Uptake Transporters of the SLC21, SLC22A, and SLC15A Families in Anticancer Therapy—Modulators of Cellular Entry or Pharmacokinetics?

Abstract
:1. Introduction
2. Organic Anion Transporting Polypeptides in Anticancer Therapy
2.1. OATP1B Transporters
2.2. OATP1A2
2.3. OATP2B1
3. Organic Cation Transporters (OCTs) in Cancer
4. Organic Cation Transporter Novel Type (OCTNs)
5. The Di- and Tripeptide Transporters (PEPTs)
6. Conclusions
Funding
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: Globocan sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, J.; Barr, R.; Shulman, L.N.; Forte, G.B.; Magrini, N. Essential medicines for cancer: Who recommendations and national priorities. Bull. World Health Organ. 2016, 94, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of abc transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Hediger, M.A.; Clemencon, B.; Burrier, R.E.; Bruford, E.A. The abcs of membrane transporters in health and disease (slc series): Introduction. Mol. Asp. Med. 2013, 34, 95–107. [Google Scholar] [CrossRef]
- Slc Tables. Available online: https://www.bioparadigms.org/slc/intro.htm (accessed on 15 July 2020).
- Roth, M.; Obaidat, A.; Hagenbuch, B. Oatps, oats and octs: The organic anion and cation transporters of the slco and slc22a gene superfamilies. Br. J. Pharmacol. 2012, 165, 1260–1287. [Google Scholar] [CrossRef] [Green Version]
- Hagenbuch, B.; Meier, P.J. Organic anion transporting polypeptides of the oatp/slc21 family: Phylogenetic classification as oatp/slco superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004, 447, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Kakyo, M.; Tokui, T.; Nakagomi, R.; Nishio, T.; Nakai, D.; Nomura, H.; Unno, M.; Suzuki, M.; Naitoh, T.; et al. Identification of a novel gene family encoding human liver-specific organic anion transporter lst-1. J. Biol. Chem. 1999, 274, 17159–17163. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Unno, M.; Onogawa, T.; Tokui, T.; Kondo, T.N.; Nakagomi, R.; Adachi, H.; Fujiwara, K.; Okabe, M.; Suzuki, T.; et al. Lst-2, a human liver-specific organic anion transporter, determines methotrexate sensitivity in gastrointestinal cancers. Gastroenterology 2001, 120, 1689–1699. [Google Scholar] [CrossRef]
- Malagnino, V.; Hussner, J.; Seibert, I.; Stolzenburg, A.; Sager, C.P.; Zu Schwabedissen, H.E.M. Lst-3tm12 is a member of the oatp1b family and a functional transporter. Biochem. Pharmacol. 2018, 148, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Obaidat, A.; Roth, M.; Hagenbuch, B. The expression and function of organic anion transporting polypeptides in normal tissues and in cancer. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 135–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirona, R.G.; Leake, B.F.; Merino, G.; Kim, R.B. Polymorphisms in oatp-c: Identification of multiple allelic variants associated with altered transport activity among european- and african-americans. J. Biol. Chem. 2001, 276, 35669–35675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.H.; Ho, R.H. Interindividual and interethnic variability in drug disposition: Polymorphisms in organic anion transporting polypeptide 1b1 (oatp1b1; slco1b1). Br. J. Clin. Pharmacol. 2017, 83, 1176–1184. [Google Scholar] [CrossRef] [Green Version]
- Niemi, M.; Pasanen, M.K.; Neuvonen, P.J. Organic anion transporting polypeptide 1b1: A genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 2011, 63, 157–181. [Google Scholar] [CrossRef]
- Kalliokoski, A.; Niemi, M. Impact of oatp transporters on pharmacokinetics. Br. J. Pharmacol. 2009, 158, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, E.I.; Hu, S.; Roberts, J.L.; Gibson, A.A.; Orwick, S.J.; Li, L.; Sparreboom, A.; Baker, S.D. Contribution of oatp1b1 and oatp1b3 to the disposition of sorafenib and sorafenib-glucuronide. Clin. Cancer Res. 2013, 19, 1458–1466. [Google Scholar] [CrossRef] [Green Version]
- Iusuf, D.; Ludwig, M.; Elbatsh, A.; van Esch, A.; van de Steeg, E.; Wagenaar, E.; van der Valk, M.; Lin, F.; van Tellingen, O.; Schinkel, A.H. Oatp1a/1b transporters affect irinotecan and sn-38 pharmacokinetics and carboxylesterase expression in knockout and humanized transgenic mice. Mol. Cancer Ther. 2014, 13, 492–503. [Google Scholar] [CrossRef] [Green Version]
- Durmus, S.; van Hoppe, S.; Schinkel, A.H. The impact of organic anion-transporting polypeptides (oatps) on disposition and toxicity of antitumor drugs: Insights from knockout and humanized mice. Drug Resist. Updates 2016, 27, 72–88. [Google Scholar] [CrossRef]
- Van de Steeg, E.; Stranecky, V.; Hartmannova, H.; Noskova, L.; Hrebicek, M.; Wagenaar, E.; van Esch, A.; de Waart, D.R.; Oude Elferink, R.P.; Kenworthy, K.E.; et al. Complete oatp1b1 and oatp1b3 deficiency causes human rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J. Clin. Investig. 2012, 122, 519–528. [Google Scholar] [CrossRef]
- Vasilyeva, A.; Durmus, S.; Li, L.; Wagenaar, E.; Hu, S.; Gibson, A.A.; Panetta, J.C.; Mani, S.; Sparreboom, A.; Baker, S.D.; et al. Hepatocellular shuttling and recirculation of sorafenib-glucuronide is dependent on abcc2, abcc3, and oatp1a/1b. Cancer Res. 2015, 75, 2729–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrissey, K.M.; Benet, L.Z.; Ware, J.A. Commentary on: “Influence of oatp1b1 function on the disposition of sorafenib-beta-d-glucuronide”. Clin. Transl. Sci. 2017, 10, 240–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bins, S.; van Doorn, L.; Phelps, M.A.; Gibson, A.A.; Hu, S.; Li, L.; Vasilyeva, A.; Du, G.; Hamberg, P.; Eskens, F.; et al. Influence of oatp1b1 function on the disposition of sorafenib-beta-d-glucuronide. Clin. Transl. Sci. 2017, 10, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bins, S.; Lenting, A.; El Bouazzaoui, S.; van Doorn, L.; Oomen-de Hoop, E.; Eskens, F.A.; van Schaik, R.H.; Mathijssen, R.H. Polymorphisms in slco1b1 and ugt1a1 are associated with sorafenib-induced toxicity. Pharmacogenomics 2016, 17, 1483–1490. [Google Scholar] [CrossRef]
- Chen, M.; Neul, C.; Schaeffeler, E.; Frisch, F.; Winter, S.; Schwab, M.; Koepsell, H.; Hu, S.; Laufer, S.; Baker, S.D.; et al. Sorafenib activity and disposition in liver cancer does not depend on organic cation transporter 1. Clin. Pharmacol. Ther. 2020, 107, 227–237. [Google Scholar] [CrossRef]
- Swift, B.; Nebot, N.; Lee, J.K.; Han, T.; Proctor, W.R.; Thakker, D.R.; Lang, D.; Radtke, M.; Gnoth, M.J.; Brouwer, K.L. Sorafenib hepatobiliary disposition: Mechanisms of hepatic uptake and disposition of generated metabolites. Drug Metab. Dispos. 2013, 41, 1179–1186. [Google Scholar] [CrossRef] [Green Version]
- Herraez, E.; Lozano, E.; Macias, R.I.; Vaquero, J.; Bujanda, L.; Banales, J.M.; Marin, J.J.; Briz, O. Expression of slc22a1 variants may affect the response of hepatocellular carcinoma and cholangiocarcinoma to sorafenib. Hepatology 2013, 58, 1065–1073. [Google Scholar] [CrossRef]
- Hu, S.; Chen, Z.; Franke, R.; Orwick, S.; Zhao, M.; Rudek, M.A.; Sparreboom, A.; Baker, S.D. Interaction of the multikinase inhibitors sorafenib and sunitinib with solute carriers and atp-binding cassette transporters. Clin. Cancer Res. 2009, 15, 6062–6069. [Google Scholar] [CrossRef] [Green Version]
- Grimm, D.; Lieb, J.; Weyer, V.; Vollmar, J.; Darstein, F.; Lautem, A.; Hoppe-Lotichius, M.; Koch, S.; Schad, A.; Schattenberg, J.M.; et al. Organic cation transporter 1 (oct1) mrna expression in hepatocellular carcinoma as a biomarker for sorafenib treatment. BMC Cancer 2016, 16, 94. [Google Scholar] [CrossRef] [Green Version]
- Geier, A.; Macias, R.I.; Bettinger, D.; Weiss, J.; Bantel, H.; Jahn, D.; Al-Abdulla, R.; Marin, J.J. The lack of the organic cation transporter oct1 at the plasma membrane of tumor cells precludes a positive response to sorafenib in patients with hepatocellular carcinoma. Oncotarget 2017, 8, 15846–15857. [Google Scholar] [CrossRef] [Green Version]
- Schaeffeler, E.; Hellerbrand, C.; Nies, A.T.; Winter, S.; Kruck, S.; Hofmann, U.; van der Kuip, H.; Zanger, U.M.; Koepsell, H.; Schwab, M. DNA methylation is associated with downregulation of the organic cation transporter oct1 (slc22a1) in human hepatocellular carcinoma. Genome Med. 2011, 3, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Abdulla, R.; Lozano, E.; Macias, R.I.R.; Monte, M.J.; Briz, O.; O’Rourke, C.J.; Serrano, M.A.; Banales, J.M.; Avila, M.A.; Martinez-Chantar, M.L.; et al. Epigenetic events involved in organic cation transporter 1-dependent impaired response of hepatocellular carcinoma to sorafenib. Br. J. Pharmacol. 2019, 176, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, B.H.; Kim, B.C.; Shin, A.; Kim, J.S.; Hong, S.H.; Hwang, J.A.; Lee, J.A.; Nam, S.; Lee, S.H.; et al. Slc15a2 genomic variation is associated with the extraordinary response of sorafenib treatment: Whole-genome analysis in patients with hepatocellular carcinoma. Oncotarget 2015, 6, 16449–16460. [Google Scholar] [CrossRef] [PubMed]
