Inhibition of the Lysophosphatidylinositol Transporter ABCC1 Reduces Prostate Cancer Cell Growth and Sensitizes to Chemotherapy

 , ,
, ,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
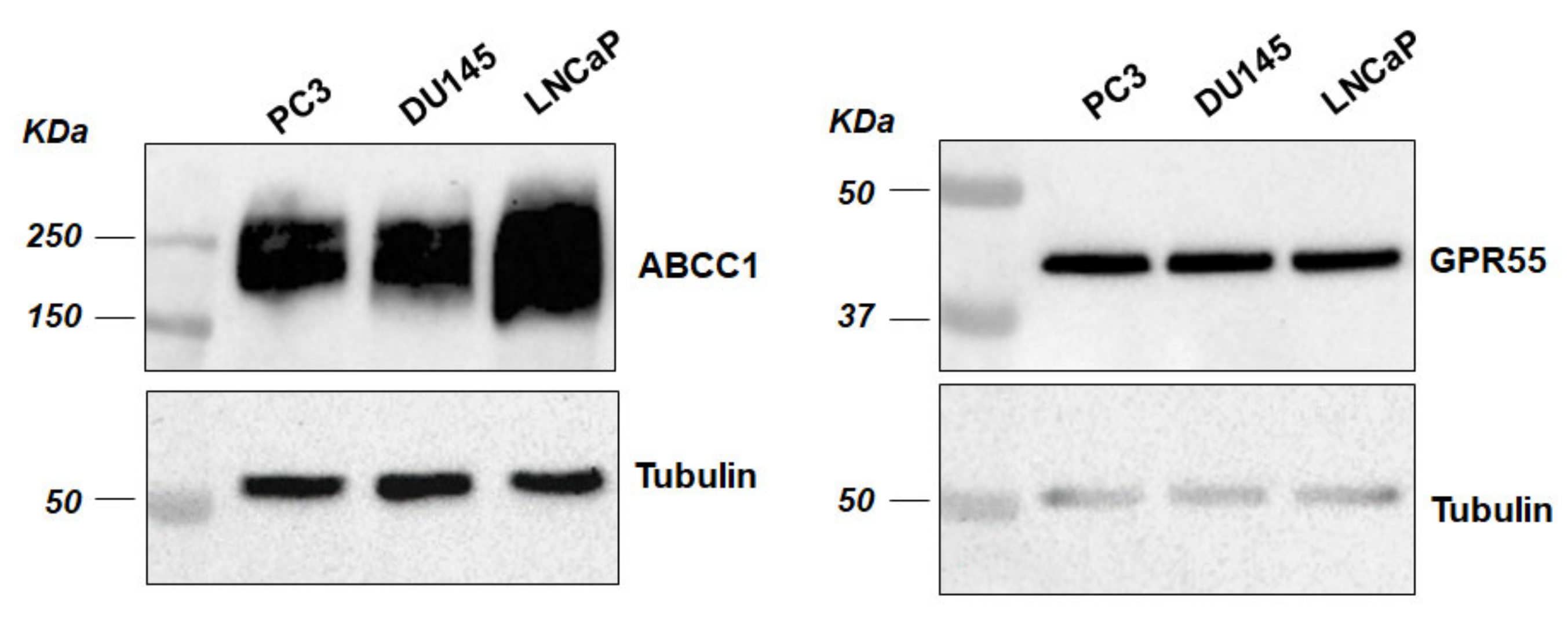
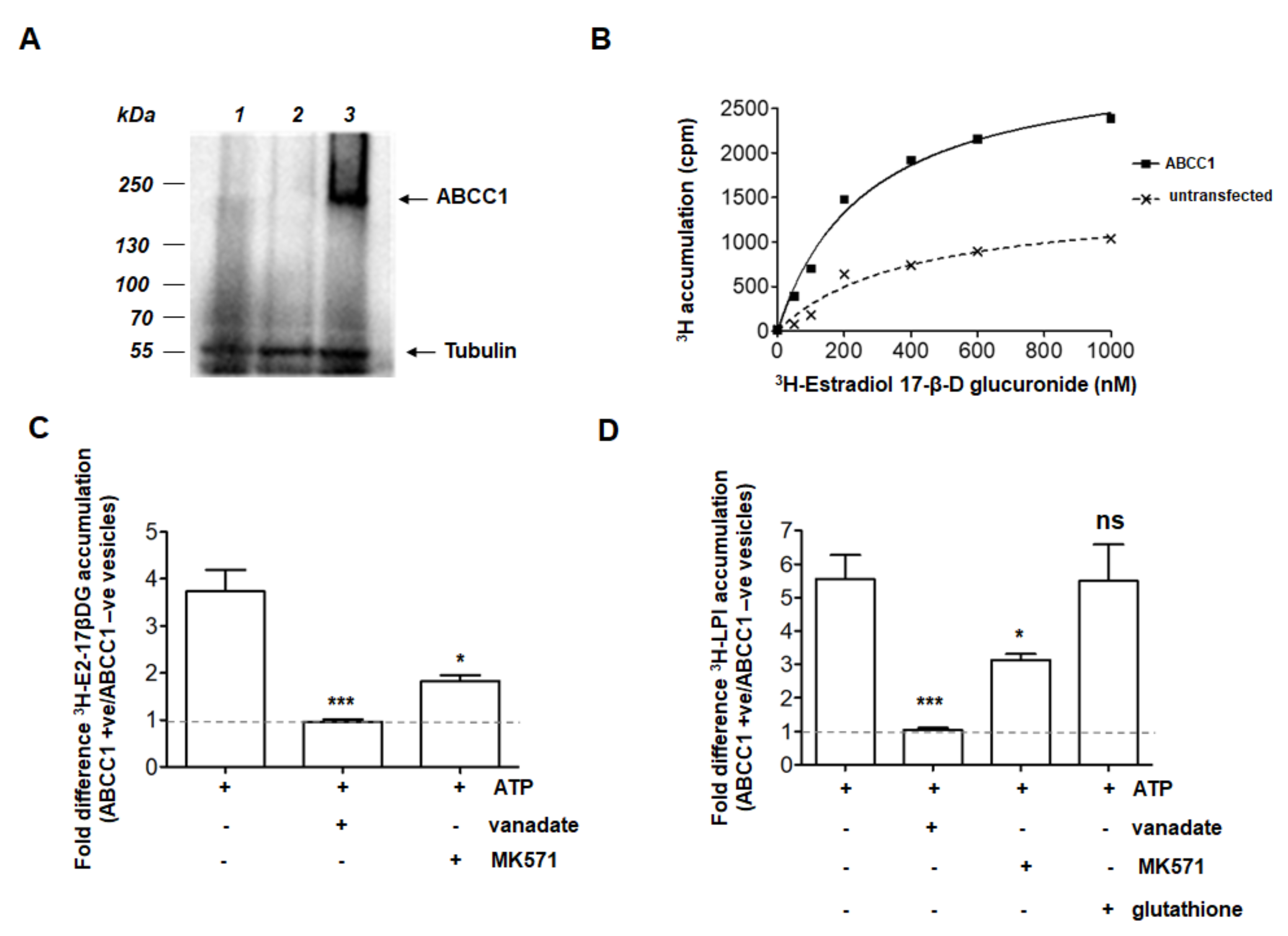
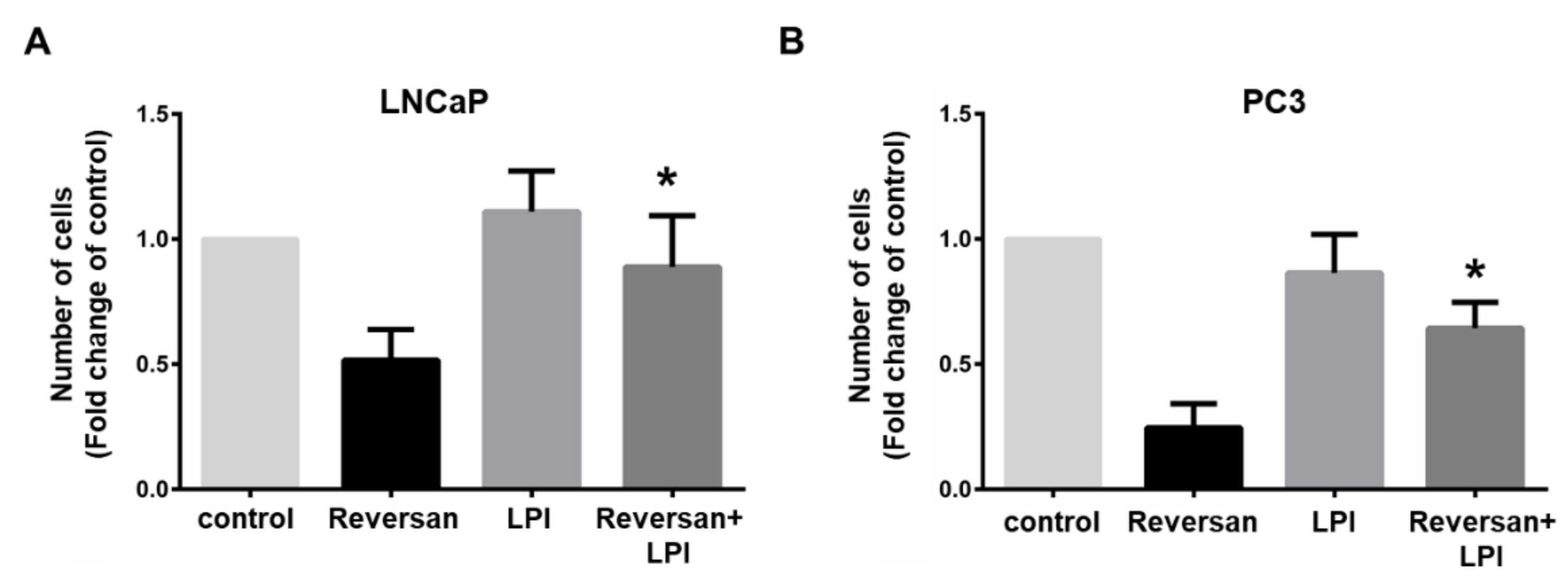
2.1. ABCC1 Can Efflux LPI
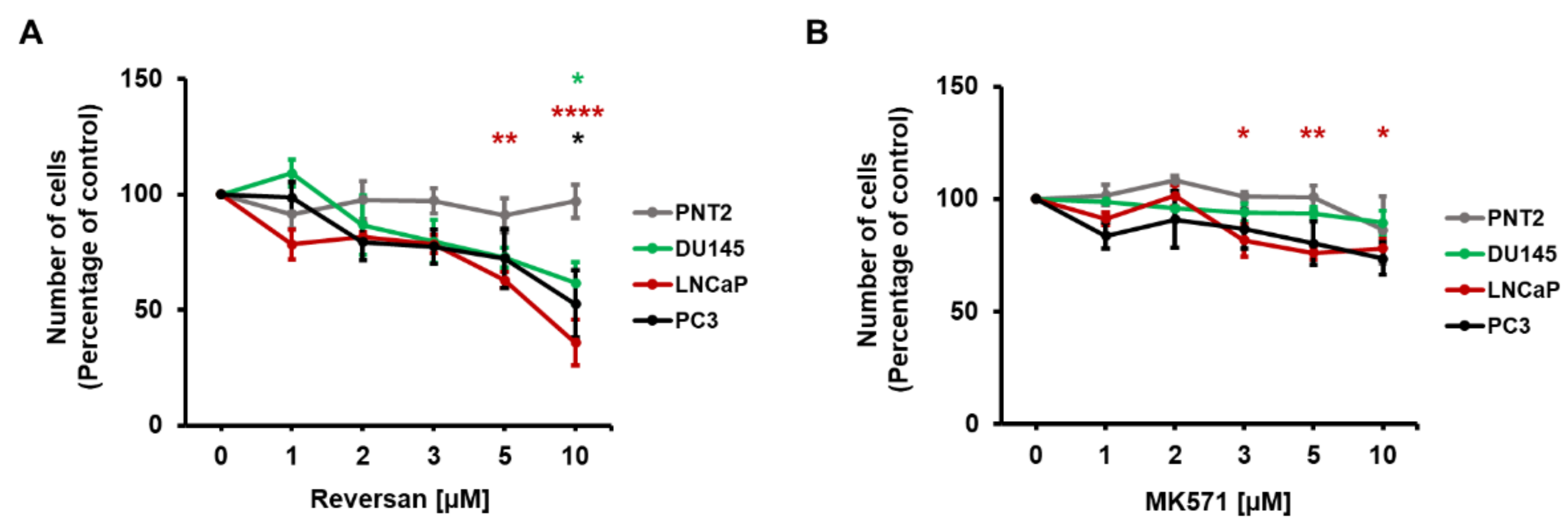
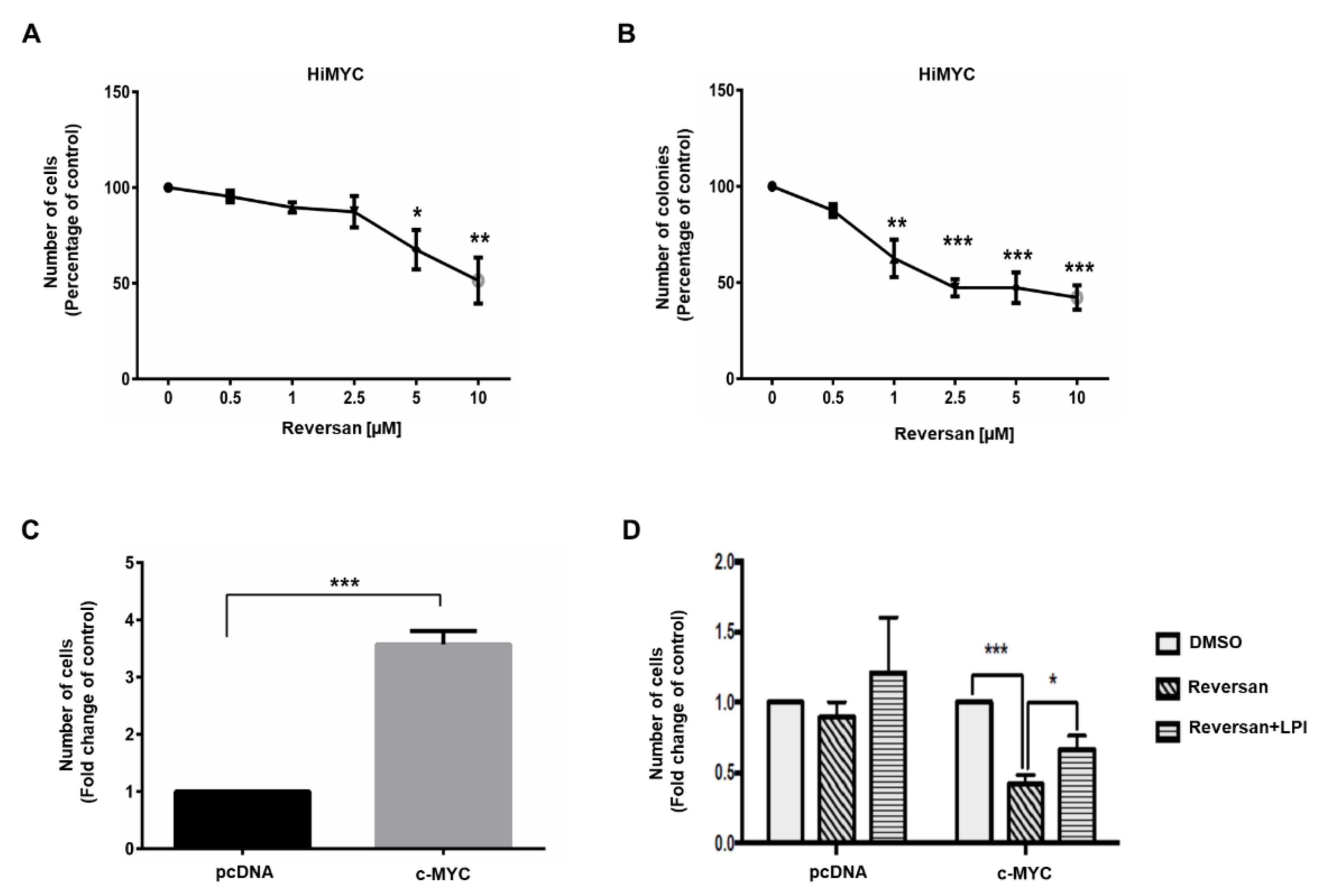
2.2. ABCC1 Inhibitors Reduce Prostate Cancer Cell Growth without Affecting Normal, Immortalised Prostate Cells
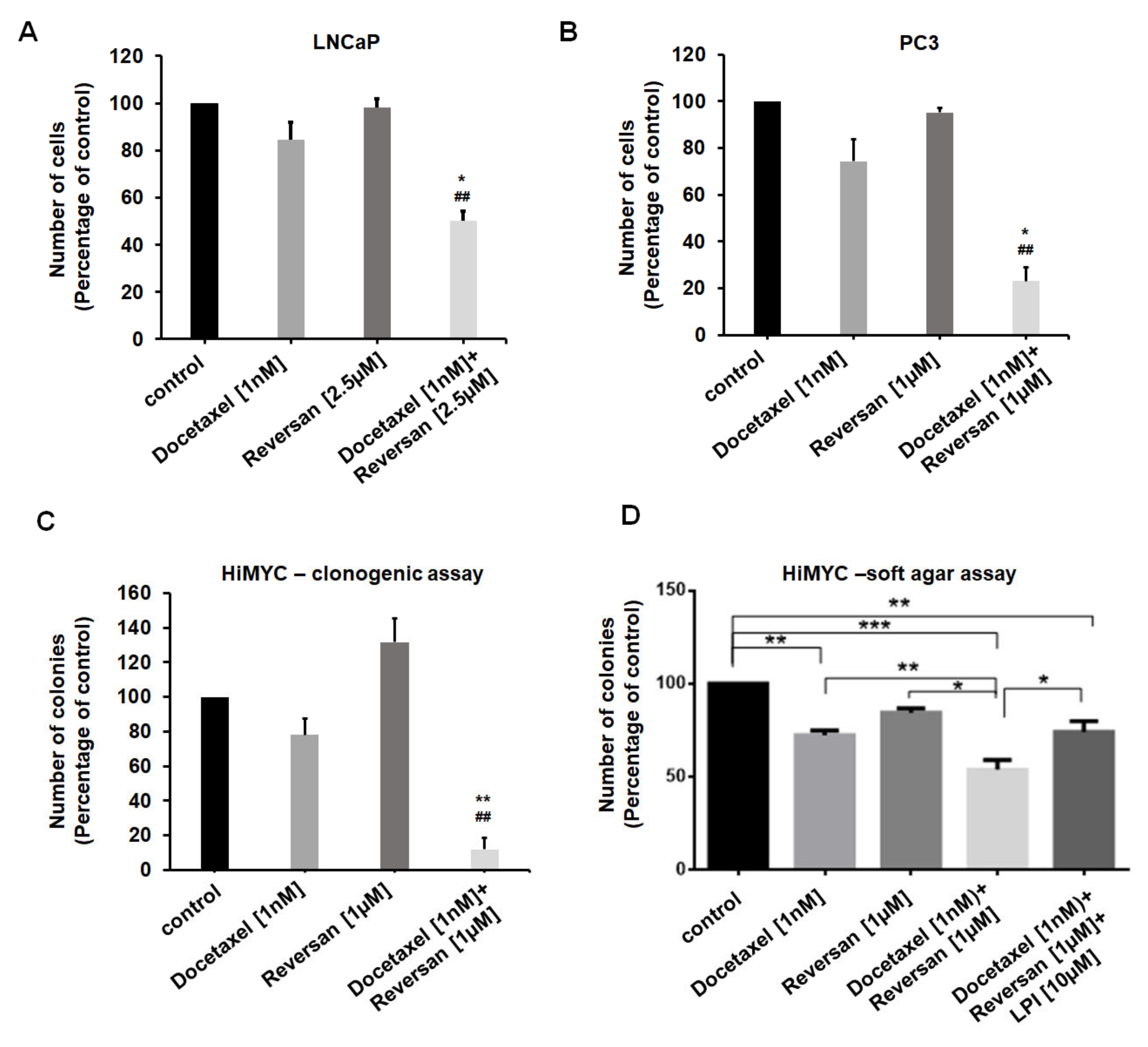
2.3. Combination of Sub-Optimal Concentrations of Reversan and Docetaxel Inhibits Prostate Cancer Cell Growth In Vitro and In Vivo
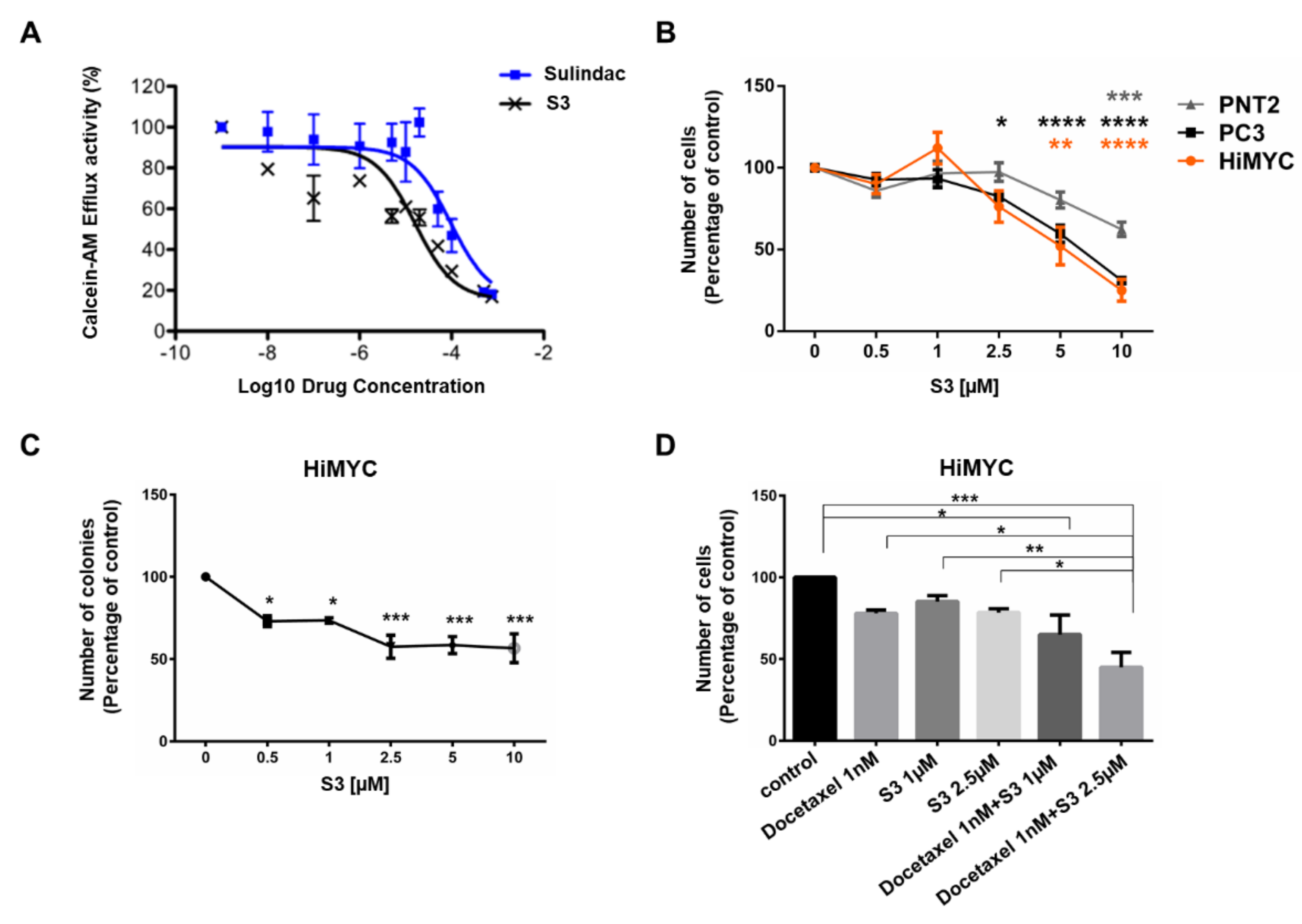
2.4. Identification of Novel ABCC1 Inhibitors and Validation of Their Activity In Vitro
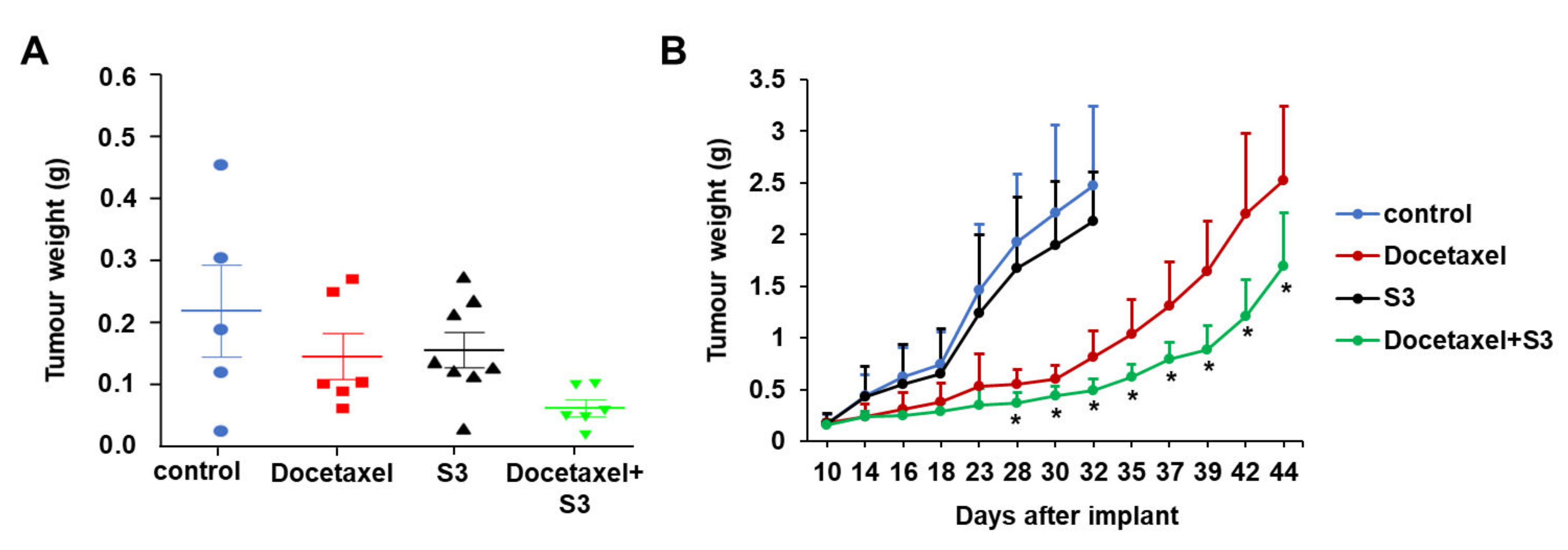
2.5. In Vivo Activity of ABCC1 Inhibitor S3
3. Discussion
4. Materials and Methods
4.1. Plasmids
4.2. Transient Transfection of HEK293T Cells for Two Colour Flow Cytometry
4.3. Transport of Radiolabelled Ligands into Inside-Out Vesicles
4.4. Drug Transport Assay by Two-Colour Flow Cytometry
4.5. Cell Culture and Treatments
4.6. Cell Growth
4.6.1. Cell Counting
4.6.2. MTT Assays
4.6.3. Clonogenic Assay
4.6.4. Anchorage Independent Growth
4.7. Quantitative Real-Time PCR Analysis
4.8. Western Blotting
4.9. In Vivo Studies
4.9.1. Docetaxel Dose Response Experiments and Orthotopic Model
4.9.2. Docetaxel and the S3 Combination in the Xenograft Model
4.10. Pharmacokinetic Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, G.; Zhao, D.; Spring, D.J.; Depinho, R.A. Genetics and biology of prostate cancer. Genes Dev. 2018, 32, 1105–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishan, A.U.; Wang, X.; Seiferheld, W.; Collette, L.; Sandler, K.A.; Sandler, H.M.; Bolla, M.; Maingon, P.; De Reijke, T.; Hanks, G.E.; et al. Association of Gleason Grade With Androgen Deprivation Therapy Duration and Survival Outcomes: A Systematic Review and Patient-Level Meta-analysis. JAMA Oncol. 