
Approaches to Enhance Natural Killer Cell-Based Immunotherapy for Pediatric Solid Tumors


Abstract
:Simple Summary
Abstract
1. Introduction
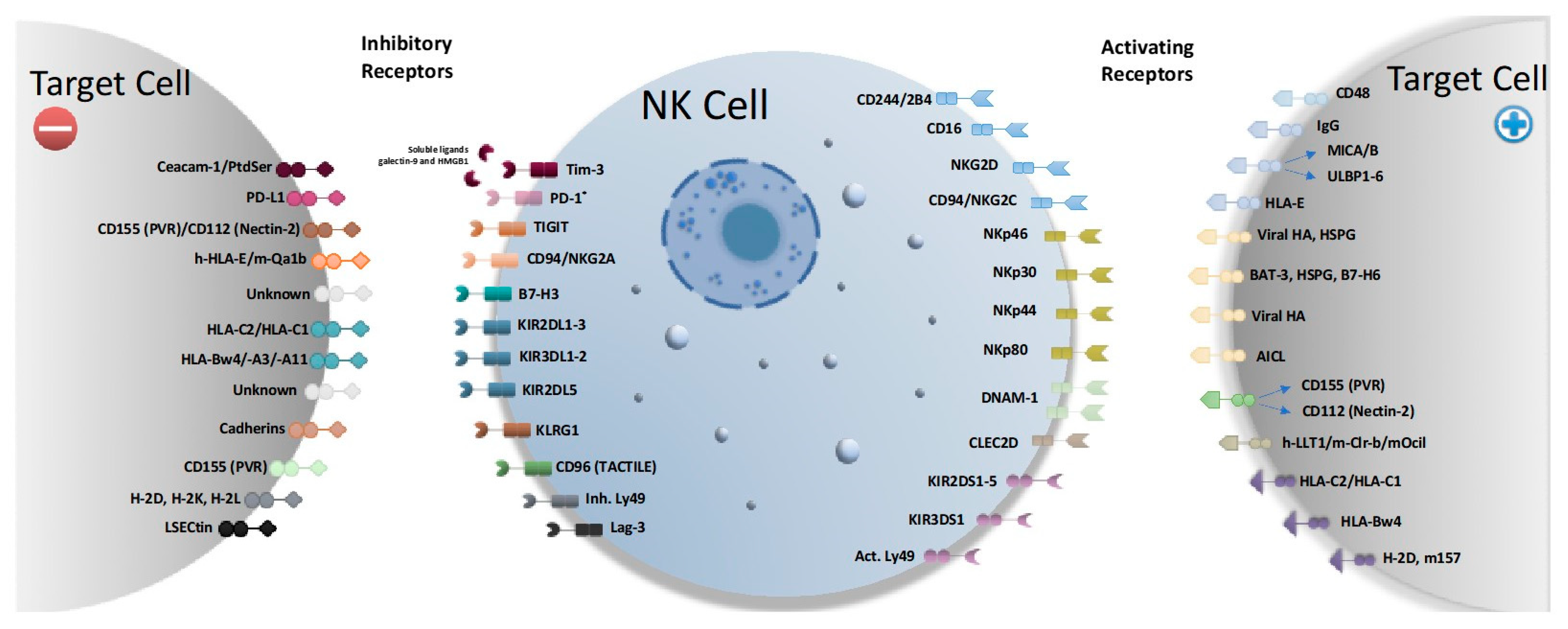
2. NK Cell Activating and Inhibitory Receptors
2.1. NK KIR Receptors
2.2. NK Activating Receptors
2.2.1. NKG2 Receptor Family
2.2.2. DNAM-1
2.2.3. CD16
2.3. NK Inhibitory Receptors
2.3.1. PD-1/PD-L1
2.3.2. CD94/NKG2A
2.3.3. TIGIT
2.3.4. TIM-3
2.3.5. LAG-3
2.3.6. B7-H3
3. Cytokines
4. Canine Models of NK Adoptive Immunotherapy for Pediatric Solid Tumors
5. Combination Therapy with Monoclonal Antibodies
6. CAR NK Cells
7. Clinical Trials Involving NK Cell Enriched Allogeneic HSCT for Pediatric Solid Tumors
8. Adoptive NK Cell Trials for Pediatric Solid Tumors
9. Combination Therapy with NK Cells and Immunotherapies
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Outzen, H.C.; Custer, R.P.; Eaton, G.J.; Prehn, R.T. Spontaneous and induced tumor incidence in germfree “nude” mice. J. Reticuloendothel. Soc. 1975, 17, 1–9. [Google Scholar] [PubMed]
- Mah, A.Y.; Cooper, M.A. Metabolic Regulation of Natural Killer Cell IFN-γ Production. Crit. Rev. Immunol. 2016, 36, 131–147. [Google Scholar] [CrossRef]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Olson, J.A.; Leveson-Gower, D.B.; Gill, S.; Baker, J.; Beilhack, A.; Negrin, R.S. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood 2010, 115, 4293–4301. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.M.; Savithri, B.; Khar, A. Activating and inhibitory receptors and their role in natural killer cell function. Indian J. Biochem. Biophys. 2003, 40, 291–299. [Google Scholar] [PubMed]
- Zelensky, A.N.; Gready, J.E. The C-type lectin-like domain superfamily. FEBS J. 2005, 272, 6179–6217. [Google Scholar] [CrossRef] [PubMed]
- Bartel, Y.; Bauer, B.; Steinle, A. Modulation of NK cell function by genetically coupled C-type lectin-like receptor/ligand pairs encoded in the human natural killer gene complex. Front. Immunol. 2013, 4, 362. [Google Scholar] [CrossRef] [Green Version]
- Hanke, T.; Takizawa, H.; McMahon, C.W.; Busch, D.H.; Pamer, E.G.; Miller, J.D.; Altman, J.D.; Liu, Y.; Cado, D.; Lemonnier, F.A.; et al. Direct assessment of MHC class I binding by seven Ly49 inhibitory NK cell receptors. Immunity 1999, 11, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, N.; Ribaudo, R.K.; Abastado, J.P.; Margulies, D.H.; Yokoyama, W.M. The lectin-like NK cell receptor Ly-49A recognizes a carbohydrate-independent epitope on its MHC class I ligand. Immunity 1998, 8, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Pende, D.; Falco, M.; Vitale, M.; Cantoni, C.; Vitale, C.; Munari, E.; Bertaina, A.; Moretta, F.; Del Zotto, G.; Pietra, G.; et al. Killer Ig-Like Receptors (KIRs): Their Role in NK Cell Modulation and Developments Leading to Their Clinical Exploitation. Front. Immunol. 2019, 10, 1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McVicar, D.W.; Burshtyn, D.N. Intracellular signaling by the killer immunoglobulin-like receptors and Ly49. Sci. STKE 2001, 2001, re1. [Google Scholar] [CrossRef]