- Knutter, I.; Wollesky, C.; Kottra, G.; Hahn, M.G.; Fischer, W.; Zebisch, K.; Neubert, R.H.; Daniel, H.; Brandsch, M. Transport of angiotensin-converting enzyme inhibitors by h+/peptide transporters revisited. J. Pharmacol. Exp. Ther. 2008, 327, 432–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolis, L.; Butler, T.H. An unusual and heavy infection of a prawn, pandalus borealis kroyer, by a nematode, Contracaecum sp. J. Parasitol. 1954, 40, 649–655. [Google Scholar] [CrossRef]
- Han, J.Y.; Lim, H.S.; Shin, E.S.; Yoo, Y.K.; Park, Y.H.; Lee, J.E.; Kim, H.T.; Lee, J.S. Influence of the organic anion-transporting polypeptide 1b1 (oatp1b1) polymorphisms on irinotecan-pharmacokinetics and clinical outcome of patients with advanced non-small cell lung cancer. Lung Cancer 2008, 59, 69–75. [Google Scholar] [CrossRef]
- Teft, W.A.; Welch, S.; Lenehan, J.; Parfitt, J.; Choi, Y.H.; Winquist, E.; Kim, R.B. Oatp1b1 and tumour oatp1b3 modulate exposure, toxicity, and survival after irinotecan-based chemotherapy. Br. J. Cancer 2015, 112, 857–865. [Google Scholar] [CrossRef]
- Fujita, D.; Saito, Y.; Nakanishi, T.; Tamai, I. Organic anion transporting polypeptide (oatp)2b1 contributes to gastrointestinal toxicity of anticancer drug sn-38, active metabolite of irinotecan hydrochloride. Drug Metab. Dispos. 2016, 44, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Mostaghel, E.A.; Cho, E.; Zhang, A.; Alyamani, M.; Kaipainen, A.; Green, S.; Marck, B.T.; Sharifi, N.; Wright, J.L.; Gulati, R.; et al. Association of tissue abiraterone levels and slco genotype with intraprostatic steroids and pathologic response in men with high-risk localized prostate cancer. Clin. Cancer Res. 2017, 23, 4592–4601. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.W.; Gill, D.M.; Poole, A.; Nussenzveig, R.H.; Wilson, S.; Farnham, J.M.; Stephenson, R.A.; Cannon-Albright, L.A.; Maughan, B.L.; Agarwal, N. Germline variant in slco2b1 and response to abiraterone acetate plus prednisone (aa) in new-onset metastatic castration-resistant prostate cancer (mcrpc). Mol. Cancer Ther. 2019, 18, 726–729. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhou, W. Ameliorative effects of slc22a2 gene polymorphism 808 g/t and cimetidine on cisplatin-induced nephrotoxicity in Chinese cancer patients. Food Chem. Toxicol. 2012, 50, 2289–2293. [Google Scholar] [CrossRef] [PubMed]
- Eechoute, K.; Franke, R.M.; Loos, W.J.; Scherkenbach, L.A.; Boere, I.; Verweij, J.; Gurney, H.; Kim, R.B.; Tirona, R.G.; Mathijssen, R.H.; et al. Environmental and genetic factors affecting transport of imatinib by oatp1a2. Clin. Pharmacol. Ther. 2011, 89, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Angelini, S.; Soverini, S.; Ravegnini, G.; Barnett, M.; Turrini, E.; Thornquist, M.; Pane, F.; Hughes, T.P.; White, D.L.; Radich, J.; et al. Association between imatinib transporters and metabolizing enzymes genotype and response in newly diagnosed chronic myeloid leukemia patients receiving imatinib therapy. Haematologica 2013, 98, 193–200. [Google Scholar] [CrossRef]
- Yamakawa, Y.; Hamada, A.; Shuto, T.; Yuki, M.; Uchida, T.; Kai, H.; Kawaguchi, T.; Saito, H. Pharmacokinetic impact of slco1a2 polymorphisms on imatinib disposition in patients with chronic myeloid leukemia. Clin. Pharmacol. Ther. 2011, 90, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Angelini, S.; Pantaleo, M.A.; Ravegnini, G.; Zenesini, C.; Cavrini, G.; Nannini, M.; Fumagalli, E.; Palassini, E.; Saponara, M.; Di Battista, M.; et al. Polymorphisms in octn1 and octn2 transporters genes are associated with prolonged time to progression in unresectable gastrointestinal stromal tumours treated with imatinib therapy. Pharmacol. Res. 2013, 68, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yin, J.; Li, W.; Xiao, C.; Han, J.; Zhou, F. Association between slco1a2 genetic variation and methotrexate toxicity in human rheumatoid arthritis treatment. J. Biochem. Mol. Toxicol. 2020, e22513. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Zheng, J.; Zhu, L.; Jodal, A.; Cui, P.H.; Wong, M.; Gurney, H.; Church, W.B.; Murray, M. Functional analysis of novel polymorphisms in the human slco1a2 gene that encodes the transporter oatp1a2. AAPS J. 2013, 15, 1099–1108. [Google Scholar] [CrossRef]
- Kloth, J.S.L.; Verboom, M.C.; Swen, J.J.; van der Straaten, T.; Sleijfer, S.; Reyners, A.K.L.; Steeghs, N.; Gelderblom, H.; Guchelaar, H.J.; Mathijssen, R.H.J. Genetic polymorphisms as predictive biomarker of survival in patients with gastrointestinal stromal tumors treated with sunitinib. Pharmacogenet. J. 2018, 18, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Callens, C.; Debled, M.; Delord, M.; Turbiez-Stalain, I.; Veyret, C.; Bieche, I.; Brain, E. High-throughput pharmacogenetics identifies slco1a2 polymorphisms as candidates to elucidate the risk of febrile neutropenia in the breast cancer rapp-01 trial. Breast Cancer Res. Treat. 2015, 153, 383–389. [Google Scholar] [CrossRef]
- Drenberg, C.D.; Paugh, S.W.; Pounds, S.B.; Shi, L.; Orwick, S.J.; Li, L.; Hu, S.; Gibson, A.A.; Ribeiro, R.C.; Rubnitz, J.E.; et al. Inherited variation in oatp1b1 is associated with treatment outcome in acute myeloid leukemia. Clin. Pharmacol. Ther. 2016, 99, 651–660. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.M.; Lin, P.M.; Chang, J.G.; Lin, H.C.; Li, S.H.; Lin, S.F.; Yang, M.Y. Upregulated slc22a3 has a potential for improving survival of patients with head and neck squamous cell carcinoma receiving cisplatin treatment. Oncotarget 2017, 8, 74348–74358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drenberg, C.D.; Gibson, A.A.; Pounds, S.B.; Shi, L.; Rhinehart, D.P.; Li, L.; Hu, S.; Du, G.; Nies, A.T.; Schwab, M.; et al. Octn1 is a high-affinity carrier of nucleoside analogues. Cancer Res. 2017, 77, 2102–2111. [Google Scholar] [CrossRef] [Green Version]
- Nishino, S.; Itoh, A.; Matsuoka, H.; Maeda, K.; Kamoshida, S. Immunohistochemical analysis of organic anion transporter 2 and reduced folate carrier 1 in colorectal cancer: Significance as a predictor of response to oral uracil/ftorafur plus leucovorin chemotherapy. Mol. Clin. Oncol. 2013, 1, 661–667. [Google Scholar]
- Tashiro, A.; Tatsumi, S.; Takeda, R.; Naka, A.; Matsuoka, H.; Hashimoto, Y.; Hatta, K.; Maeda, K.; Kamoshida, S. High expression of organic anion transporter 2 and organic cation transporter 2 is an independent predictor of good outcomes in patients with metastatic colorectal cancer treated with folfox-based chemotherapy. Am. J. Cancer Res. 2014, 4, 528–536. [Google Scholar]
- Le Roy, B.; Tixier, L.; Pereira, B.; Sauvanet, P.; Buc, E.; Petorin, C.; Dechelotte, P.; Pezet, D.; Balayssac, D. Assessment of the relation between the expression of oxaliplatin transporters in colorectal cancer and response to folfox-4 adjuvant chemotherapy: A case control study. PLoS ONE 2016, 11, e0148739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsumi, S.; Matsuoka, H.; Hashimoto, Y.; Hatta, K.; Maeda, K.; Kamoshida, S. Organic cation transporter 2 and tumor budding as independent prognostic factors in metastatic colorectal cancer patients treated with oxaliplatin-based chemotherapy. Int. J. Clin. Exp. Pathol. 2014, 7, 204–212. [Google Scholar] [PubMed]
- Hashimoto, Y.; Tatsumi, S.; Takeda, R.; Naka, A.; Ogane, N.; Kameda, Y.; Kawachi, K.; Shimizu, S.; Sakai, M.; Kamoshida, S. Expression of organic anion-transporting polypeptide 1a2 and organic cation transporter 6 as a predictor of pathologic response to neoadjuvant chemotherapy in triple negative breast cancer. Breast Cancer Res. Treat. 2014, 145, 101–111. [Google Scholar] [CrossRef]
- Van de Steeg, E.; van Esch, A.; Wagenaar, E.; Kenworthy, K.E.; Schinkel, A.H. Influence of human oatp1b1, oatp1b3, and oatp1a2 on the pharmacokinetics of methotrexate and paclitaxel in humanized transgenic mice. Clin. Cancer Res. 2013, 19, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Iusuf, D.; Hendrikx, J.J.; van Esch, A.; van de Steeg, E.; Wagenaar, E.; Rosing, H.; Beijnen, J.H.; Schinkel, A.H. Human oatp1b1, oatp1b3 and oatp1a2 can mediate the in vivo uptake and clearance of docetaxel. Int. J. Cancer 2015, 136, 225–233. [Google Scholar] [CrossRef]
- Lee, H.H.; Leake, B.F.; Teft, W.; Tirona, R.G.; Kim, R.B.; Ho, R.H. Contribution of hepatic organic anion-transporting polypeptides to docetaxel uptake and clearance. Mol. Cancer Ther. 2015, 14, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.H.; Leake, B.F.; Kim, R.B.; Ho, R.H. Contribution of organic anion-transporting polypeptides 1a/1b to doxorubicin uptake and clearance. Mol. Pharmacol. 2017, 91, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, S.D.; Verweij, J.; Cusatis, G.A.; van Schaik, R.H.; Marsh, S.; Orwick, S.J.; Franke, R.M.; Hu, S.; Schuetz, E.G.; Lamba, V.; et al. Pharmacogenetic pathway analysis of docetaxel elimination. Clin. Pharmacol. Ther. 2009, 85, 155–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Graan, A.J.; Lancaster, C.S.; Obaidat, A.; Hagenbuch, B.; Elens, L.; Friberg, L.E.; de Bruijn, P.; Hu, S.; Gibson, A.A.; Bruun, G.H.; et al. Influence of polymorphic oatp1b-type carriers on the disposition of docetaxel. Clin. Cancer Res. 2012, 18, 4433–4440. [Google Scholar] [CrossRef] [Green Version]
