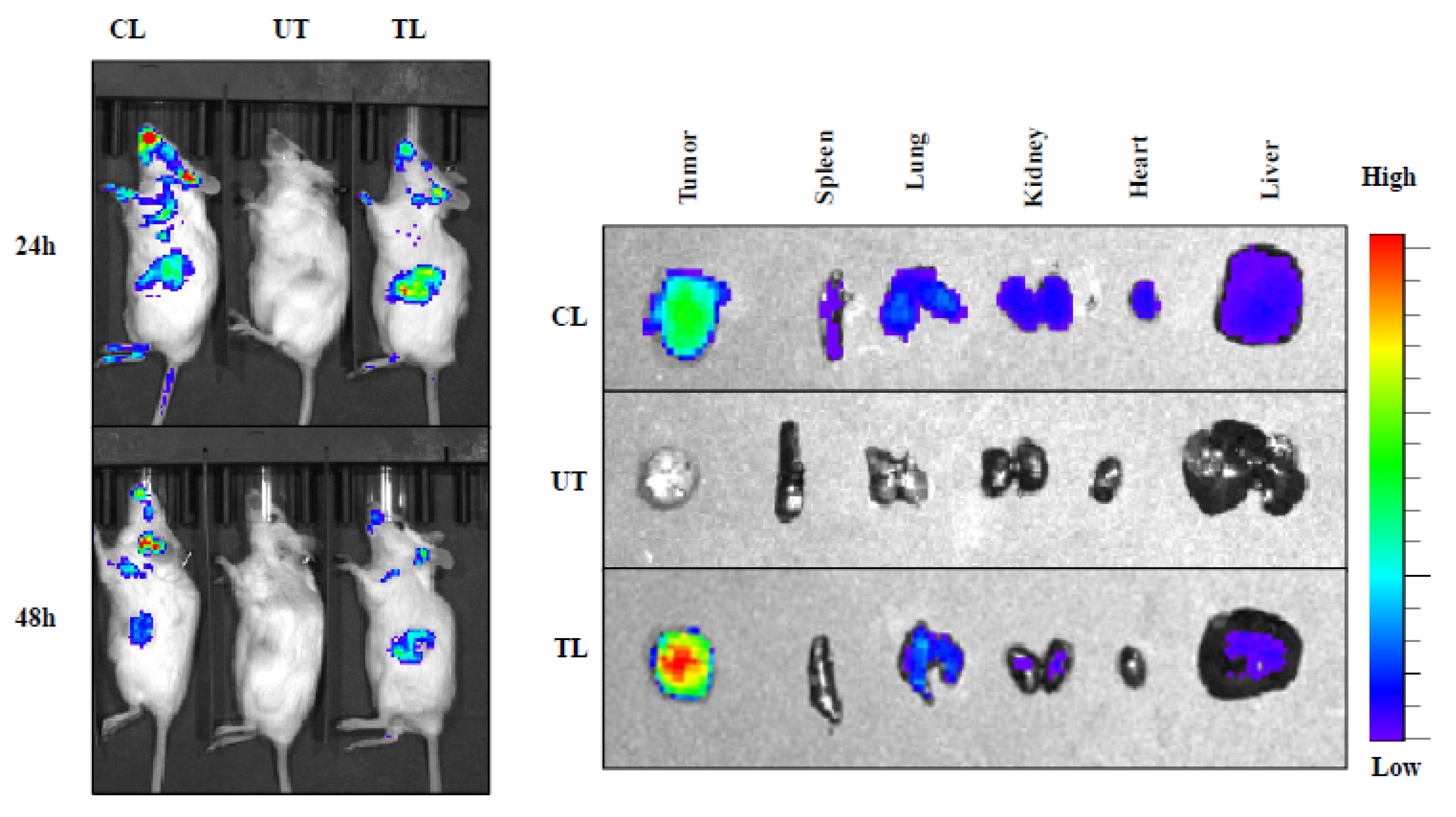
2.4. In Vivo Tumor Uptake Study of NIR-Dye-Loaded Liposomes in Pancreatic Cancer Xenograft
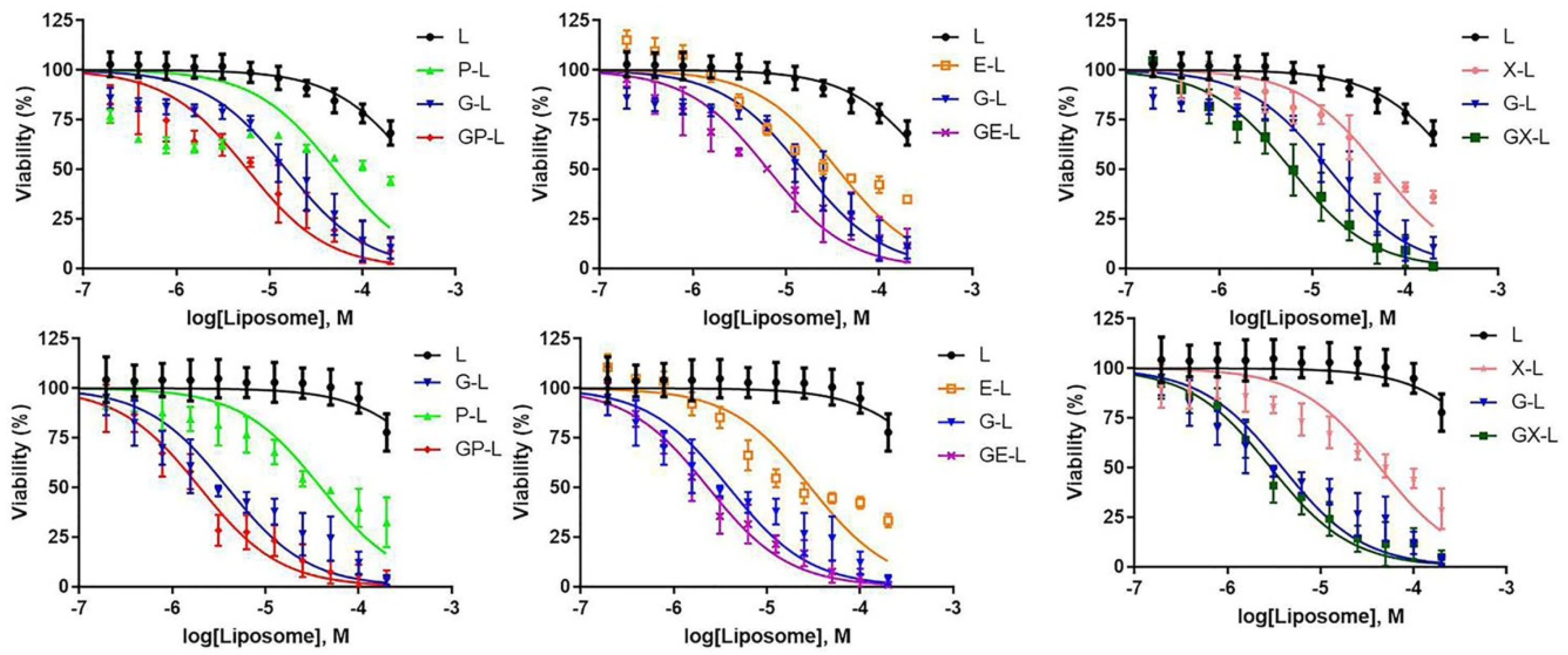
2.5. In Vitro Cytotoxicity of Drug-Loaded Liposomes in Pancreatic Cancer Cell Lines
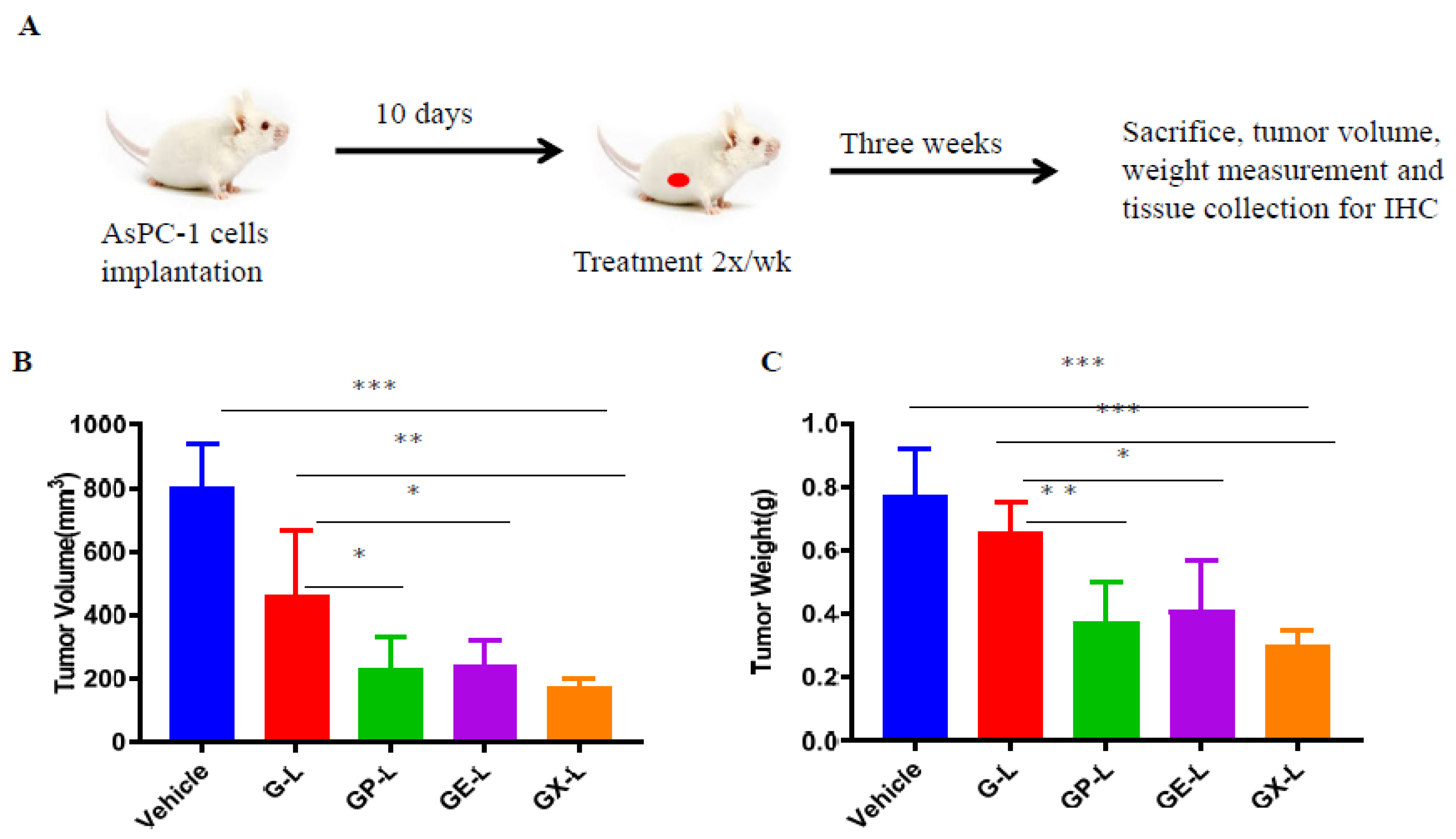
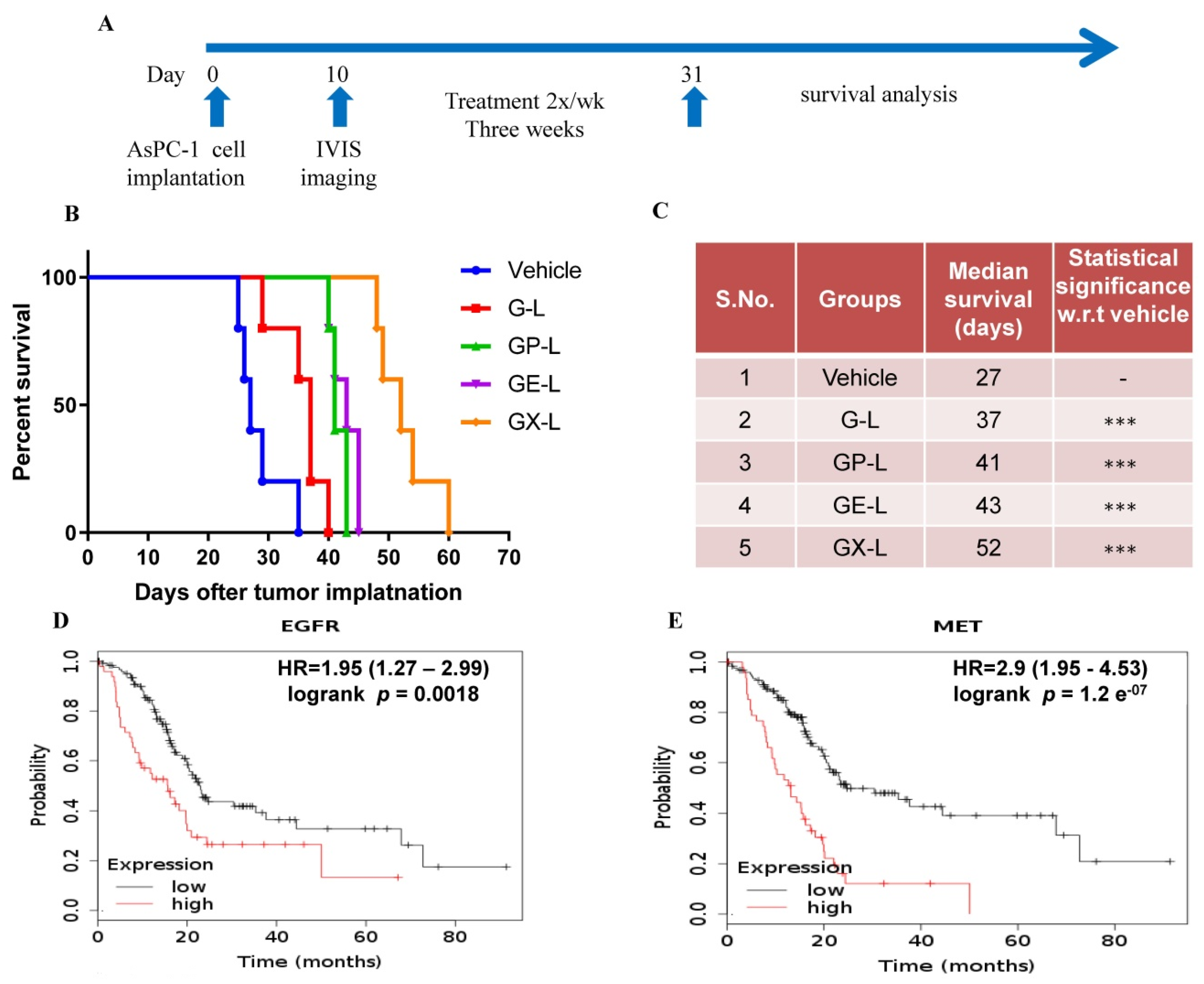
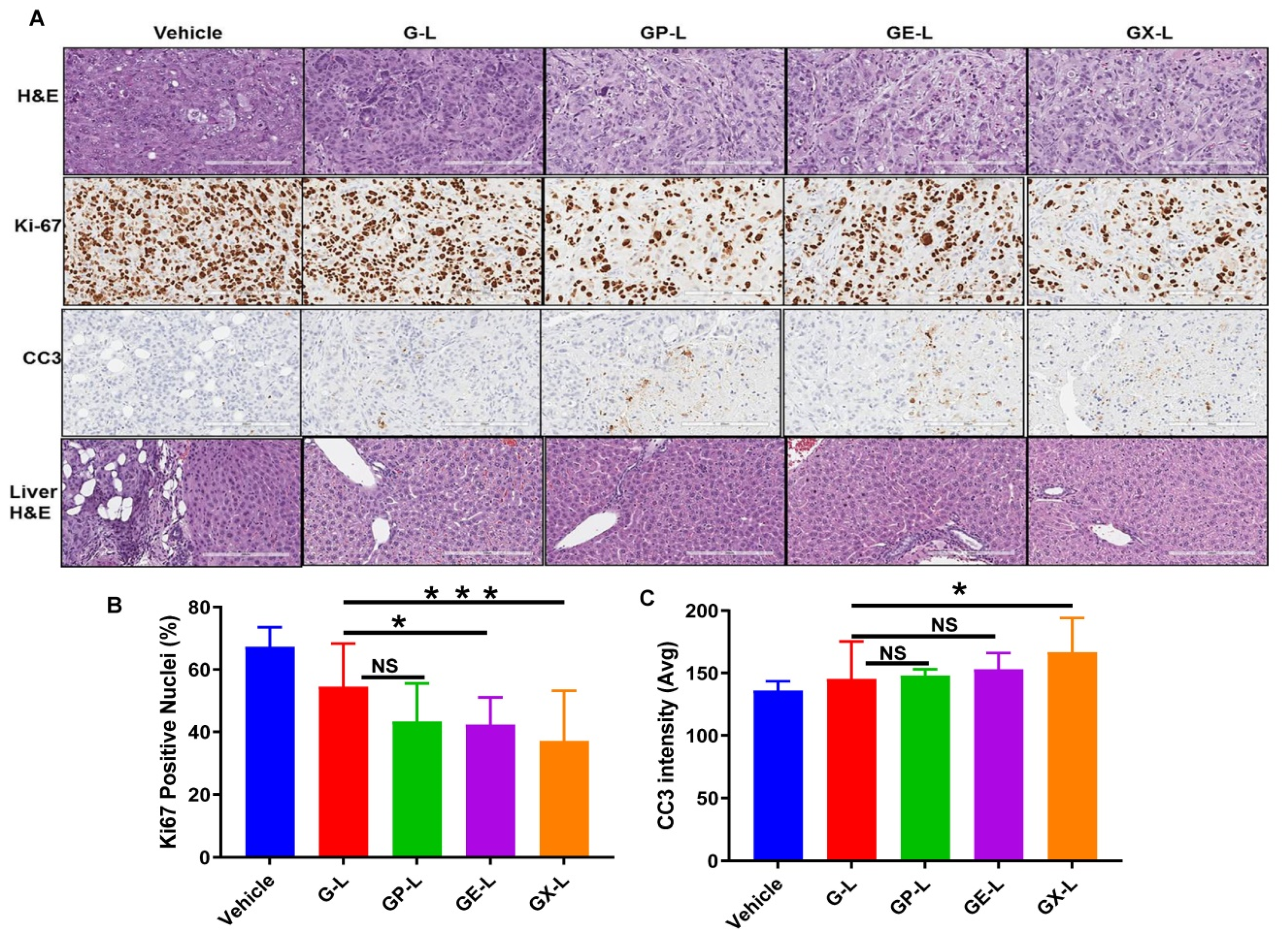
2.6. In Vivo Efficacy of Drug-Loaded Liposomes in Pancreatic Cancer Xenograft
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Preparation Drug/Drugs-Loaded Liposomes
4.4. Physical Characterization and Drug Loading of Liposomes
4.5. Cellular Uptake Assay of Rhodamine-PE-Labeled Liposomes
4.6. In Vitro Cytotoxicity Assay
4.7. Animal Experiments
4.8. Ethics Committee Approval
4.9. In Vivo Tumor-Targeting Evaluation
4.10. In Vivo Antitumor Efficacy and Survival Study
4.11. Immunohistochemistry
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bang, J.Y.; Rösch, T. 14—Endoscopic Ultrasound and Pancreatic Tumors. In Endosonography, 4th ed.; Hawes, R.H., Fockens, P., Varadarajulu, S., Eds.; Content Repository Only!: Philadelphia, PA, USA, 2019; pp. 171–184.e175. [Google Scholar]
- Blando, J.; Sharma, A.; Higa, M.G.; Zhao, H.; Vence, L.; Yadav, S.S.; Kim, J.; Sepulveda, A.M.; Sharp, M.; Maitra, A.; et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 1692–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.A.M.A.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elahi-Gedwillo, K.Y.; Carlson, M.; Zettervall, J.; Provenzano, P.P. Antifibrotic Therapy Disrupts Stromal Barriers and Modulates the Immune Landscape in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2019, 79, 372–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef] [PubMed]
- Dimou, A.; Syrigos, K.N.; Saif, M.W. Overcoming the stromal barrier: Technologies to optimize drug delivery in pancreatic cancer. Ther. Adv. Med. Oncol. 2012, 4, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Miao, L.; Lin, C.M.; Huang, L. Stromal barriers and strategies for the delivery of nanomedicine to desmoplastic tumors. J. Control. Release Off. J. Control. Release Soc. 2015, 219, 192–204. [Google Scholar] [CrossRef] [Green Version]