- Rahim, M.M.A.; Makrigiannis, A.P. Ly49 receptors: Evolution, genetic diversity, and impact on immunity. Immunol. Rev. 2015, 267, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. How ITAMs Inhibit Signaling. Sci. Signal. 2011, 4, pe20. [Google Scholar] [CrossRef] [Green Version]
- Burshtyn, D.N.; Scharenberg, A.M.; Wagtmann, N.; Rajagopalan, S.; Berrada, K.; Yi, T.; Kinet, J.P.; Long, E.O. Recruitment of tyrosine phosphatase HCP by the killer cell inhibitor receptor. Immunity 1996, 4, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Bolland, S.; Tempst, P.; Ravetch, J.V. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature 1996, 383, 263–266. [Google Scholar] [CrossRef]
- Olcese, L.; Lang, P.; Vély, F.; Cambiaggi, A.; Marguet, D.; Bléry, M.; Hippen, K.L.; Biassoni, R.; Moretta, A.; Moretta, L.; et al. Human and mouse killer-cell inhibitory receptors recruit PTP1C and PTP1D protein tyrosine phosphatases. J. Immunol. 1996, 156, 4531–4534. [Google Scholar] [PubMed]
- Burshtyn, D.N.; Yang, W.; Yi, T.; Long, E.O. A novel phosphotyrosine motif with a critical amino acid at position -2 for the SH2 domain-mediated activation of the tyrosine phosphatase SHP-1. J. Biol. Chem. 1997, 272, 13066–13072. [Google Scholar] [CrossRef] [Green Version]
- Vyas, Y.M.; Maniar, H.; Lyddane, C.E.; Sadelain, M.; Dupont, B. Ligand binding to inhibitory killer cell Ig-like receptors induce colocalization with Src homology domain 2-containing protein tyrosine phosphatase 1 and interruption of ongoing activation signals. J. Immunol. 2004, 173, 1571–1578. [Google Scholar] [CrossRef]
- Winter, C.C.; Gumperz, J.E.; Parham, P.; Long, E.O.; Wagtmann, N. Direct binding and functional transfer of NK cell inhibitory receptors reveal novel patterns of HLA-C allotype recognition. J. Immunol. 1998, 161, 571–577. [Google Scholar]
- Delgado, D.; Webster, D.E.; DeSantes, K.B.; Durkin, E.T.; Shaaban, A.F. KIR Receptor-Ligand Incompatibility Predicts Killing of Osteosarcoma Cell Lines by Allogeneic NK Cells. Pediatr. Blood Cancer 2010, 55, 1300–1305. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Poursine-Laurent, J.; Truscott, S.M.; Lybarger, L.; Song, Y.-J.; Yang, L.; French, A.R.; Sunwoo, J.B.; Lemieux, S.; Hansen, T.H.; et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 2005, 436, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, N.; André, P.; Guia, S.; Falk, C.S.; Roetynck, S.; Stewart, C.A.; Breso, V.; Frassati, C.; Reviron, D.; Middleton, D.; et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity 2006, 25, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, N.C.; Treiner, E.; Vance, R.E.; Jamieson, A.M.; Lemieux, S.; Raulet, D.H. A subset of natural killer cells achieves self-tolerance without expressing inhibitory receptors specific for self-MHC molecules. Blood 2005, 105, 4416–4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, C.Y.; Blazar, B.R.; George, T.; Welniak, L.A.; Capitini, C.M.; Raziuddin, A.; Murphy, W.J.; Bennett, M. Augmentation of antitumor effects by NK cell inhibitory receptor blockade in vitro and in vivo. Blood 2001, 97, 3132–3137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vey, N.; Dumas, P.