- Chew, S.C.; Sandanaraj, E.; Singh, O.; Chen, X.; Tan, E.H.; Lim, W.T.; Lee, E.J.; Chowbay, B. Influence of slco1b3 haplotype-tag snps on docetaxel disposition in Chinese nasopharyngeal cancer patients. Br. J. Clin. Pharmacol. 2012, 73, 606–618. [Google Scholar] [CrossRef] [Green Version]
- Schulte, R.R.; Ho, R.H. Organic anion transporting polypeptides: Emerging roles in cancer pharmacology. Mol. Pharmacol. 2019, 95, 490–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhuri, S.; Klaassen, C.D. Elucidation of oatp1b1 and 1b3 transporter function using transgenic rodent models and commonly known single nucleotide polymorphisms. Toxicol. Appl. Pharmacol. 2020, 399, 115039. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, U.I.; zu Schwabedissen, H.E.M.; Tirona, R.G.; Suzuki, A.; Leake, B.F.; Mokrab, Y.; Mizuguchi, K.; Ho, R.H.; Kim, R.B. Identification of novel functional organic anion-transporting polypeptide 1b3 polymorphisms and assessment of substrate specificity. Pharmacogenet. Genom. 2011, 21, 103–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada, A.; Sissung, T.; Price, D.K.; Danesi, R.; Chau, C.H.; Sharifi, N.; Venzon, D.; Maeda, K.; Nagao, K.; Sparreboom, A.; et al. Effect of slco1b3 haplotype on testosterone transport and clinical outcome in caucasian patients with androgen-independent prostatic cancer. Clin. Cancer Res. 2008, 14, 3312–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nies, A.T.; Niemi, M.; Burk, O.; Winter, S.; Zanger, U.M.; Stieger, B.; Schwab, M.; Schaeffeler, E. Genetics is a major determinant of expression of the human hepatic uptake transporter oatp1b1, but not of oatp1b3 and oatp2b1. Genome Med. 2013, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakkar, N.; Kim, K.; Jang, E.R.; Han, S.; Kim, K.; Kim, D.; Merchant, N.; Lockhart, A.C.; Lee, W. A cancer-specific variant of the slco1b3 gene encodes a novel human organic anion transporting polypeptide 1b3 (oatp1b3) localized mainly in the cytoplasm of colon and pancreatic cancer cells. Mol. Pharm. 2013, 10, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Kikuchi, R.; Tsuruya, Y.; Naoi, S.; Nishida, S.; Kusuhara, H.; Sugiyama, Y. Epigenetic regulation of organic anion transporting polypeptide 1b3 in cancer cell lines. Pharm. Res. 2013, 30, 2880–2890. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Furihata, T.; Matsumoto, S.; Ishii, S.; Motohashi, S.; Yoshino, I.; Ugajin, M.; Miyajima, A.; Matsumoto, S.; Chiba, K. Identification of a new organic anion transporting polypeptide 1b3 mrna isoform primarily expressed in human cancerous tissues and cells. Biochem. Biophys. Res. Commun. 2012, 418, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Furihata, T.; Ishii, S.; Nagai, M.; Harada, M.; Shimozato, O.; Kamijo, T.; Motohashi, S.; Yoshino, I.; Kamiichi, A.; et al. Unique expression features of cancer-type organic anion transporting polypeptide 1b3 mrna expression in human colon and lung cancers. Clin. Transl. Med. 2014, 3, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Yuan, J.; Li, Z.; Wang, Z.; Cheng, D.; Du, Y.; Li, W.; Kan, Q.; Zhang, W. Genetic polymorphisms and function of the organic anion-transporting polypeptide 1a2 and its clinical relevance in drug disposition. Pharmacology 2015, 95, 201–208. [Google Scholar] [CrossRef]
- Liedauer, R.; Svoboda, M.; Wlcek, K.; Arrich, F.; Ja, W.; Toma, C.; Thalhammer, T. Different expression patterns of organic anion transporting polypeptides in osteosarcomas, bone metastases and aneurysmal bone cysts. Oncol. Rep. 2009, 22, 1485–1492. [Google Scholar]
- Ballestero, M.R.; Monte, M.J.; Briz, O.; Jimenez, F.; Gonzalez-San Martin, F.; Marin, J.J. Expression of transporters potentially involved in the targeting of cytostatic bile acid derivatives to colon cancer and polyps. Biochem. Pharmacol. 2006, 72, 729–738. [Google Scholar] [CrossRef]
- Miki, Y.; Suzuki, T.; Kitada, K.; Yabuki, N.; Shibuya, R.; Moriya, T.; Ishida, T.; Ohuchi, N.; Blumberg, B.; Sasano, H. Expression of the steroid and xenobiotic receptor and its possible target gene, organic anion transporting polypeptide-a, in human breast carcinoma. Cancer Res. 2006, 66, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Zu Schwabedissen, H.E.M.; Tirona, R.G.; Yip, C.S.; Ho, R.H.; Kim, R.B. Interplay between the nuclear receptor pregnane x receptor and the uptake transporter organic anion transporter polypeptide 1a2 selectively enhances estrogen effects in breast cancer. Cancer Res. 2008, 68, 9338–9347. [Google Scholar] [CrossRef] [Green Version]
- Justenhoven, C.; Schaeffeler, E.; Winter, S.; Baisch, C.; Hamann, U.; Harth, V.; Rabstein, S.; Spickenheuer, A.; Pesch, B.; Bruning, T.; et al. Polymorphisms of the nuclear receptor pregnane x receptor and organic anion transporter polypeptides 1a2, 1b1, 1b3, and 2b1 are not associated with breast cancer risk. Breast Cancer Res. Treat. 2011, 125, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, N.; Allen, C.; Bendayan, R. Differential role of organic anion-transporting polypeptides in estrone-3-sulphate uptake by breast epithelial cells and breast cancer cells. J. Pharmacol. Exp. Ther. 2012, 342, 510–519. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, N.; Fonge, H.; Mikhail, A.; Reilly, R.M.; Bendayan, R.; Allen, C. Estrone-3-sulphate, a potential novel ligand for targeting breast cancers. PLoS ONE 2013, 8, e64069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenady, D.E.; Pavlik, E.J.; Nelson, K.; van Nagell, J.R.; Gallion, H.; DePriest, P.D.; Ryo, U.Y.; Baranczuk, R.J. Images of estrogen-receptor-positive breast tumors produced by estradiol labeled with iodine i 123 at 16 alpha. Arch. Surg. 1993, 128, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Benard, F.; Ahmed, N.; Beauregard, J.M.; Rousseau, J.; Aliaga, A.; Dubuc, C.; Croteau, E.; van Lier, J.E. [18f]fluorinated estradiol derivatives for oestrogen receptor imaging: Impact of substituents, formulation and specific activity on the biodistribution in breast tumour-bearing mice. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1473–1479. [Google Scholar] [CrossRef]
- Lal, S.; Wong, Z.W.; Jada, S.R.; Xiang, X.; Chen, S.X.; Ang, P.C.; Figg, W.D.; Lee, E.J.; Chowbay, B. Novel slc22a16 polymorphisms and influence on doxorubicin pharmacokinetics in asian breast cancer patients. Pharmacogenomics 2007, 8, 567–575. [Google Scholar] [CrossRef]
- Arakawa, H.; Nakanishi, T.; Yanagihara, C.; Nishimoto, T.; Wakayama, T.; Mizokami, A.; Namiki, M.; Kawai, K.; Tamai, I. Enhanced expression of organic anion transporting polypeptides (oatps) in androgen receptor-positive prostate cancer cells: Possible role of oatp1a2 in adaptive cell growth under androgen-depleted conditions. Biochem. Pharmacol. 2012, 84, 1070–1077. [Google Scholar] [CrossRef] [Green Version]
- Wright, J.L.; Kwon, E.M.; Ostrander, E.A.; Montgomery, R.B.; Lin, D.W.; Vessella, R.; Stanford, J.L.; Mostaghel, E.A. Expression of slco transport genes in castration-resistant prostate cancer and impact of genetic variation in slco1b3 and slco2b1 on prostate cancer outcomes. Cancer Epidemiol. Biomark. Prev. 2011, 20, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Kullak-Ublick, G.A.; Hagenbuch, B.; Stieger, B.; Schteingart, C.D.; Hofmann, A.F.; Wolkoff, A.W.; Meier, P.J. Molecular and functional characterization of an organic anion transporting polypeptide cloned from human liver. Gastroenterology 1995, 109, 1274–1282. [Google Scholar] [CrossRef]
- Tamai, I.; Nezu, J.; Uchino, H.; Sai, Y.; Oku, A.; Shimane, M.; Tsuji, A. Molecular identification and characterization of novel members of the human organic anion transporter (oatp) family. Biochem. Biophys. Res. Commun. 2000, 273, 251–260. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, Z.; Tay-Sontheimer, J.; Levy, R.H.; Ragueneau-Majlessi, I. Intestinal drug interactions mediated by oatps: A systematic review of preclinical and clinical findings. J. Pharm. Sci. 2017, 106, 2312–2325. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Hagenbuch, B.; Kullak-Ublick, G.A.; Benke, D.; Aguzzi, A.; Meier, P.J. Organic anion-transporting polypeptides mediate transport of opioid peptides across blood-brain barrier. J. Pharmacol. Exp. Ther. 2000, 294, 73–79. [Google Scholar]