2019, 5, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzmaurice, C. Global Burden of Disease Cancer Collaboration Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 2006 to 2016: A systematic analysis for the Global Burden of Disease study. J. Clin. Oncol. 2018, 36, 1568. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Tampé, R. Multifaceted structures and mechanisms of ABC transport systems in health and disease. Curr. Opin. Struct. Boil. 2018, 51, 116–128. [Google Scholar] [CrossRef]
- Nobili, S.; Lapucci, A.; Landini, I.; Coronnello, M.; Roviello, G.; Mini, E. Role of ATP-binding cassette transporters in cancer initiation and progression. Semin. Cancer Boil. 2020, 60, 72–95. [Google Scholar] [CrossRef]
- Cole, S. Targeting Multidrug Resistance Protein 1 (MRP1,ABCC1): Past, Present, and Future. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 95–117. [Google Scholar] [CrossRef]
- Falasca, M.; Linton, K.J. Investigational ABC transporter inhibitors. Expert Opin. Investig. Drugs 2012, 21, 657–666. [Google Scholar] [CrossRef]
- Rice, A.J.; Alvarez, F.J.; Davidson, A.L.; Pinkett, H.W. Effects of lipid environment on the conformational changes of an ABC importer. Channels 2014, 8, 327–333. [Google Scholar] [CrossRef] [Green Version]
- Gulati, S.; Jamshad, M.; Knowles, T.; Morrison, K.; Downing, R.; Cant, N.; Collins, R.; Koenderink, J.B.; Ford, R.C.; Overduin, M.; et al. Detergent-free purification of ABC (ATP-binding-cassette) transporters. Biochem. J. 2014, 461, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Cole, S. Multidrug Resistance Protein 1 (MRP1, ABCC1), a “Multitasking” ATP-binding Cassette (ABC) Transporter*. J. Boil. Chem. 2014, 289, 30880–30888. [Google Scholar] [CrossRef] [Green Version]