-Y.; Recher, C.; Gastaud, L.; Lioure, B.; Bulabois, C.-E.; Pautas, C.; Marolleau, J.-P.; Leprêtre, S.; Raffoux, E.; et al. Randomized Phase 2 Trial of Lirilumab (anti-KIR monoclonal antibody, mAb) As Maintenance Treatment in Elderly Patients (pts) with Acute Myeloid Leukemia (AML): Results of the Effikir Trial. Blood 2017, 130, 889. [Google Scholar] [CrossRef]
- Carlsten, M.; Korde, N.; Kotecha, R.; Reger, R.; Bor, S.; Kazandjian, D.; Landgren, O.; Childs, R.W. Checkpoint Inhibition of KIR2D with the Monoclonal Antibody IPH2101 Induces Contraction and Hyporesponsiveness of NK Cells in Patients with Myeloma. Clin. Cancer Res. 2016, 22, 5211–5222. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Wang, X.; Jasinski, D.L.; Medina, J.L.; Spencer, D.M.; Foster, A.E.; Bayle, J.H. Inducible MyD88/CD40 synergizes with IL-15 to enhance antitumor efficacy of CAR-NK cells. Blood Adv. 2020, 4, 1950–1964. [Google Scholar] [CrossRef]
- Calvo, T.; Reina-Ortiz, C.; Giraldos, D.; Gascón, M.; Woods, D.; Asenjo, J.; Marco-Brualla, J.; Azaceta, G.; Izquierdo, I.; Palomera, L.; et al. Expanded and activated allogeneic NK cells are cytotoxic against B-chronic lymphocytic leukemia (B-CLL) cells with sporadic cases of resistance. Sci. Rep. 2020, 10, 19398. [Google Scholar] [CrossRef] [PubMed]
- Meazza, R.; Falco, M.; Loiacono, F.; Canevali, P.; Della Chiesa, M.; Bertaina, A.; Pagliara, D.; Merli, P.; Indio, V.; Galaverna, F.; et al. Phenotypic and Functional Characterization of NK Cells in αβT-Cell and B-Cell Depleted Haplo-HSCT to Cure Pediatric Patients with Acute Leukemia. Cancers 2020, 12, 2187. [Google Scholar] [CrossRef]
- Nguyen, R.; Wu, H.; Pounds, S.; Inaba, H.; Ribeiro, R.C.; Cullins, D.; Rooney, B.; Bell, T.; Lacayo, N.J.; Heym, K.; et al. A phase II clinical trial of adoptive transfer of haploidentical natural killer cells for consolidation therapy of pediatric acute myeloid leukemia. J. Immunother. Cancer 2019, 7, 81. [Google Scholar] [CrossRef]
- Rubnitz, J.E.; Inaba, H.; Kang, G.; Gan, K.; Hartford, C.; Triplett, B.M.; Dallas, M.; Shook, D.; Gruber, T.; Pui, C.H.; et al. Natural killer cell therapy in children with relapsed leukemia. Pediatr. Blood Cancer 2015, 62, 1468–1472. [Google Scholar] [CrossRef] [Green Version]
- Rettinger, E.; Kuçi, S.; Naumann, I.; Becker, P.; Kreyenberg, H.; Anzaghe, M.; Willasch, A.; Koehl, U.; Bug, G.; Ruthardt, M.; et al. The cytotoxic potential of interleukin-15-stimulated cytokine-induced killer cells against leukemia cells. Cytotherapy 2012, 14, 91–103. [Google Scholar] [CrossRef]