- Yoo, B.; Jordan, V.C.; Sheedy, P.; Billig, A.-M.; Ross, A.; Pantazopoulos, P.; Medarova, Z. RNAi-Mediated PD-L1 Inhibition for Pancreatic Cancer Immunotherapy. Sci. Rep. 2019, 9, 4712. [Google Scholar] [CrossRef]
- Borazanci, E.; Dang, C.V.; Robey, R.W.; Bates, S.E.; Chabot, J.A.; Von Hoff, D.D. Pancreatic Cancer: “A Riddle Wrapped in a Mystery inside an Enigma”. Clin. Cancer Res. 2017, 23, 1629–1637. [Google Scholar] [CrossRef] [Green Version]
- Leary, M.; Heerboth, S.; Lapinska, K.; Sarkar, S. Sensitization of Drug Resistant Cancer Cells: A Matter of Combination Therapy. Cancers 2018, 10, 483. [Google Scholar] [CrossRef] [Green Version]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomstrand, H.; Scheibling, U.; Bratthäll, C.; Green, H.; Elander, N.O. Real world evidence on gemcitabine and nab-paclitaxel combination chemotherapy in advanced pancreatic cancer. BMC Cancer 2019, 19, 40. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.; Kim, D.W.; Kim, K.; Kim, R.D.; Bottiglieri, S.M. Dose intensity of nab-paclitaxel and gemcitabine chemotherapy in metastatic pancreatic cancer. J. Clin. Oncol. 2019, 37, 251. [Google Scholar] [CrossRef]
- Haas, M.; Siveke, J.T.; Schenk, M.; Lerch, M.M.; Caca, K.; Freiberg-Richter, J.; Fischer von Weikersthal, L.; Kullmann, F.; Reinacher-Schick, A.; Fuchs, M.; et al. Efficacy of gemcitabine plus erlotinib in rash-positive patients with metastatic pancreatic cancer selected according to eligibility for FOLFIRINOX: A prospective phase II study of the ‘Arbeitsgemeinschaft Internistische Onkologie’. Eur. J. Cancer 2018, 94, 95–103. [Google Scholar] [CrossRef]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib Plus Gemcitabine Compared With Gemcitabine Alone in Patients With Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Li, C.; Wu, J.J.; Hynes, M.; Dosch, J.; Sarkar, B.; Welling, T.H.; Pasca di Magliano, M.; Simeone, D.M. c-Met Is a Marker of Pancreatic Cancer Stem Cells and Therapeutic Target. Gastroenterology 2011, 141, 2218–2227.e2215. [Google Scholar] [CrossRef]