- Mitra, P.; Oskeritzian, C.A.; Payne, S.G.; Beaven, M.A.; Milstien, S.; Spiegel, S. Role of ABCC1 in export of sphingosine-1-phosphate from mast cells. Proc. Natl. Acad. Sci. 2006, 103, 16394–16399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piñeiro, R.; Maffucci, T.; Falasca, M. The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene 2010, 30, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, A.; Nagahashi, M.; Aoyagi, T.; Huang, W.-C.; Lima, S.; Hait, N.C.; Maiti, A.; Kida, K.; Terracina, K.P.; Miyazaki, H.; et al. ABCC1-Exported Sphingosine-1-phosphate, Produced by Sphingosine Kinase 1, Shortens Survival of Mice and Patients with Breast Cancer. Mol. Cancer Res. 2018, 16, 1059–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takabe, K.; Kim, R.H.; Allegood, J.C.; Mitra, P.; Ramachandran, S.; Nagahashi, M.; Harikumar, K.B.; Hait, N.C.; Milstien, S.; Spiegel, S. Estradiol Induces Export of Sphingosine 1-Phosphate from Breast Cancer Cells via ABCC1 and ABCG. J. Boil. Chem. 2010, 285, 10477–10486. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updat. 2016, 26, 1–9. [Google Scholar] [CrossRef]
- Domenichini, A.; Adamska, A.; Falasca, M. ABC transporters as cancer drivers: Potential functions in cancer development. Biochim. Biophys. Acta BBA Gen. Subj. 2019, 1863, 52–60. [Google Scholar] [CrossRef]
- Tan, K.W.; Sampson, A.; Osa-Andrews, B.; Iram, S.H. Calcitriol and Calcipotriol Modulate Transport Activity of ABC Transporters and Exhibit Selective Cytotoxicity in MRP1-overexpressing Cells. Drug Metab. Dispos. 2018, 46, 1856–1866. [Google Scholar] [CrossRef] [Green Version]
- Ellwood-Yen, K.; Graeber, T.G.; Wongvipat, J.; Iruela-Arispe, M.; Zhang, J.; Matusik, R.; Thomas, G.V.; Sawyers, C.L. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 2003, 4, 223–238. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.W.; Bin Im, Y.; Go, W.-J.; Han, D.-W. c-Myc Amplification Altered the Gene Expression of ABC- and SLC-Transporters in Human Breast Epithelial Cells. Mol. Pharm. 2009, 6, 627–633. [Google Scholar] [CrossRef]