- Lee, D.A.; Denman, C.J.; Rondon, G.; Woodworth, G.; Chen, J.; Fisher, T.; Kaur, I.; Fernandez-Vina, M.; Cao, K.; Ciurea, S.; et al. Haploidentical Natural Killer Cells Infused before Allogeneic Stem Cell Transplantation for Myeloid Malignancies: A Phase I Trial. Biol. Blood Marrow Transplant. 2016, 22, 1290–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.L.; Béziat, V.; Oei, V.Y.S.; Pfefferle, A.; Schaffer, M.; Lehmann, S.; Hellström-Lindberg, E.; Söderhäll, S.; Heyman, M.; Grandér, D.; et al. Ex Vivo Expanded Adaptive NK Cells Effectively Kill Primary Acute Lymphoblastic Leukemia Cells. Cancer Immunol. Res. 2017, 5, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.K.; Somanchi, S.S.; Dastgheyb, N.; Aquino-Lopez, A.; Cobanoglu, Z.E.; Geier, B.; Lee, D.A. Expression of carcinoma, apoptosis, and cell-death–related genes are determinants for sensitivity of pediatric cancer cell lines to lysis by natural killer cells. Pediatr. Blood Cancer 2019, 66, e27783. [Google Scholar] [CrossRef] [PubMed]
- Ljunggren, H.G.; Kärre, K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef]
- Haworth, K.B.; Leddon, J.L.; Chen, C.Y.; Horwitz, E.M.; Mackall, C.L.; Cripe, T.P. Going back to class I: MHC and immunotherapies for childhood cancer. Pediatr. Blood Cancer 2015, 62, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Mechtersheimer, G.; Staudter, M.; Majdic, O.; Dörken, B.; Moldenhauer, G.; Möller, P. Expression of HLA-A,B,C, beta 2-microglobulin (beta 2m), HLA-DR, -DP, -DQ and of HLA-D-associated invariant chain (Ii) in soft-tissue tumors. Int. J. Cancer 1990, 46, 813–823. [Google Scholar] [CrossRef]
- Castriconi, R.; Dondero, A.; Corrias, M.V.; Lanino, E.; Pende, D.; Moretta, L.; Bottino, C.; Moretta, A. Natural killer cell-mediated killing of freshly isolated neuroblastoma cells: Critical role of DNAX accessory molecule-1-poliovirus receptor interaction. Cancer Res. 2004, 64, 9180–9184. [Google Scholar] [CrossRef] [Green Version]
- Hadad, U.; Thauland, T.J.; Martinez, O.M.; Butte, M.J.; Porgador, A.; Krams, S.M. NKp46 Clusters at the Immune Synapse and Regulates NK Cell Polarization. Front. Immunol. 2015, 6, 495. [Google Scholar] [CrossRef] [Green Version]
- Elboim, M.; Gazit, R.; Gur, C.; Ghadially, H.; Betser-Cohen, G.; Mandelboim, O. Tumor Immunoediting by NKp46. J. Immunol. 2010, 184, 5637–5644. [Google Scholar] [CrossRef] [PubMed]
- Glasner, A.; Levi, A.; Enk, J.; Isaacson, B.; Viukov, S.; Orlanski, S.; Scope, A.; Neuman, T.; Enk, C.D.; Hanna, J.H.; et al. NKp46 Receptor-Mediated Interferon-γ Production by Natural Killer Cells Increases Fibronectin 1 to Alter Tumor Architecture and Control Metastasis. Immunity 2018, 48, 107–119.e4. [Google Scholar] [CrossRef] [PubMed]
- Halfteck, G.G.; Elboim, M.; Gur, C.; Achdout, H.; Ghadially, H.; Mandelboim, O. Enhanced in vivo growth of lymphoma tumors in the absence of the NK-activating receptor NKp46/NCR1. J. Immunol. 2009, 182, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Donatelli, S.S.; Djeu, J.Y. Chapter 9—Immunological Sculpting: Natural Killer-Cell Receptors and Ligands. In Cancer Immunotherapy, 2nd ed.; Prendergast, G.C., Jaffee, E.M., Eds.; Academic Press: San Diego, CA, USA, 2013; pp. 115–127. [Google Scholar] [CrossRef]