- Hage, C.; Rausch, V.; Giese, N.; Giese, T.; Schönsiegel, F.; Labsch, S.; Nwaeburu, C.; Mattern, J.; Gladkich, J.; Herr, I. The novel c-Met inhibitor cabozantinib overcomes gemcitabine resistance and stem cell signaling in pancreatic cancer. Cell Death Dis. 2013, 4, e627. [Google Scholar] [CrossRef]
- Zhen, D.B.; Griffith, K.A.; Ruch, J.M.; Camphausen, K.; Savage, J.E.; Kim, E.J.; Sahai, V.; Simeone, D.M.; Zalupski, M.M. A phase I trial of cabozantinib and gemcitabine in advanced pancreatic cancer. Investig. New Drugs 2016, 34, 733–739. [Google Scholar] [CrossRef]
- Kutova, O.M.; Guryev, E.L.; Sokolova, E.A.; Alzeibak, R.; Balalaeva, I.V. Targeted Delivery to Tumors: Multidirectional Strategies to Improve Treatment Efficiency. Cancers 2019, 11, 68. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Huang, P.; Chen, X. Hierarchical Targeting Strategy for Enhanced Tumor Tissue Accumulation/Retention and Cellular Internalization. Adv. Mater. 2016, 28, 7340–7364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabral, H.; Matsumoto, Y.; Mizuno, K.; Chen, Q.; Murakami, M.; Kimura, M.; Terada, Y.; Kano, M.R.; Miyazono, K.; Uesaka, M.; et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat. Nanotechnol. 2011, 6, 815. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Chen, Z.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, N.; Udayakumar, T.; D’Souza, W.; Simone Ii, C.; Raghavan, S.; Polf, J.; Mahmood, J. Liposomes: Clinical Applications and Potential for Image-Guided Drug Delivery. Molecules 2018, 23, 288. [Google Scholar] [CrossRef] [Green Version]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015. [Google Scholar] [CrossRef] [Green Version]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, A.R.; Crooke, S.T. RNA Therapeutics in Oncology: Advances, Challenges, and Future Directions. J. Clin. Pharmacol. 2017, 57, S43–S59. [Google Scholar] [CrossRef]