- Wolpaw, A.; Dang, C.V. MYC-induced metabolic stress and tumorigenesis. Biochim. Biophys. Acta BBA Rev. Cancer 2018, 1870, 43–50. [Google Scholar] [CrossRef]
- A Tomlins, S.; Mehra, R.; Rhodes, D.R.; Cao, X.; Wang, L.; Han, S.; Kalyana-Sundaram, S.; Wei, J.T.; Rubin, M.A.; Pienta, K.J.; et al. Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 2006, 39, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Peng, G.; Sahgal, N.; Fazli, L.; Gleave, M.; Zhang, Y.; Hussain, A.; Qi, J. Regulation of c-Myc expression by the histone demethylase JMJD1A is essential for prostate cancer cell growth and survival. Oncogene 2015, 35, 2441–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebello, R.; Kusnadi, E.; Cameron, D.; Pearson, H.; Lesmana, A.; Devlin, J.; Drygin, D.; Clark, A.; Porter, L.; Pedersen, J.; et al. The dual inhibition of RNA Pol I transcription and PIM kinase as a new therapeutic approach to treat advanced prostate cancer. Eur. J. Cancer 2016, 61, S167. [Google Scholar] [CrossRef]
- Whitt, J.D.; Keeton, A.B.; Gary, B.D.; Sklar, L.A.; Sodani, K.; Chen, Z.-S.; Piazza, G.A. Sulindac sulfide selectively increases sensitivity of ABCC1 expressing tumor cells to doxorubicin and glutathione depletion. J. Biomed. Res. 2016, 30, 120–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Chen, X.; Zhu, B.; Ramírez-Alcántara, V.; Canzoneri, J.C.; Lee, K.; Sigler, S.; Gary, B.; Li, Y.; Zhang, W.; et al. Suppression of β-catenin/TCF transcriptional activity and colon tumor cell growth by dual inhibition of PDE5 and 10. Oncotarget 2015, 6, 27403–27415. [Google Scholar] [CrossRef] [Green Version]
- Feller, N.; Kuiper, C.; Lankelma, J.; Ruhdal, J.; Scheper, R.; Pinedo, H.; Broxterman, H. Functional detection of MDR1/P170 and MRP/P190-mediated multidrug resistance in tumour cells by flow cytometry. Br. J. Cancer 1995, 72, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Hedditch, E.L.; Gao, B.; Russell, A.J.; Lu, Y.; Emmanuel, C.; Beesley, J.; Johnatty, S.E.; Chen, X.; Harnett, P.R.; George, J.; et al. ABCA Transporter Gene Expression and Poor Outcome in Epithelial Ovarian Cancer. J. Natl. Cancer Inst. 2014, 106, 149. [Google Scholar] [CrossRef]