- Dukovska, D.; Fernández-Soto, D.; Valés-Gómez, M.; Reyburn, H.T. NKG2H-Expressing T Cells Negatively Regulate Immune Responses. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orbelyan, G.A.; Tang, F.; Sally, B.; Solus, J.; Meresse, B.; Ciszewski, C.; Grenier, J.C.; Barreiro, L.B.; Lanier, L.L.; Jabri, B. Human NKG2E is expressed and forms an intracytoplasmic complex with CD94 and DAP12. J. Immunol. 2014, 193, 610–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlums, H.; Cichocki, F.; Tesi, B.; Theorell, J.; Beziat, V.; Holmes, T.D.; Han, H.; Chiang, S.C.; Foley, B.; Mattsson, K.; et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 2015, 42, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.C.; Beilke, J.N.; Lanier, L.L. Adaptive immune features of natural killer cells. Nature 2009, 457, 557–561. [Google Scholar] [CrossRef]
- Muccio, L.; Bertaina, A.; Falco, M.; Pende, D.; Meazza, R.; Lopez-Botet, M.; Moretta, L.; Locatelli, F.; Moretta, A.; Della Chiesa, M. Analysis of memory-like natural killer cells in human cytomegalovirus-infected children undergoing αβ+T and B cell-depleted hematopoietic stem cell transplantation for hematological malignancies. Haematologica 2016, 101, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Cichocki, F.; Taras, E.; Chiuppesi, F.; Wagner, J.E.; Blazar, B.R.; Brunstein, C.; Luo, X.; Diamond, D.J.; Cooley, S.; Weisdorf, D.J.; et al. Adaptive NK cell reconstitution is associated with better clinical outcomes. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.R.; Chakraborty, S.; Lakhchaura, R.; Shashi, P.; Mehta, A.; Soni, M.; Chakrabarti, S. Early and Sustained Expansion of Adaptive Natural Killer Cells Following Haploidentical Transplantation and CTLA4Ig-Primed Donor Lymphocyte Infusions Dissociate Graft-versus-Leukemia and Graft-versus-Host Effects. Transplant. Cell. Ther. 2021, 27, 144–151. [Google Scholar] [CrossRef]
- Vo, D.N.; Constantinides, M.; Allende-Vega, N.; Alexia, C.; Cartron, G.; Villalba, M. Dissecting the NK Cell Population in Hematological Cancers Confirms the Presence of Tumor Cells and Their Impact on NK Population Function. Vaccines 2020, 8, 727. [Google Scholar] [CrossRef]
- Schlegel, P.; Feuchtinger, T.; Nitschke-Gérard, C.; Seidel, U.J.; Lang, A.M.; Kyzirakos, C.; Teltschik, H.M.; Ebinger, M.; Schumm, M.; Koscielniak, E.; et al. Favorable NK cell activity after haploidentical hematopoietic stem cell transplantation in stage IV relapsed Ewing’s sarcoma patients. Bone Marrow Transplant. 2015, 50 (Suppl. 2), S72–S76. [Google Scholar] [CrossRef] [Green Version]
- Kloess, S.; Huenecke, S.; Piechulek, D.; Esser, R.; Koch, J.; Brehm, C.; Soerensen, J.; Gardlowski, T.; Brinkmann, A.; Bader, P.; et al. IL-2-activated haploidentical NK cells restore NKG2D-mediated NK-cell cytotoxicity in neuroblastoma patients by scavenging of plasma MICA. Eur. J. Immunol. 2010, 40, 3255–3267. [Google Scholar] [CrossRef] [PubMed]