- Meng, H.; Wang, M.; Liu, H.; Liu, X.; Situ, A.; Wu, B.; Ji, Z.; Chang, C.H.; Nel, A.E. Use of a Lipid-Coated Mesoporous Silica Nanoparticle Platform for Synergistic Gemcitabine and Paclitaxel Delivery to Human Pancreatic Cancer in Mice. ACS Nano 2015, 9, 3540–3557. [Google Scholar] [CrossRef] [Green Version]
- Spring, B.Q.; Bryan Sears, R.; Zheng, L.Z.; Mai, Z.; Watanabe, R.; Sherwood, M.E.; Schoenfeld, D.A.; Pogue, B.W.; Pereira, S.P.; Villa, E.; et al. A photoactivable multi-inhibitor nanoliposome for tumour control and simultaneous inhibition of treatment escape pathways. Nat. Nanotechnol. 2016, 11, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Lekka, K.; Tzitzi, E.; Giakoustidis, A.; Papadopoulos, V.; Giakoustidis, D. Contemporary management of borderline resectable pancreatic ductal adenocarcinoma. Ann. Hepato-Biliary-Pancreat. Surg. 2019, 23. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Yang, G.; Qiu, J.; Luan, J.; Zhang, Y.; You, L.; Feng, M.; Zhao, F.; Liu, Y.; Cao, Z.; et al. Novel discoveries targeting gemcitabine-based chemoresistance and new therapies in pancreatic cancer: How far are we from the destination? Cancer Med. 2019, 8, 6403–6413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, K.; Setua, S.; Kumari, S.; Jaggi, M.; Yallapu, M.M.; Chauhan, S.C. Gemcitabine Combination Nano Therapies for Pancreatic Cancer. Pharmaceutics 2019, 11, 574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2019, 17, 108–123. [Google Scholar] [CrossRef]
- Kalaydina, R.-V.; Bajwa, K.; Qorri, B.; DeCarlo, A.; Szewczuk, M.R. Recent advances in “smart” delivery systems for extended drug release in cancer therapy. Int. J. Nanomed. 2018, 13, 4727–4745. [Google Scholar] [CrossRef] [Green Version]
- Brachi, G.; Bussolino, F.; Ciardelli, G.; Mattu, C. Nanomedicine for Imaging and Therapy of Pancreatic Adenocarcinoma. Front. Bioeng. Biotechnol. 2019, 7. [Google Scholar] [CrossRef]
- Riaz, M.; Riaz, M.; Zhang, X.; Lin, C.; Wong, K.; Chen, X.; Zhang, G.; Lu, A.; Yang, Z. Surface Functionalization and Targeting Strategies of Liposomes in Solid Tumor Therapy: A Review. Int. J. Mol. Sci. 2018, 19, 195. [Google Scholar] [CrossRef] [Green Version]