- Bronger, H.; Konig, J.; Kopplow, K.; Steiner, H.H.; Ahmadi, R.; Herold-Mende, C.; Keppler, D.; Nies, A.T. Abcc drug efflux pumps and organic anion uptake transporters in human gliomas and the blood-tumor barrier. Cancer Res. 2005, 65, 11419–11428. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, A.M.; Zu Schwabedissen, H.E.M.; Bien-Moller, S.; Hubeny, A.; Vogelgesang, S.; Oswald, S.; Grube, M. Oatp1a2 and oatp2b1 are interacting with dopamine-receptor agonists and antagonists. Mol. Pharm. 2020, 17, 1987–1995. [Google Scholar] [CrossRef] [PubMed]
- Billington, S.; Salphati, L.; Hop, C.; Chu, X.; Evers, R.; Burdette, D.; Rowbottom, C.; Lai, Y.; Xiao, G.; Humphreys, W.G.; et al. Interindividual and regional variability in drug transporter abundance at the human blood-brain barrier measured by quantitative targeted proteomics. Clin. Pharmacol. Ther. 2019, 106, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Wu, J.; Xie, Y.; Kim, S.; Michelhaugh, S.; Jiang, J.; Mittal, S.; Sanai, N.; Li, J. Protein expression and functional relevance of efflux and uptake drug transporters at the blood-brain barrier of human brain and glioblastoma. Clin. Pharmacol. Ther. 2020, 107, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Franke, R.M.; Filipski, K.K.; Hu, C.; Orwick, S.J.; de Bruijn, E.A.; Burger, H.; Baker, S.D.; Sparreboom, A. Interaction of imatinib with human organic ion carriers. Clin. Cancer Res. 2008, 14, 3141–3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huennekens, F.M. The methotrexate story: A paradigm for development of cancer chemotherapeutic agents. Adv. Enzyme Regul. 1994, 34, 397–419. [Google Scholar] [CrossRef]
- Badagnani, I.; Castro, R.A.; Taylor, T.R.; Brett, C.M.; Huang, C.C.; Stryke, D.; Kawamoto, M.; Johns, S.J.; Ferrin, T.E.; Carlson, E.J.; et al. Interaction of methotrexate with organic-anion transporting polypeptide 1a2 and its genetic variants. J. Pharmacol. Exp. Ther. 2006, 318, 521–529. [Google Scholar] [CrossRef]
- Gschwind, H.P.; Pfaar, U.; Waldmeier, F.; Zollinger, M.; Sayer, C.; Zbinden, P.; Hayes, M.; Pokorny, R.; Seiberling, M.; Ben-Am, M.; et al. Metabolism and disposition of imatinib mesylate in healthy volunteers. Drug Metab. Dispos. 2005, 33, 1503–1512. [Google Scholar] [CrossRef] [Green Version]
- Jaruskova, M.; Curik, N.; Hercog, R.; Polivkova, V.; Motlova, E.; Benes, V.; Klamova, H.; Pecherkova, P.; Belohlavkova, P.; Vrbacky, F.; et al. Genotypes of slc22a4 and slc22a5 regulatory loci are predictive of the response of chronic myeloid leukemia patients to imatinib treatment. J. Exp. Clin. Cancer Res. 2017, 36, 55. [Google Scholar] [CrossRef]
- Thomas, J.; Wang, L.; Clark, R.E.; Pirmohamed, M. Active transport of imatinib into and out of cells: Implications for drug resistance. Blood 2004, 104, 3739–3745. [Google Scholar] [CrossRef] [Green Version]
- Nies, A.T.; Schaeffeler, E.; van der Kuip, H.; Cascorbi, I.; Bruhn, O.; Kneba, M.; Pott, C.; Hofmann, U.; Volk, C.; Hu, S.; et al. Cellular uptake of imatinib into leukemic cells is independent of human organic cation transporter 1 (oct1). Clin. Cancer Res. 2014, 20, 985–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, D.L.; Saunders, V.A.; Dang, P.; Engler, J.; Venables, A.; Zrim, S.; Zannettino, A.; Lynch, K.; Manley, P.W.; Hughes, T. Most cml patients who have a suboptimal response to imatinib have low oct-1 activity: Higher doses of imatinib may overcome the negative impact of low oct-1 activity. Blood 2007, 110, 4064–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, D.L.; Radich, J.; Soverini, S.; Saunders, V.A.; Frede, A.K.; Dang, P.; Cilloni, D.; Lin, P.; Mongay, L.; Woodman, R.; et al. Chronic phase chronic myeloid leukemia patients with low oct-1 activity randomized to high-dose imatinib achieve better responses and have lower failure rates than those randomized to standard-dose imatinib. Haematologica 2012, 97, 907–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, D.B.; Hughes, T.P.; White, D.L. Oct1 and imatinib transport in cml: Is it clinically relevant? Leukemia 2015, 29, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Farag, S.; Verheijen, R.B.; Martijn Kerst, J.; Cats, A.; Huitema, A.D.; Steeghs, N. Imatinib pharmacokinetics in a large observational cohort of gastrointestinal stromal tumour patients. Clin. Pharmacokinet. 2017, 56, 287–292. [Google Scholar] [CrossRef]
- Gong, I.Y.; Kim, R.B. Impact of genetic variation in oatp transporters to drug disposition and response. Drug Metab. Pharmacokinet. 2013, 28, 4–18. [Google Scholar] [CrossRef] [Green Version]
- Van de Steeg, E.; Wagenaar, E.; van der Kruijssen, C.M.; Burggraaff, J.E.; de Waart, D.R.; Elferink, R.P.; Kenworthy, K.E.; Schinkel, A.H. Organic anion transporting polypeptide 1a/1b-knockout mice provide insights into hepatic handling of bilirubin, bile acids, and drugs. J. Clin. Investig. 2010, 120, 2942–2952. [Google Scholar] [CrossRef]
- Cipriani, P.; Ruscitti, P.; Carubbi, F.; Liakouli, V.; Giacomelli, R. Methotrexate: An old new drug in autoimmune disease. Expert Rev. Clin. Immunol. 2014, 10, 1519–1530. [Google Scholar] [CrossRef]
- Uwai, Y.; Taniguchi, R.; Motohashi, H.; Saito, H.; Okuda, M.; Inui, K. Methotrexate-loxoprofen interaction: Involvement of human organic anion transporters hoat1 and hoat3. Drug Metab. Pharmacokinet. 2004, 19, 369–374. [Google Scholar] [CrossRef]
- Aherne, G.W.; Piall, E.; Marks, V.; Mould, G.; White, W.F. Prolongation and enhancement of serum methotrexate concentrations by probenecid. Br. Med. J. 1978, 1, 1097–1099. [Google Scholar] [CrossRef] [Green Version]
- Frenia, M.L.; Long, K.S. Methotrexate and nonsteroidal antiinflammatory drug interactions. Ann. Pharmacother. 1992, 26, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Doki, K.; Homma, M.; Tamaki, H.; Hori, S.; Ohtani, H.; Sawada, Y.; Kohda, Y. Co-administration of proton pump inhibitors delays elimination of plasma methotrexate in high-dose methotrexate therapy. Br. J. Clin. Pharmacol. 2009, 67, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narumi, K.; Sato, Y.; Kobayashi, M.; Furugen, A.; Kasashi, K.; Yamada, T.; Teshima, T.; Iseki, K. Effects of proton pump inhibitors and famotidine on elimination of plasma methotrexate: Evaluation of drug-drug interactions mediated by organic anion transporter 3. Biopharm. Drug Dispos. 2017, 38, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Kullak-Ublick, G.A.; Ismair, M.G.; Stieger, B.; Landmann, L.; Huber, R.; Pizzagalli, F.; Fattinger, K.; Meier, P.J.; Hagenbuch, B. Organic anion-transporting polypeptide b (oatp-b) and its functional comparison with three other oatps of human liver. Gastroenterology 2001, 120, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Bleasby, K.; Castle, J.C.; Roberts, C.J.; Cheng, C.; Bailey, W.J.; Sina, J.F.; Kulkarni, A.V.; Hafey, M.J.; Evers, R.; Johnson, J.M.; et al. Expression profiles of 50 xenobiotic transporter genes in humans and pre-clinical species: A resource for investigations into drug disposition. Xenobiotica 2006, 36, 963–988. [Google Scholar] [CrossRef]
- Drozdzik, M.; Busch, D.; Lapczuk, J.; Muller, J.; Ostrowski, M.; Kurzawski, M.; Oswald, S. Protein abundance of clinically relevant drug transporters in the human liver and intestine: A comparative analysis in paired tissue specimens. Clin. Pharmacol. Ther. 2019, 105, 1204–1212. [Google Scholar] [CrossRef]
- Knauer, M.J.; Urquhart, B.L.; zu Schwabedissen, H.E.M.; Schwarz, U.I.; Lemke, C.J.; Leake, B.F.; Kim, R.B.; Tirona, R.G. Human skeletal muscle drug transporters determine local exposure and toxicity of statins. Circ. Res. 2010, 106, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, C.; Hagen, P.; Stern, M.; Hussner, J.; Zimmermann, U.; Grube, M.; Zu Schwabedissen, H.E.M. The scaffold protein pdzk1 modulates expression and function of the organic anion transporting polypeptide 2b1. Eur. J. Pharm. Sci. 2018, 120, 181–190. [Google Scholar] [CrossRef]
- Hussner, J.; Begunk, R.; Boettcher, K.; Gliesche, D.G.; Prestin, K.; Zu Schwabedissen, H.E.M. Expression of oatp2b1 as determinant of drug effects in the microcompartment of the coronary artery. Vasc. Pharmacol. 2015, 72, 25–34. [Google Scholar] [CrossRef]
- Bauer, M.; Traxl, A.; Matsuda, A.; Karch, R.; Philippe, C.; Nics, L.; Klebermass, E.M.; Wulkersdorfer, B.; Weber, M.; Poschner, S.; et al. Effect of rifampicin on the distribution of [(11)c]erlotinib to the liver, a translational pet study in humans and in mice. Mol. Pharm. 2018, 15, 4589–4598. [Google Scholar] [CrossRef]
- Schaefer, A.M.; Bock, T.; Zu Schwabedissen, H.E.M. Establishment and validation of competitive counterflow as a method to detect substrates of the organic anion transporting polypeptide 2b1. Mol. Pharm. 2018, 15, 5501–5513. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Harshman, L.C.; Xie, W.; Nakabayashi, M.; Qu, F.; Pomerantz, M.M.; Lee, G.S.; Kantoff, P.W. Association of slco2b1 genotypes with time to progression and overall survival in patients receiving androgen-deprivation therapy for prostate cancer. J. Clin. Oncol. 2016, 34, 352–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alex, A.B.; Pal, S.K.; Agarwal, N. Cyp17 inhibitors in prostate cancer: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2016, 8, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kou, L.; Yao, Q.; Sivaprakasam, S.; Luo, Q.; Sun, Y.; Fu, Q.; He, Z.; Sun, J.; Ganapathy, V. Dual targeting of l-carnitine-conjugated nanoparticles to octn2 and atb(0,+) to deliver chemotherapeutic agents for colon cancer therapy. Drug Deliv. 2017, 24, 1338–1349. [Google Scholar] [CrossRef] [Green Version]