- Henderson, M.J.; Haber, M.; Porro, A.; Munoz, M.A.; Iraci, N.; Xue, C.; Murray, J.; Flemming, C.L.; Smith, J.; Fletcher, J.I.; et al. ABCC Multidrug Transporters in Childhood Neuroblastoma: Clinical and Biological Effects Independent of Cytotoxic Drug Efflux. J. Natl. Cancer Inst. 2011, 103, 1236–1251. [Google Scholar] [CrossRef] [Green Version]
- Adamska, A.; Falasca, M. ATP-binding cassette transporters in progression and clinical outcome of pancreatic cancer: What is the way forward? World J. Gastroenterol. 2018, 24, 3222–3238. [Google Scholar] [CrossRef]
- Karatas, O.F.; Tanoglu, E.G.; Duz, M.B.; Ittmann, M.; Ozen, M. The role of ATP-binding cassette transporter genes in the progression of prostate cancer. Prostate 2015, 76, 434–444. [Google Scholar] [CrossRef]
- Robey, R.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Piñeiro, R.; Falasca, M. Lysophosphatidylinositol signalling: New wine from an old bottle. Biochim. Biophys. Acta BBA Mol. Cell Boil. Lipids 2012, 1821, 694–705. [Google Scholar] [CrossRef] [Green Version]
- Falasca, M.; Ferro, R. Role of the lysophosphatidylinositol/GPR55 axis in cancer. Adv. Boil. Regul. 2016, 60, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Ruban, E.L.; Ferro, R.; Arifin, S.A.; Falasca, M. Lysophosphatidylinositol: A novel link between ABC transporters and G-protein-coupled receptors. Biochem. Soc. Trans. 2014, 42, 1372–1377. [Google Scholar] [CrossRef]
- Adamska, A.; Ferro, R.; Lattanzio, R.; Capone, E.; Domenichini, A.; Damiani, V.; Chiorino, G.; Akkaya, B.G.; Linton, K.J.; De Laurenzi, V.; et al. ABCC3 is a novel target for the treatment of pancreatic cancer. Adv. Boil. Regul. 2019, 73, 100634. [Google Scholar] [CrossRef]