- Madamsetty, V.S.; Pal, K.; Dutta, S.K.; Wang, E.; Thompson, J.R.; Banerjee, R.K.; Caulfield, T.R.; Mody, K.; Yen, Y.; Mukhopadhyay, D.; et al. Design and Evaluation of PEGylated Liposomal Formulation of a Novel Multikinase Inhibitor for Enhanced Chemosensitivity and Inhibition of Metastatic Pancreatic Ductal Adenocarcinoma. Bioconjugate Chem. 2019, 30, 2703–2713. [Google Scholar] [CrossRef] [PubMed]
- Nag, O.; Awasthi, V. Surface Engineering of Liposomes for Stealth Behavior. Pharmaceutics 2013, 5, 542–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naderinezhad, S.; Amoabediny, G.; Haghiralsadat, F. Co-delivery of hydrophilic and hydrophobic anticancer drugs using biocompatible pH-sensitive lipid-based nano-carriers for multidrug-resistant cancers. RSC Adv. 2017, 7, 30008–30019. [Google Scholar] [CrossRef] [Green Version]
- Zununi Vahed, S.; Salehi, R.; Davaran, S.; Sharifi, S. Liposome-based drug co-delivery systems in cancer cells. Mater. Sci. Eng. C 2017, 71, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Bareschino, M.A.; Schettino, C.; Troiani, T.; Martinelli, E.; Morgillo, F.; Ciardiello, F. Erlotinib in cancer treatment. Ann. Oncol. 2007, 18, vi35–vi41. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Parsels, L.A.; Kollar, L.E.; Normolle, D.P.; Maybaum, J.; Lawrence, T.S. The Combination of Epidermal Growth Factor Receptor Inhibitors with Gemcitabine and Radiation in Pancreatic Cancer. Clin. Cancer Res. 2008, 14, 5142–5149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhou, D.a.; Liu, Z.; Huang, X.; Liu, Q.; Kang, Y.; Chen, Z.; Guo, Y.; Zhu, H.; Sun, C. Combination of gemcitabine and erlotinib inhibits recurrent pancreatic cancer growth in mice via the JAK-STAT pathway. Oncol. Rep. 2018. [Google Scholar] [CrossRef] [Green Version]
- Torres, C.; Linares, A.; Alejandre, M.J.; Palomino-Morales, R.J.; Delgado, J.R.; Perales, S. Interplay Between Gemcitabine and Erlotinib Over Pancreatic Adenocarcinoma Cells. Pancreas 2016, 45, 269–280. [Google Scholar] [CrossRef]