- Criado, J.J.; Dominguez, M.F.; Medarde, M.; Fernandez, E.R.; Macias, R.I.; Marin, J.J. Structural characterization, kinetic studies, and in vitro biological activity of new cis-diamminebis-cholylglycinate(o,o’) pt(ii) and cis-diamminebis-ursodeoxycholate(o,o’) pt(ii) complexes. Bioconjug. Chem. 2000, 11, 167–174. [Google Scholar] [CrossRef]
- Viennois, E.; Ingersoll, S.A.; Ayyadurai, S.; Zhao, Y.; Wang, L.; Zhang, M.; Han, M.K.; Garg, P.; Xiao, B.; Merlin, D. Critical role of pept1 in promoting colitis-associated cancer and therapeutic benefits of the anti-inflammatory pept1-mediated tripeptide kpv in a murine model. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 340–357. [Google Scholar] [CrossRef] [Green Version]
- Pusztai, L.; Wagner, P.; Ibrahim, N.; Rivera, E.; Theriault, R.; Booser, D.; Symmans, F.W.; Wong, F.; Blumenschein, G.; Fleming, D.R.; et al. Phase ii study of tariquidar, a selective p-glycoprotein inhibitor, in patients with chemotherapy-resistant, advanced breast carcinoma. Cancer 2005, 104, 682–691. [Google Scholar] [CrossRef]
- Furihata, T.; Sun, Y.; Chiba, K. Cancer-type organic anion transporting polypeptide 1b3: Current knowledge of the gene structure, expression profile, functional implications and future perspectives. Curr. Drug Metab. 2015, 16, 474–485. [Google Scholar] [CrossRef]
- Ateyya, H.; Hassan, Z.A.; El-Sherbeeny, N.A. The selective tyrosine kinase-inhibitor nilotinib alleviates experimentally induced cisplatin nephrotoxicity and heptotoxicity. Environ. Toxicol. Pharmacol. 2017, 55, 60–67. [Google Scholar] [CrossRef]
- Falkowski, S.; Woillard, J.B.; Postil, D.; Tubiana-Mathieu, N.; Terrebonne, E.; Pariente, A.; Smith, D.; Guimbaud, R.; Thalamas, C.; Rouguieg-Malki, K.; et al. Common variants in glucuronidation enzymes and membrane transporters as potential risk factors for colorectal cancer: A case control study. BMC Cancer 2017, 17, 901. [Google Scholar] [CrossRef]
- Kleberg, K.; Jensen, G.M.; Christensen, D.P.; Lundh, M.; Grunnet, L.G.; Knuhtsen, S.; Poulsen, S.S.; Hansen, M.B.; Bindslev, N. Transporter function and cyclic amp turnover in normal colonic mucosa from patients with and without colorectal neoplasia. BMC Gastroenterol. 2012, 12, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wlcek, K.; Svoboda, M.; Riha, J.; Zakaria, S.; Olszewski, U.; Dvorak, Z.; Sellner, F.; Ellinger, I.; Jager, W.; Thalhammer, T. The analysis of organic anion transporting polypeptide (oatp) mrna and protein patterns in primary and metastatic liver cancer. Cancer Biol. Ther. 2011, 11, 801–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billington, S.; Ray, A.S.; Salphati, L.; Xiao, G.; Chu, X.; Humphreys, W.G.; Liao, M.; Lee, C.A.; Mathias, A.; Hop, C.; et al. Transporter expression in noncancerous and cancerous liver tissue from donors with hepatocellular carcinoma and chronic hepatitis c infection quantified by lc-ms/ms proteomics. Drug Metab. Dispos. 2018, 46, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Wlcek, K.; Svoboda, M.; Thalhammer, T.; Sellner, F.; Krupitza, G.; Jaeger, W. Altered expression of organic anion transporter polypeptide (oatp) genes in human breast carcinoma. Cancer Biol. Ther. 2008, 7, 1450–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kindla, J.; Rau, T.T.; Jung, R.; Fasching, P.A.; Strick, R.; Stoehr, R.; Hartmann, A.; Fromm, M.F.; Konig, J. Expression and localization of the uptake transporters oatp2b1, oatp3a1 and oatp5a1 in non-malignant and malignant breast tissue. Cancer Biol. Ther. 2011, 11, 584–591. [Google Scholar] [CrossRef] [Green Version]
- Knauer, M.J.; Girdwood, A.J.; Kim, R.B.; Tirona, R.G. Transport function and transcriptional regulation of a liver-enriched human organic anion transporting polypeptide 2b1 transcriptional start site variant. Mol. Pharmacol. 2013, 83, 1218–1228. [Google Scholar] [CrossRef] [Green Version]
- McFeely, S.J.; Wu, L.; Ritchie, T.K.; Unadkat, J. Organic anion transporting polypeptide 2b1—More than a glass-full of drug interactions. Pharmacol. Ther. 2019, 196, 204–215. [Google Scholar] [CrossRef]
- Chen, M.; Hu, S.; Li, Y.; Gibson, A.A.; Fu, Q.; Baker, S.D.; Sparreboom, A. Role of oatp2b1 in drug absorption and drug-drug interactions. Drug Metab. Dispos. 2020, 48, 419–425. [Google Scholar] [CrossRef]
- Medwid, S.; Li, M.M.J.; Knauer, M.J.; Lin, K.; Mansell, S.E.; Schmerk, C.L.; Zhu, C.; Griffin, K.E.; Yousif, M.D.; Dresser, G.K.; et al. Fexofenadine and rosuvastatin pharmacokinetics in mice with targeted disruption of organic anion transporting polypeptide 2b1. Drug Metab. Dispos. 2019, 47, 832–842. [Google Scholar] [CrossRef]
- Bauer, M.; Matsuda, A.; Wulkersdorfer, B.; Philippe, C.; Traxl, A.; Ozvegy-Laczka, C.; Stanek, J.; Nics, L.; Klebermass, E.M.; Poschner, S.; et al. Influence of oatps on hepatic disposition of erlotinib measured with positron emission tomography. Clin. Pharmacol. Ther. 2018, 104, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Johnston, R.A.; Rawling, T.; Chan, T.; Zhou, F.; Murray, M. Selective inhibition of human solute carrier transporters by multikinase inhibitors. Drug Metab. Dispos. 2014, 42, 1851–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koepsell, H. Organic cation transporters in health and disease. Pharmacol. Rev. 2020, 72, 253–319. [Google Scholar] [CrossRef] [PubMed]
- Al-Abdulla, R.; Perez-Silva, L.; Abete, L.; Romero, M.R.; Briz, O.; Marin, J.J.G. Unraveling ‘the cancer genome atlas’ information on the role of slc transporters in anticancer drug uptake. Expert Rev. Clin. Pharmacol. 2019, 12, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; Herrmann, E.; Brom, M.; Keppler, D. Vectorial transport of the plant alkaloid berberine by double-transfected cells expressing the human organic cation transporter 1 (oct1, slc22a1) and the efflux pump mdr1 p-glycoprotein (abcb1). Naunyn Schmiedebergs Arch. Pharmacol. 2008, 376, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Sakurai, Y.; Saito, H.; Masuda, S.; Urakami, Y.; Goto, M.; Fukatsu, A.; Ogawa, O.; Inui, K. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J. Am. Soc. Nephrol. 2002, 13, 866–874. [Google Scholar]
- Lautem, A.; Heise, M.; Grasel, A.; Hoppe-Lotichius, M.; Weiler, N.; Foltys, D.; Knapstein, J.; Schattenberg, J.M.; Schad, A.; Zimmermann, A.; et al. Downregulation of organic cation transporter 1 (slc22a1) is associated with tumor progression and reduced patient survival in human cholangiocellular carcinoma. Int. J. Oncol. 2013, 42, 1297–1304. [Google Scholar] [CrossRef] [Green Version]
- Heise, M.; Lautem, A.; Knapstein, J.; Schattenberg, J.M.; Hoppe-Lotichius, M.; Foltys, D.; Weiler, N.; Zimmermann, A.; Schad, A.; Grundemann, D.; et al. Downregulation of organic cation transporters oct1 (slc22a1) and oct3 (slc22a3) in human hepatocellular carcinoma and their prognostic significance. BMC Cancer 2012, 12, 109. [Google Scholar] [CrossRef] [Green Version]
- Burger, H.; Loos, W.J.; Eechoute, K.; Verweij, J.; Mathijssen, R.H.; Wiemer, E.A. Drug transporters of platinum-based anticancer agents and their clinical significance. Drug Resist. Updates 2011, 14, 22–34. [Google Scholar] [CrossRef]
- Yonezawa, A.; Masuda, S.; Yokoo, S.; Katsura, T.; Inui, K. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (slc22a1-3 and multidrug and toxin extrusion family). J. Pharmacol. Exp. Ther. 2006, 319, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Peng, X.; Yang, H.; Rodriguez, J.A.; Shu, Y. Contribution of organic cation transporter 3 to cisplatin cytotoxicity in human cervical cancer cells. J. Pharm. Sci. 2012, 101, 394–404. [Google Scholar] [CrossRef]