- Begicevic, R.-R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [Green Version]
- Begicevic, R.-R.; Arfuso, F.; Falasca, M. Bioactive lipids in cancer stem cells. World J. Stem Cells 2019, 11, 693–704. [Google Scholar] [CrossRef]
- Andradas, C.; Caffarel, M.; Perez, M.D.M.D.M.L.P.; Salazar-Roa, M.; Lorente, M.; Velasco, G.; Guzmán, M.; Sanchez, C. The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene 2010, 30, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Andradas, C.; Blasco-Benito, S.; Castillo-Lluva, S.; Pilla, P.D.-; Alarcia, R.D.; Garcia, A.J.; García-Taboada, E.; Hernando-Llorente, R.; Soriano, J.; Hamann, S.; et al. Activation of the orphan receptor GPR55 by lysophosphatidylinositol promotes metastasis in triple-negative breast cancer. Oncotarget 2016, 7, 47565–47575. [Google Scholar] [CrossRef]
- Ferro, R.; Adamska, A.; Lattanzio, R.; Mavrommati, I.; Edling, C.E.; Arifin, S.A.; Fyffe, C.A.; Sala, G.; Sacchetto, L.; Chiorino, G.; et al. GPR55 signalling promotes proliferation of pancreatic cancer cells and tumour growth in mice, and its inhibition increases effects of gemcitabine. Oncogene 2018, 37, 6368–6382. [Google Scholar] [CrossRef]
- Adamska, A.; Domenichini, A.; Capone, E.; Damiani, V.; Akkaya, B.G.; Linton, K.J.; Di Sebastiano, P.; Chen, X.; Keeton, A.B.; Ramirez-Alcantara, V.; et al. Pharmacological inhibition of ABCC3 slows tumour progression in animal models of pancreatic cancer. J. Exp. Clin. Cancer Res. 2019, 38, 312–314. [Google Scholar] [CrossRef]
- Kool, M.; Van Der Linden, M.; De Haas, M.; Scheffer, G.L.; De Vree, J.M.L.; Smith, A.J.; Jansen, G.; Peters, G.J.; Ponne, N.; Scheper, R.J.; et al. MRP3, an organic anion transporter able to transport anti-cancer drugs. Proc. Natl. Acad. Sci. 1999, 96, 6914–6919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Vo, T.; Hajar, A.; Li, S.; Chen, X.; Parissenti, A.M.; Brindley, D.N.; Wang, Z. Multiple mechanisms underlying acquired resistance to taxanes in selected docetaxel-resistant MCF-7 breast cancer cells. BMC Cancer 2014, 14, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamaki, A.; Ierano’, C.; Szakács, G.; Robey, R.; Bates, S.E. The controversial role of ABC transporters in clinical oncology. Essays Biochem. 