- Cohen, S.J.; O’Neil, B.H.; Berlin, J.; Ames, P.; McKinley, M.; Horan, J.; Catalano, P.M.; Davies, A.; Weekes, C.D.; Leichman, L. A phase 1b study of erlotinib in combination with gemcitabine and nab-paclitaxel in patients with previously untreated advanced pancreatic cancer: An Academic Oncology GI Cancer Consortium study. Cancer Chemother. Pharmacol. 2016, 77, 693–701. [Google Scholar] [CrossRef]
- Xu, J.; Wang, J.; Zhang, S. Mechanisms of resistance to irreversible epidermal growth factor receptor tyrosine kinase inhibitors and therapeutic strategies in non-small cell lung cancer. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, C.-W.D.; Frolov, A.; Frolova, N.; Jhala, N.C.; Howard, J.H.; Buchsbaum, D.J.; Vickers, S.M.; Heslin, M.J.; Arnoletti, J.P. Epidermal growth factor receptor (EGFR) is highly conserved in pancreatic cancer. Surgery 2007, 141, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Delitto, D. c-Met signaling in the development of tumorigenesis and chemoresistance: Potential applications in pancreatic cancer. World J. Gastroenterol. 2014, 20. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hu, Q.; Xu, Y.; Liu, H.; Zhong, L. Antibody fragment-conjugated gemcitabine and paclitaxel-based liposome for effective therapeutic efficacy in pancreatic cancer. Mater. Sci. Eng. C 2018, 89, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, P.; Zou, Q.; Li, X.; Fu, J.; Luo, Y.; Liang, X.; Jin, Y. Co-Delivery of Gemcitabine and Paclitaxel in cRGD-Modified Long Circulating Nanoparticles with Asymmetric Lipid Layers for Breast Cancer Treatment. Molecules 2018, 23, 906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, S.; Guo, Y.; Duan, Y.; Li, Z.; Wang, C.; Niu, L.; Wang, N.; Ma, M.; Shi, Y.; Zhang, M. Co-delivery of paclitaxel and gemcitabine by methoxy poly(ethylene glycol)–poly(lactide-coglycolide)-polypeptide nanoparticles for effective breast cancer therapy. Anti-Cancer Drugs 2018, 29, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Di, Y.; Gao, Y.; Gai, X.; Wang, D.; Wang, Y.; Yang, X.; Zhang, D.; Pan, W.; Yang, X. Co-delivery of hydrophilic gemcitabine and hydrophobic paclitaxel into novel polymeric micelles for cancer treatment. RSC Adv. 2017, 7, 24030–24039. [Google Scholar] [CrossRef] [Green Version]