- Zhang, S.; Lovejoy, K.S.; Shima, J.E.; Lagpacan, L.L.; Shu, Y.; Lapuk, A.; Chen, Y.; Komori, T.; Gray, J.W.; Chen, X.; et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 2006, 66, 8847–8857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, H.; Zoumaro-Djayoon, A.; Boersma, A.W.; Helleman, J.; Berns, E.M.; Mathijssen, R.H.; Loos, W.J.; Wiemer, E.A. Differential transport of platinum compounds by the human organic cation transporter hoct2 (hslc22a2). Br. J. Pharmacol. 2010, 159, 898–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- More, S.S.; Li, S.; Yee, S.W.; Chen, L.; Xu, Z.; Jablons, D.M.; Giacomini, K.M. Organic cation transporters modulate the uptake and cytotoxicity of picoplatin, a third-generation platinum analogue. Mol. Cancer Ther. 2010, 9, 1058–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoo, S.; Yonezawa, A.; Masuda, S.; Fukatsu, A.; Katsura, T.; Inui, K. Differential contribution of organic cation transporters, oct2 and mate1, in platinum agent-induced nephrotoxicity. Biochem. Pharmacol. 2007, 74, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Filipski, K.K.; Mathijssen, R.H.; Mikkelsen, T.S.; Schinkel, A.H.; Sparreboom, A. Contribution of organic cation transporter 2 (oct2) to cisplatin-induced nephrotoxicity. Clin. Pharmacol. Ther. 2009, 86, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Katsuda, H.; Yamashita, M.; Katsura, H.; Yu, J.; Waki, Y.; Nagata, N.; Sai, Y.; Miyamoto, K. Protecting cisplatin-induced nephrotoxicity with cimetidine does not affect antitumor activity. Biol. Pharm. Bull. 2010, 33, 1867–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprowl, J.A.; van Doorn, L.; Hu, S.; van Gerven, L.; de Bruijn, P.; Li, L.; Gibson, A.A.; Mathijssen, R.H.; Sparreboom, A. Conjunctive therapy of cisplatin with the oct2 inhibitor cimetidine: Influence on antitumor efficacy and systemic clearance. Clin. Pharmacol. Ther. 2013, 94, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Zazuli, Z.; Otten, L.S.; Drogemoller, B.I.; Medeiros, M.; Monzon, J.G.; Wright, G.E.B.; Kollmannsberger, C.K.; Bedard, P.L.; Chen, Z.; Gelmon, K.A.; et al. Outcome definition influences the relationship between genetic polymorphisms of ercc1, ercc2, slc22a2 and cisplatin nephrotoxicity in adult testicular cancer patients. Genes 2019, 10, 364. [Google Scholar] [CrossRef] [Green Version]
- Nieskens, T.T.G.; Peters, J.G.P.; Dabaghie, D.; Korte, D.; Jansen, K.; Van Asbeck, A.H.; Tavraz, N.N.; Friedrich, T.; Russel, F.G.M.; Masereeuw, R.; et al. Expression of organic anion transporter 1 or 3 in human kidney proximal tubule cells reduces cisplatin sensitivity. Drug Metab. Dispos. 2018, 46, 592–599. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Leblanc, A.F.; Gibson, A.A.; Hong, K.W.; Kim, J.Y.; Janke, L.J.; Li, L.; Vasilyeva, A.; Finkelstein, D.B.; Sprowl, J.A.; et al. Identification of oat1/oat3 as contributors to cisplatin toxicity. Clin. Transl. Sci. 2017, 10, 412–420. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Ohshiro, N.; Sakai, R.; Ohbayashi, M.; Kohyama, N.; Yamamoto, T. Transport mechanism and substrate specificity of human organic anion transporter 2 (hoat2 [slc22a7]). J. Pharm. Pharmacol. 2005, 57, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Marada, V.V.; Florl, S.; Kuhne, A.; Muller, J.; Burckhardt, G.; Hagos, Y. Interaction of human organic anion transporter 2 (oat2) and sodium taurocholate cotransporting polypeptide (ntcp) with antineoplastic drugs. Pharmacol. Res. 2015, 91, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, S.; Masuda, S.; Yonezawa, A.; Terada, T.; Katsura, T.; Inui, K. Significance of organic cation transporter 3 (slc22a3) expression for the cytotoxic effect of oxaliplatin in colorectal cancer. Drug Metab. Dispos. 2008, 36, 2299–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, C.S.; Sprowl, J.A.; Walker, A.L.; Hu, S.; Gibson, A.A.; Sparreboom, A. Modulation of oatp1b-type transporter function alters cellular uptake and disposition of platinum chemotherapeutics. Mol. Cancer Ther. 2013, 12, 1537–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briz, O.; Serrano, M.A.; Rebollo, N.; Hagenbuch, B.; Meier, P.J.; Koepsell, H.; Marin, J.J. Carriers involved in targeting the cytostatic bile acid-cisplatin derivatives cis-diammine-chloro-cholylglycinate-platinum(ii) and cis-diammine-bisursodeoxycholate-platinum(ii) toward liver cells. Mol. Pharmacol. 2002, 61, 853–860. [Google Scholar] [CrossRef] [Green Version]
- McBride, B.F.; Yang, T.; Liu, K.; Urban, T.J.; Giacomini, K.M.; Kim, R.B.; Roden, D.M. The organic cation transporter, octn1, expressed in the human heart, potentiates antagonism of the herg potassium channel. J. Cardiovasc. Pharmacol. 2009, 54, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Grube, M.; zu Schwabedissen, H.E.M.; Prager, D.; Haney, J.; Moritz, K.U.; Meissner, K.; Rosskopf, D.; Eckel, L.; Bohm, M.; Jedlitschky, G.; et al. Uptake of cardiovascular drugs into the human heart: Expression, regulation, and function of the carnitine transporter octn2 (slc22a5). Circulation 2006, 113, 1114–1122. [Google Scholar] [CrossRef] [Green Version]
- Glaeser, H.; Bailey, D.G.; Dresser, G.K.; Gregor, J.C.; Schwarz, U.I.; McGrath, J.S.; Jolicoeur, E.; Lee, W.; Leake, B.F.; Tirona, R.G.; et al. Intestinal drug transporter expression and the impact of grapefruit juice in humans. Clin. Pharmacol. Ther. 2007, 81, 362–370. [Google Scholar] [CrossRef]
- Tamai, I.; Yabuuchi, H.; Nezu, J.; Sai, Y.; Oku, A.; Shimane, M.; Tsuji, A. Cloning and characterization of a novel human ph-dependent organic cation transporter, octn1. FEBS Lett. 1997, 419, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Tamai, I.; Ohashi, R.; Nezu, J.; Yabuuchi, H.; Oku, A.; Shimane, M.; Sai, Y.; Tsuji, A. Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter octn2. J. Biol. Chem. 1998, 273, 20378–20382. [Google Scholar] [CrossRef] [Green Version]
- Juraszek, B.; Nalecz, K.A. Slc22a5 (octn2) carnitine transporter-indispensable for cell metabolism, a jekyll and hyde of human cancer. Molecules 2019, 25, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabuuchi, H.; Tamai, I.; Nezu, J.; Sakamoto, K.; Oku, A.; Shimane, M.; Sai, Y.; Tsuji, A. Novel membrane transporter octn1 mediates multispecific, bidirectional, and ph-dependent transport of organic cations. J. Pharmacol. Exp. Ther. 1999, 289, 768–773. [Google Scholar] [PubMed]
- Tschirka, J.; Kreisor, M.; Betz, J.; Grundemann, D. Substrate selectivity check of the ergothioneine transporter. Drug Metab. Dispos. 2018, 46, 779–785. [Google Scholar] [CrossRef]
- Jong, N.N.; Nakanishi, T.; Liu, J.J.; Tamai, I.; McKeage, M.J. Oxaliplatin transport mediated by organic cation/carnitine transporters octn1 and octn2 in overexpressing human embryonic kidney 293 cells and rat dorsal root ganglion neurons. J. Pharmacol. Exp. Ther. 2011, 338, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houten, S.M.; Wanders, R.J. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Uray, I.P.; Mazumdar, A.; Mayer, J.A.; Brown, P.H. Slc22a5/octn2 expression in breast cancer is induced by estrogen via a novel intronic estrogen-response element (ere). Breast Cancer Res. Treat. 2012, 134, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Fink, M.A.; Paland, H.; Herzog, S.; Grube, M.; Vogelgesang, S.; Weitmann, K.; Bialke, A.; Hoffmann, W.; Rauch, B.H.; Schroeder, H.W.S.; et al. L-carnitine-mediated tumor cell protection and poor patient survival associated with octn2 overexpression in glioblastoma multiforme. Clin. Cancer Res. 2019, 25, 2874–2886. [Google Scholar] [CrossRef]
- Martini, M.; Ferrara, A.M.; Giachelia, M.; Panieri, E.; Siminovitch, K.; Galeotti, T.; Larocca, L.M.; Pani, G. Association of the octn1/1672t variant with increased risk for colorectal cancer in young individuals and ulcerative colitis patients. Inflamm. Bowel Dis. 2012, 18, 439–448. [Google Scholar] [CrossRef]
- Zou, D.; Lou, J.; Ke, J.; Mei, S.; Li, J.; Gong, Y.; Yang, Y.; Zhu, Y.; Tian, J.; Chang, J.; et al. Integrative expression quantitative trait locus-based analysis of colorectal cancer identified a functional polymorphism regulating slc22a5 expression. Eur. J. Cancer 2018, 93, 1–9. [Google Scholar] [CrossRef]
- Sankaranarayanan, R.; Valiveti, C.K.; Dachineni, R.; Kumar, D.R.; Lick, T.; Bhat, G.J. Aspirin metabolites 2,3dhba and 2,5dhba inhibit cancer cell growth: Implications in colorectal cancer prevention. Mol. Med. Rep. 2020, 21, 20–34. [Google Scholar]
- Dachineni, R.; Kumar, D.R.; Callegari, E.; Kesharwani, S.S.; Sankaranarayanan, R.; Seefeldt, T.; Tummala, H.; Bhat, G.J. Salicylic acid metabolites and derivatives inhibit cdk activity: Novel insights into aspirin’s chemopreventive effects against colorectal cancer. Int. J. Oncol. 2017, 51, 1661–1673. [Google Scholar] [CrossRef] [Green Version]
- Barry, E.L.; Fedirko, V.; Uppal, K.; Ma, C.; Liu, K.; Mott, L.A.; Peacock, J.L.; Passarelli, M.N.; Baron, J.A.; Jones, D.P. Metabolomics analysis of aspirin’s effects in human colon tissue and associations with adenoma risk. Cancer Prev. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Haschke, M.; Vitins, T.; Lude, S.; Todesco, L.; Novakova, K.; Herrmann, R.; Krahenbuhl, S. Urinary excretion of carnitine as a marker of proximal tubular damage associated with platin-based antineoplastic drugs. Nephrol. Dial. Transplant. 2010, 25, 426–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancinelli, A.; D’Iddio, S.; Bisonni, R.; Graziano, F.; Lippe, P.; Calvani, M. Urinary excretion of l-carnitine and its short-chain acetyl-l-carnitine in patients undergoing carboplatin treatment. Cancer Chemother. Pharmacol. 2007, 60, 19–26. [Google Scholar] [CrossRef]