2011, 50, 209–332. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Bonafé, P.; Goma, A.; Cantos, R.; Martinez, F.; Tortosa, A.; Vidal, A.; Condom, E.; Tomás, R.P.; Mir, R. Multidrug resistance protein 1 localization in lipid raft domains and prostasomes in prostate cancer cell lines. OncoTargets Ther. 2014, 7, 2215–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingwood, D.; Simons, K. Lipid Rafts As a Membrane-Organizing Principle. Science 2009, 327, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Ren, D. Calcium Signaling in Sperm: Help from Prostasomes. Sci. Signal. 2011, 4, pe27. [Google Scholar] [CrossRef]
- Sullivan, G.F.; Yang, J.-M.; Vassil, A.; Yang, J.; Bash-Babula, J.; Hait, W.N. Regulation of expression of the multidrug resistance protein MRP1 by p53 in human prostate cancer cells. J. Clin. Investig. 2000, 105, 1261–1267. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Li, Z.; Bi, L.; Li, K.; Zhou, B.; Xu, C.; Huang, J.; Xu, K. NOTCH1 signaling promotes chemoresistance via regulating ABCC1 expression in prostate cancer stem cells. Mol. Cell. Biochem. 2014, 393, 265–270. [Google Scholar] [CrossRef]
- Porro, A.; Haber, M.; Diolaiti, D.; Iraci, N.; Henderson, M.; Gherardi, S.; Valli, E.; Munoz, M.A.; Xue, C.; Flemming, C.; et al. Direct and Coordinate Regulation of ATP-binding Cassette Transporter Genes by Myc Factors Generates Specific Transcription Signatures That Significantly Affect the Chemoresistance Phenotype of Cancer Cells. J. Boil. Chem. 2010, 285, 19532–19543. [Google Scholar] [CrossRef] [Green Version]