- Noorani, M.; Azarpira, N.; Karimian, K.; Heli, H. Erlotinib-loaded albumin nanoparticles: A novel injectable form of erlotinib and its in vivo efficacy against pancreatic adenocarcinoma ASPC-1 and PANC-1 cell lines. Int. J. Pharm. 2017, 531, 299–305. [Google Scholar] [CrossRef]
- Zhou, X.; Shi, K.; Hao, Y.; Yang, C.; Zha, R.; Yi, C.; Qian, Z. Advances in nanotechnology-based delivery systems for EGFR tyrosine kinases inhibitors in cancer therapy. Asian J. Pharm. Sci. 2019. [Google Scholar] [CrossRef]
- Yang, Q.; Moulder, K.R.; Cohen, M.S.; Cai, S.; Forrest, L.M. Cabozantinib Loaded DSPE-PEG(2000) Micelles as Delivery System: Formulation, Characterization and Cytotoxicity Evaluation. BAOJ Pharm. Sci. 2015, 1, 001. [Google Scholar]
- Pal, K.; Madamsetty, V.S.; Dutta, S.K.; Mukhopadhyay, D. Co-delivery of everolimus and vinorelbine via a tumor-targeted liposomal formulation inhibits tumor growth and metastasis in RCC. Int. J. Nanomed. 2019, 14, 5109–5123. [Google Scholar] [CrossRef] [Green Version]
- Gentine, P.; Bourel-Bonnet, L.; Frisch, B. Modified and derived ethanol injection toward liposomes: Development of the process. J. Liposome Res. 2013, 23, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Madamsetty, V.S.; Kiers, S.; Alekhina, O.; Ugolkov, A.; Dube, J.; Zhang, Y.; Zhang, J.-S.; Wang, E.; Dutta, S.K.; et al. Glycogen Synthase Kinase-3 Inhibition Sensitizes Pancreatic Cancer Cells to Chemotherapy by Abrogating the TopBP1/ATR-Mediated DNA Damage Response. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Nagy, Á.; Lánczky, A.; Menyhárt, O.; Győrffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LIPOSOME | SIZE (nm) | PDI | ZETA (mV) |
|---|---|---|---|
| L | 69.15 ± 0.98 | 0.170 ± 0.007 | 23.5 ± 4.08 |
| P-L | 67.92 ± 0.65 | 0.184 ± 0.009 | 16.3 ± 2.7 |
| E-L | 68.60 ± 0.79 | 0.172 ± 0.003 | 15.9 ± 0.95 |
| X-L | 71.06 ± 0.38 | 0.145 ± 0.003 | 17.4 ± 2.56 |