- Lancaster, C.S.; Hu, C.; Franke, R.M.; Filipski, K.K.; Orwick, S.J.; Chen, Z.; Zuo, Z.; Loos, W.J.; Sparreboom, A. Cisplatin-induced downregulation of octn2 affects carnitine wasting. Clin. Cancer Res. 2010, 16, 4789–4799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Lancaster, C.S.; Zuo, Z.; Hu, S.; Chen, Z.; Rubnitz, J.E.; Baker, S.D.; Sparreboom, A. Inhibition of octn2-mediated transport of carnitine by etoposide. Mol. Cancer Ther. 2012, 11, 921–929. [Google Scholar] [CrossRef] [Green Version]
- Fei, Y.J.; Kanai, Y.; Nussberger, S.; Ganapathy, V.; Leibach, F.H.; Romero, M.F.; Singh, S.K.; Boron, W.F.; Hediger, M.A. Expression cloning of a mammalian proton-coupled oligopeptide transporter. Nature 1994, 368, 563–566. [Google Scholar] [CrossRef]
- Liu, W.; Liang, R.; Ramamoorthy, S.; Fei, Y.J.; Ganapathy, M.E.; Hediger, M.A.; Ganapathy, V.; Leibach, F.H. Molecular cloning of pept 2, a new member of the h+/peptide cotransporter family, from human kidney. Biochim. Biophys. Acta 1995, 1235, 461–466. [Google Scholar] [CrossRef] [Green Version]
- Mrsny, R.J. Oligopeptide transporters as putative therapeutic targets for cancer cells. Pharm. Res. 1998, 15, 816–818. [Google Scholar] [CrossRef]
- Terada, T.; Sawada, K.; Irie, M.; Saito, H.; Hashimoto, Y.; Inui, K. Structural requirements for determining the substrate affinity of peptide transporters pept1 and pept2. Pflugers Arch. 2000, 440, 679–684. [Google Scholar] [CrossRef]
- Ganapathy, M.E.; Brandsch, M.; Prasad, P.D.; Ganapathy, V.; Leibach, F.H. Differential recognition of beta -lactam antibiotics by intestinal and renal peptide transporters, pept 1 and pept 2. J. Biol. Chem. 1995, 270, 25672–25677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagiya, Y.; Fukuhara, H.; Matsumoto, K.; Endo, Y.; Nakajima, M.; Tanaka, T.; Okura, I.; Kurabayashi, A.; Furihata, M.; Inoue, K.; et al. Expression levels of pept1 and abcg2 play key roles in 5-aminolevulinic acid (ala)-induced tumor-specific protoporphyrin ix (ppix) accumulation in bladder cancer. Photodiagn. Photodyn. Ther. 2013, 10, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, H.; Heinrich, M.; Birkemeyer, C.; Meixensberger, J.; Gaunitz, F. The proton-coupled oligopeptide transporters pept2, pht1 and pht2 mediate the uptake of carnosine in glioblastoma cells. Amino Acids 2019, 51, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; Chen, Z.; Cheng, K. Expression profile and functional activity of peptide transporters in prostate cancer cells. Mol. Pharm. 2013, 10, 477–487. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Terada, T.; Okuda, M.; Inui, K. Regulation of human peptide transporter 1 (pept1) in gastric cancer cells by anticancer drugs. Cancer Lett. 2005, 230, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.E.; Covitz, K.M.; Sadee, W.; Mrsny, R.J. An oligopeptide transporter is expressed at high levels in the pancreatic carcinoma cell lines aspc-1 and capan-2. Cancer Res. 1998, 58, 519–525. [Google Scholar]
- Gong, Y.; Wu, X.; Wang, T.; Zhao, J.; Liu, X.; Yao, Z.; Zhang, Q.; Jian, X. Targeting pept1: A novel strategy to improve the antitumor efficacy of doxorubicin in human hepatocellular carcinoma therapy. Oncotarget 2017, 8, 40454–40468. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, C.; Liu, Q.; Meng, Q.; Huo, X.; Sun, P.; Yang, X.; Sun, H.; Zhen, Y.; Peng, J.; et al. Pept1- and oat1/3-mediated drug-drug interactions between bestatin and cefixime in vivo and in vitro in rats, and in vitro in human. Eur. J. Pharm. Sci. 2014, 63, 77–86. [Google Scholar] [CrossRef]
- Nakanishi, T.; Tamai, I.; Takaki, A.; Tsuji, A. Cancer cell-targeted drug delivery utilizing oligopeptide transport activity. Int. J. Cancer 2000, 88, 274–280. [Google Scholar] [CrossRef]
- Rutenburg, A.M.; Goldbarg, J.A.; Pineda, E.P. Leucine aminopeptidase activity; observations in patients with cancer of the pancreas and other diseases. N. Engl. J. Med. 1958, 259, 469–472. [Google Scholar] [CrossRef]
- Scornik, O.A.; Botbol, V. Bestatin as an experimental tool in mammals. Curr. Drug Metab. 2001, 2, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Willighagen, R.G.; Planteydt, H.T. Aminopeptidase activity in cancer cells. Nature 1959, 183, 263–264. [Google Scholar] [CrossRef] [PubMed]
- Hitzerd, S.M.; Verbrugge, S.E.; Ossenkoppele, G.; Jansen, G.; Peters, G.J. Positioning of aminopeptidase inhibitors in next generation cancer therapy. Amino Acids 2014, 46, 793–808. [Google Scholar] [CrossRef] [PubMed]
- Takano, M.; Tomita, Y.; Katsura, T.; Yasuhara, M.; Inui, K.; Hori, R. Bestatin transport in rabbit intestinal brush-border membrane vesicles. Biochem. Pharmacol. 1994, 47, 1089–1090. [Google Scholar] [CrossRef]
- Hori, R.; Tomita, Y.; Katsura, T.; Yasuhara, M.; Inui, K.; Takano, M. Transport of bestatin in rat renal brush-border membrane vesicles. Biochem. Pharmacol. 1993, 45, 1763–1768. [Google Scholar]
- Terada, T.; Saito, H.; Inui, K. Interaction of beta-lactam antibiotics with histidine residue of rat h+/peptide cotransporters, pept1 and pept2. J. Biol. Chem. 1998, 273, 5582–5585. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Meng, Q.; Wang, C.; Liu, Q.; Sun, H.; Kaku, T.; Liu, K. Organic anion transporters involved in the excretion of bestatin in the kidney. Peptides 2012, 33, 265–271. [Google Scholar] [CrossRef]
- Ota, K.; Uzuka, Y. Clinical trials of bestatin for leukemia and solid tumors. Biotherapy 1992, 4, 205–214. [Google Scholar] [CrossRef]
- Ichinose, Y.; Genka, K.; Koike, T.; Kato, H.; Watanabe, Y.; Mori, T.; Iioka, S.; Sakuma, A.; Ohta, M.; NK421 Lung Cancer Surgery Group. Randomized double-blind placebo-controlled trial of bestatin in patients with resected stage i squamous-cell lung carcinoma. J. Natl. Cancer Inst. 2003, 95, 605–610. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.B.; Brown, E.A.; Walker, I. The present and future role of photodynamic therapy in cancer treatment. Lancet Oncol. 2004, 5, 497–508. [Google Scholar] [CrossRef]
- Allison, R.R.; Bagnato, V.S.; Sibata, C.H. Future of oncologic photodynamic therapy. Future Oncol. 2010, 6, 929–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, R.; Kawabata, S.; Ikeda, N.; Nonoguchi, N.; Furuse, M.; Katayama, Y.; Kajimoto, Y.; Kuroiwa, T. Intraoperative 5-aminolevulinic acid-induced photodynamic diagnosis of metastatic brain tumors with histopathological analysis. World J. Surg. Oncol. 2017, 15, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishi, K.; Fujiwara, Y.; Yano, M.; Motoori, M.; Sugimura, K.; Takahashi, H.; Ohue, M.; Sakon, M. Usefulness of diagnostic laparoscopy with 5-aminolevulinic acid (ala)-mediated photodynamic diagnosis for the detection of peritoneal micrometastasis in advanced gastric cancer after chemotherapy. Surg. Today 2016, 46, 1427–1434. [Google Scholar] [CrossRef]
- Hillemanns, P.; Wimberger, P.; Reif, J.; Stepp, H.; Klapdor, R. Photodynamic diagnosis with 5-aminolevulinic acid for intraoperative detection of peritoneal metastases of ovarian cancer: A feasibility and dose finding study. Lasers Surg. Med. 2017, 49, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Murayama, Y.; Kubo, H.; Matsuo, H.; Morimura, R.; Ikoma, H.; Fujiwara, H.; Okamoto, K.; Tanaka, T.; Otsuji, E. Photodynamic diagnosis of peritoneal metastasis in human pancreatic cancer using 5-aminolevulinic acid during staging laparoscopy. Oncol. Lett. 2018, 16, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.C.; Pottier, R.H. Endogenous protoporphyrin ix, a clinically useful photosensitizer for photodynamic therapy. J. Photochem. Photobiol. B 1992, 14, 275–292. [Google Scholar] [CrossRef]
- Doring, F.; Walter, J.; Will, J.; Focking, M.; Boll, M.; Amasheh, S.; Clauss, W.; Daniel, H. Delta-aminolevulinic acid transport by intestinal and renal peptide transporters and its physiological and clinical implications. J. Clin. Investig. 1998, 101, 2761–2767. [Google Scholar] [CrossRef]
- Irie, M.; Terada, T.; Sawada, K.; Saito, H.; Inui, K. Recognition and transport characteristics of nonpeptidic compounds by basolateral peptide transporter in caco-2 cells. J. Pharmacol. Exp. Ther. 2001, 298, 711–717. [Google Scholar]
- Baglo, Y.; Gabrielsen, M.; Sylte, I.; Gederaas, O.A. Homology modeling of human gamma-butyric acid transporters and the binding of pro-drugs 5-aminolevulinic acid and methyl aminolevulinic acid used in photodynamic therapy. PLoS ONE 2013, 8, e65200. [Google Scholar] [CrossRef] [Green Version]
- Miyabe, J.; Ohgaki, R.; Saito, K.; Wei, L.; Quan, L.; Jin, C.; Liu, X.; Okuda, S.; Nagamori, S.; Ohki, H.; et al. Boron delivery for boron neutron capture therapy targeting a cancer-upregulated oligopeptide transporter. J. Pharmacol. Sci. 2019, 139, 215–222. [Google Scholar] [CrossRef]