- Rebello, R.J.; Pearson, R.B.; Hannan, R.D.; Furic, L. Therapeutic Approaches Targeting MYC-Driven Prostate Cancer. Genes 2017, 8, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Chou, T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G.A.; Keeton, A.B.; Tinsley, H.; Gary, B.D.; Whitt, J.D.; Mathew, B.; Thaiparambil, J.; Coward, L.; Gorman, G.; Li, Y.; et al. A novel sulindac derivative that does not inhibit cyclooxygenases but potently inhibits colon tumor cell growth and induces apoptosis with antitumor activity. Cancer Prev. Res. 2009, 2, 572–580. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emmanouilidi, A.; Casari, I.; Gokcen Akkaya, B.; Maffucci, T.; Furic, L.; Guffanti, F.; Broggini, M.; Chen, X.; Maxuitenko, Y.Y.; Keeton, A.B.; et al. Inhibition of the Lysophosphatidylinositol Transporter ABCC1 Reduces Prostate Cancer Cell Growth and Sensitizes to Chemotherapy. Cancers 2020, 12, 2022. https://doi.org/10.3390/cancers12082022
Emmanouilidi A, Casari I, Gokcen Akkaya B, Maffucci T, Furic L, Guffanti F, Broggini M, Chen X, Maxuitenko YY, Keeton AB, et al. Inhibition of the Lysophosphatidylinositol Transporter ABCC1 Reduces Prostate Cancer Cell Growth and Sensitizes to Chemotherapy. Cancers. 2020; 12(8):2022. https://doi.org/10.3390/cancers12082022
Chicago/Turabian StyleEmmanouilidi, Aikaterini, Ilaria Casari, Begum Gokcen Akkaya, Tania Maffucci, Luc Furic, Federica Guffanti, Massimo Broggini, Xi Chen, Yulia Y. Maxuitenko, Adam B. Keeton, and et al. 2020. "Inhibition of the Lysophosphatidylinositol Transporter ABCC1 Reduces Prostate Cancer Cell Growth and Sensitizes to Chemotherapy" Cancers 12, no. 8: 2022. https://doi.org/10.3390/cancers12082022
APA StyleEmmanouilidi, A., Casari, I., Gokcen Akkaya, B., Maffucci, T., Furic, L., Guffanti, F., Broggini, M., Chen, X., Maxuitenko, Y. Y., Keeton, A. B., Piazza, G. A., Linton, K. J., & Falasca, M. (2020). Inhibition of the Lysophosphatidylinositol Transporter ABCC1 Reduces Prostate Cancer Cell Growth and Sensitizes to Chemotherapy. Cancers, 12(8), 2022. https://doi.org/10.3390/cancers12082022