| G-L | 69.99 ± 0.69 | 0.151 ± 0.011 | 10.2 ± 2.45 |
| GP-L | 68.85 ± 0.76 | 0.157 ± 0.001 | 13.6 ± 0.60 |
| GE-L | 101.12 ± 1.47 | 0.321 ± 0.033 | 11.8 ± 1.10 |
| GX-L | 69.82 ± 1.004 | 0.168 ± 0.010 | 17.6 ± 1.73 |
| Liposome | Total Lipid (mg/mL) | Initial Drug Added (mg/mL) | DLE (%) | EE (%) | |||
|---|---|---|---|---|---|---|---|
| G | P | E | X | ||||
| L | 5.487 | – | – | – | – | – | – |
| P-L | 5.487 | – | 0.1 | 1.5 ± 0.02 | 81.1 ± 1.5 | ||
| E-L | 5.487 | – | – | 0.2 | – | 2.6 ± 0.12 | 70.7 ± 3.9 |
| X-L | 5.487 | – | – | – | 0.150 | 1.82± 0.07 | 66.5 ± 2.9 |
| G-L | 5.487 | 1 | – | – | 3.3 ± 0.39 | 18.1 ± 2.1 | |
| GP-L | 5.487 | 1 | 0.1 | 4.5 ± 0.15(G); 1.45 ± 0.07(P) | 24.6 ± 0.8(G); 79 ± 3.8(P) | ||
| GE-L | 5.487 | 1 | – | 0.2 | 3.1 ± 0.32(G); 2.8 ± 0.08(E) | 16.9 ± 1.7(G); 77.3 ± 2.3(E) | |
| GX-L | 5.487 | 1 | – | – | 0.150 | 4.8 ± 0.06(G); 2 ± 0.06(X) | 26.6 ± 0.37 (G); 75.4 ± 2.2(X) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madamsetty, V.S.; Pal, K.; Dutta, S.K.; Wang, E.; Mukhopadhyay, D. Targeted Dual Intervention-Oriented Drug-Encapsulated (DIODE) Nanoformulations for Improved Treatment of Pancreatic Cancer. Cancers 2020, 12, 1189. https://doi.org/10.3390/cancers12051189
Madamsetty VS, Pal K, Dutta SK, Wang E, Mukhopadhyay D. Targeted Dual Intervention-Oriented Drug-Encapsulated (DIODE) Nanoformulations for Improved Treatment of Pancreatic Cancer. Cancers. 2020; 12(5):1189. https://doi.org/10.3390/cancers12051189
Chicago/Turabian StyleMadamsetty, Vijay Sagar, Krishnendu Pal, Shamit Kumar Dutta, Enfeng Wang, and Debabrata Mukhopadhyay. 2020. "Targeted Dual Intervention-Oriented Drug-Encapsulated (DIODE) Nanoformulations for Improved Treatment of Pancreatic Cancer" Cancers 12, no. 5: 1189. https://doi.org/10.3390/cancers12051189
APA StyleMadamsetty, V. S., Pal, K., Dutta, S. K., Wang, E., & Mukhopadhyay, D. (2020). Targeted Dual Intervention-Oriented Drug-Encapsulated (DIODE) Nanoformulations for Improved Treatment of Pancreatic Cancer. Cancers, 12(5), 1189. https://doi.org/10.3390/cancers12051189