- Wongthai, P.; Hagiwara, K.; Miyoshi, Y.; Wiriyasermkul, P.; Wei, L.; Ohgaki, R.; Kato, I.; Hamase, K.; Nagamori, S.; Kanai, Y. Boronophenylalanine, a boron delivery agent for boron neutron capture therapy, is transported by atb0,+, lat1 and lat2. Cancer Sci. 2015, 106, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuoka, K.; Miyoshi, S.; Kato, Y.; Murakami, Y.; Utsumi, R.; Kubo, Y.; Noda, A.; Nakamura, Y.; Nishimura, S.; Tsuji, A. Cancer detection using a pet tracer, 11c-glycylsarcosine, targeted to h+/peptide transporter. J. Nucl. Med. 2008, 49, 615–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molotkov, A.; Castrillon, J.W.; Santha, S.; Harris, P.E.; Leung, D.K.; Mintz, A.; Carberry, P. The radiolabeling of a gly-sar dipeptide derivative with flourine-18 and its use as a potential peptide transporter pet imaging agent. Molecules 2020, 25, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Smith, D.E.; Ma, K.; Jappar, D.; Thomas, W.; Hillgren, K.M. Targeted disruption of peptide transporter pept1 gene in mice significantly reduces dipeptide absorption in intestine. Mol. Pharm. 2008, 5, 1122–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio-Aliaga, I.; Frey, I.; Boll, M.; Groneberg, D.A.; Eichinger, H.M.; Balling, R.; Daniel, H. Targeted disruption of the peptide transporter pept2 gene in mice defines its physiological role in the kidney. Mol. Cell. Biol. 2003, 23, 3247–3252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.Y.; Veal, G.J.; Zhou, F.; Boddy, A.V. The role of solute carrier (slc) transporters in actinomycin d pharmacokinetics in paediatric cancer patients. Eur. J. Clin. Pharmacol. 2018, 74, 1575–1584. [Google Scholar] [CrossRef]
- Goldberg, I.H.; Rabinowitz, M.; Reich, E. Basis of actinomycin action. I. DNA binding and inhibition of rna-polymerase synthetic reactions by actinomycin. Proc. Natl. Acad. Sci. USA 1962, 48, 2094–2101. [Google Scholar] [CrossRef] [Green Version]
- Arndt, C.A.S.; Bisogno, G.; Koscielniak, E. Fifty years of rhabdomyosarcoma studies on both sides of the pond and lessons learned. Cancer Treat. Rev. 2018, 68, 94–101. [Google Scholar] [CrossRef]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing sarcoma: Current management and future approaches through collaboration. J. Clin. Oncol. 2015, 33, 3036–3046. [Google Scholar] [CrossRef]
- Phelps, H.M.; Kaviany, S.; Borinstein, S.C.; Lovvorn, H.N., 3rd. Biological drivers of wilms tumor prognosis and treatment. Children 2018, 5, 145. [Google Scholar] [CrossRef] [Green Version]


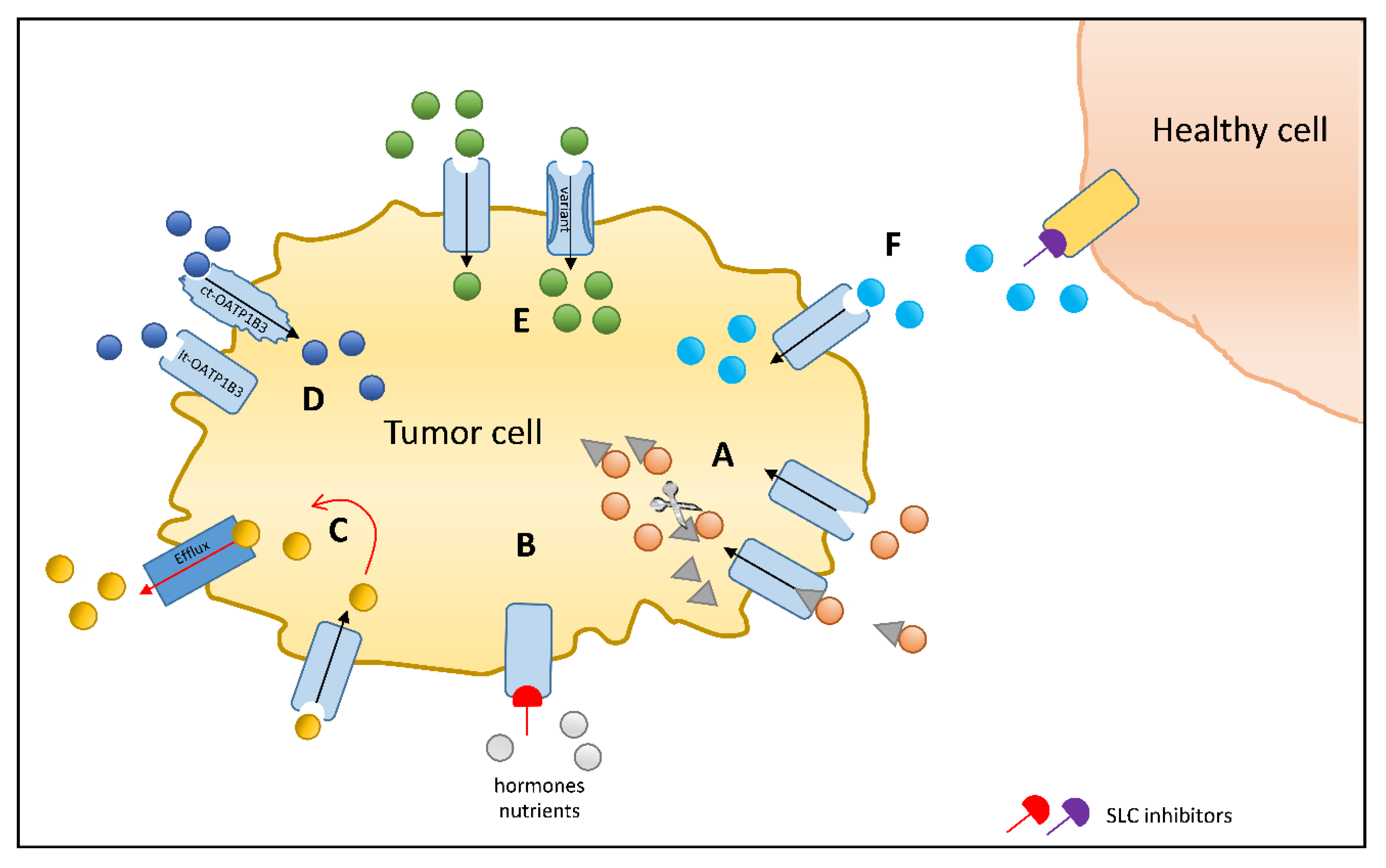{kind=link}
| Chemotherapeutic Alone or in Combination (Administration) | Tumor Indication | Transporter | SNP | Response to Treatment (Defined Criteria) | Study Design and Patient Number |
|---|---|---|---|---|---|
| Abiraterone (p.o.) | Prostate cancer | OATP2B1 | rs12422149 rs1789693 | Higher mean drug tissue levels Lower mean drug tissue levels | Clinical trial, 58 patients [40] |
| Prostate cancer | OATP2B1 | rs12422149 | Improved progression-free survival (less increase in PSA and/or radiographic or clinical progression) | Retrospective, 79 patients [41] | |
| Cisplatin (i.v.) | Different malignant solid tumors | OCT2 | rs316019 | Attenuated nephrotoxicity | Retrospective, 123 patients [42] |
| Imatinib (i.v.) | GIST | OATP1A2 | rs11568563, c.516A>C, p.E172D, OATP1A2*3 | No significant changes in imatinib plasma levels | Retrospective, 94 patients [43] |
| CML | OCT1, OCTN1, OATP1A2 | Combination of SNPs | Major (defined as a 3-log reduction in Bcr-Abl transcript level from a standardized baseline value) and complete (defined as at least a 4-log reduction corresponding to undetectable Bcr-Abl transcript by PCR) molecular response | Retrospective, 189 patients [44] | |
| CML | OATP1A2 | −361GG | Lower clearance | Retrospective, 34 patients [45] | |
| GIST | OCTN1 OCTN2 | rs1050152 rs2631367 rs2631372 | Improved time to progression (calculated from the start of imatinib therapy to the date of disease progression documented by a CT scan) | Retrospective, 54 patients [46] | |
| Irinotecan (i.v.) | NSCLC | OATP1B1 | 521CC, 521TC, −11187GG | Increased exposure of metabolite SN-38 | Phase II study, 81 patients [37] |
| Metastatic colorectal cancer and advanced/metastatic pancreatic cancer | OATP1B1 OATP1B1 | 521C c.388A>G | Increased exposure of metabolite SN-38 Longer progression-free survival (elongated time from initiation of irinotecan-based chemotherapy to the date of progression, death, last contact, or censor date) | Retrospective, 127 patients [38] | |
| Methotrexate (p.o./i.v.) | Rheumatoid arthritis | OATP1A2 | c.550G>A, p.E184K | Delayed MTX elimination, increased MTX related toxicity | Clinical trial, 60 patients [47,48] |
| c.553G>A, p.D185N c.775A>C, p.V255I c.862G>A, p.T259P | No association with MTX related PK or toxicity | ||||
| Sorafenib (p.o.) | Different tumors | OATP1B1 | rs2306283 rs4149056 | Associated with diarrhea and thrombocytopenia | Retrospective, 114 patients [25] |
| Hepatocellular carcinoma | PEPT2 | rs2257212 | Prolonged progression-free survival (Hazard ratios) | Retrospective, 174 patients [34] | |
| Sunitinib (p.o.) | GIST | OCTN2 | rs2631367 + rs2631370 + rs2631372 | Longer overall survival (Hazard ratios) | Retrospective, 127 patients [49] |
| OATP1B3 | rs4149117 | Longer overall survival (Hazard ratios) | Retrospective, 127 patients [49] | ||
| Docetaxel and Doxorubicin (i.v.) | Breast cancer | OATP1A2 | rs4762699 rs2857468 | High risk for febrile neutropenia | Clinical trial, 155 patients [50] |
| Cytarabine, Daunorubicin, Etoposide, Mitoxantrone (i.v.) | AML | OATP1B1 | rs2291075 | Association with event-free and overall survival (Hazard ratios) | Retrospective, 165 pediatric patients [51] |
| Chemotherapeutic Alone or in Combination (Administration) | Tumor Indication | Transporter, Expression Level (Quality of Measure) | Response to Treatment (Defined Criteria) | Study Design and Patient Number |
|---|---|---|---|---|
| Cisplatin (i.v.) | Head and neck squamous cell carcinoma | OCT3, high (protein) | Higher 2-year survival rate | Retrospective, 42 patients [52] |
| Cytarabine (i.v.) | AML | OCTN1, high (mRNA) | Improved event-free and overall survival (Hazard ratios) | Retrospective, 172 patients [53] |
| 5-FU (i.v.) | Colorectal cancer | OAT2, high (protein) | Good histological response | Pre-treatment biopsy, 45 patients [54] |
| FOLFOX (i.v.) | Metastatic colorectal cancer | OAT2, high (protein) | Good objective tumor response (RECIST) | Retrospective, 90 patients [55] |
| OCT2, high (protein) | Longer progression-free survival (less radiological progression or death) | Retrospective, 90 patients [55] | ||
| OCT3, high (protein) | Non-respond to therapy (cancer recurrence within one year) | Retrospective, 31 patients [56] | ||
| Irinotecan (i.v.) | Metastatic colorectal cancer and advanced/metastatic pancreatic cancer | OATP1B3, high (protein) | Reduced progression-free survival (reduced time from initiation of irinotecan-based chemotherapy to the date of progression, death, last contact, or censor date) | Prospective, 127 patients [38] |
| Oxaliplatin (i.v.) | Metastatic colorectal cancer | OCT2, high (protein) | Longer progression-free survival (longer period from the start of first-line chemotherapy until the first evidence of radiological progression or death) | Retrospective, 80 patients [57] |
| Sorafenib (p.o.) | Hepatocellular carcinoma | OCT1 (mRNA) | Positive association with survival (Hazard ratios) | Retrospective, 60 patients [30] |
| Hepatocellular carcinoma | OCT1, expression at the plasma membrane (protein) | Longer overall survival | Retrospective, 39 patients [31] | |
| Anthracycline and taxane (i.v.) | Breast cancer | OATP1A2, OCT6, high expression of both (protein) | Pathologic good and complete response | Retrospective, 124 patients [58] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brecht, K.; Schäfer, A.M.; Meyer zu Schwabedissen, H.E. Uptake Transporters of the SLC21, SLC22A, and SLC15A Families in Anticancer Therapy—Modulators of Cellular Entry or Pharmacokinetics? Cancers 2020, 12, 2263. https://doi.org/10.3390/cancers12082263
Brecht K, Schäfer AM, Meyer zu Schwabedissen HE. Uptake Transporters of the SLC21, SLC22A, and SLC15A Families in Anticancer Therapy—Modulators of Cellular Entry or Pharmacokinetics? Cancers. 2020; 12(8):2263. https://doi.org/10.3390/cancers12082263
Chicago/Turabian StyleBrecht, Karin, Anima Magdalena Schäfer, and Henriette E. Meyer zu Schwabedissen. 2020. "Uptake Transporters of the SLC21, SLC22A, and SLC15A Families in Anticancer Therapy—Modulators of Cellular Entry or Pharmacokinetics?" Cancers 12, no. 8: 2263. https://doi.org/10.3390/cancers12082263
APA StyleBrecht, K., Schäfer, A. M., & Meyer zu Schwabedissen, H. E. (2020). Uptake Transporters of the SLC21, SLC22A, and SLC15A Families in Anticancer Therapy—Modulators of Cellular Entry or Pharmacokinetics? Cancers, 12(8), 2263. https://doi.org/10.3390/cancers12082263