- Veneziani, I.; Infante, P.; Ferretti, E.; Melaiu, O.; Battistelli, C.; Lucarini, V.; Compagnone, M.; Nicoletti, C.; Castellano, A.; Petrini, S.; et al. Nutlin-3a Enhances Natural Killer Cell-Mediated Killing of Neuroblastoma by Restoring p53-Dependent Expression of Ligands for NKG2D and DNAM-1 Receptors. Cancer Immunol. Res. 2021, 9, 170–183. [Google Scholar] [CrossRef]
- Fernández, L.; Valentín, J.; Zalacain, M.; Leung, W.; Patiño-García, A.; Pérez-Martínez, A. Activated and expanded natural killer cells target osteosarcoma tumor initiating cells in an NKG2D-NKG2DL dependent manner. Cancer Lett. 2015, 368, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Boerman, G.H.; van Ostaijen-ten Dam, M.M.; Kraal, K.C.; Santos, S.J.; Ball, L.M.; Lankester, A.C.; Schilham, M.W.; Egeler, R.M.; van Tol, M.J. Role of NKG2D, DNAM-1 and natural cytotoxicity receptors in cytotoxicity toward rhabdomyosarcoma cell lines mediated by resting and IL-15-activated human natural killer cells. Cancer Immunol. Immunother. 2015, 64, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrity, D.; Call, M.E.; Feng, J.; Wucherpfennig, K.W. The activating NKG2D receptor assembles in the membrane with two signaling dimers into a hexameric structure. Proc. Natl. Acad. Sci. USA 2005, 102, 7641–7646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horng, T.; Bezbradica, J.S.; Medzhitov, R. NKG2D signaling is coupled to the interleukin 15 receptor signaling pathway. Nat. Immunol. 2007, 8, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Gilfillan, S.; Ho, E.L.; Cella, M.; Yokoyama, W.M.; Colonna, M. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat. Immunol. 2002, 3, 1150–1155. [Google Scholar] [CrossRef]
- Bálint, Š.; Lopes, F.B.; Davis, D.M. A nanoscale reorganization of the IL-15 receptor is triggered by NKG2D in a ligand-dependent manner. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Molfetta, R.; Quatrini, L.; Capuano, C.; Gasparrini, F.; Zitti, B.; Zingoni, A.; Galandrini, R.; Santoni, A.; Paolini, R. c-Cbl regulates MICA- but not ULBP2-induced NKG2D down-modulation in human NK cells. Eur. J. Immunol. 2014, 44, 2761–2770. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, N.; Lu, Y.; Davidson, D.; Colonna, M.; Veillette, A. DNAM-1 controls NK cell activation via an ITT-like motif. J. Exp. Med. 2015, 212, 2165–2182. [Google Scholar] [CrossRef]
- de Andrade, L.F.; Smyth, M.J.; Martinet, L. DNAM-1 control of natural killer cells functions through nectin and nectin-like proteins. Immunol. Cell Biol. 2014, 92, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Martinet, L.; Ferrari De Andrade, L.; Guillerey, C.; Lee, J.S.; Liu, J.; Souza-Fonseca-Guimaraes, F.; Hutchinson, D.S.; Kolesnik, T.B.; Nicholson, S.E.; Huntington, N.D.; et al. DNAM-1 expression marks an alternative program of NK cell maturation. Cell Rep. 2015, 11, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Verhoeven, D.H.; de Hooge, A.S.; Mooiman, E.C.; Santos, S.J.; ten Dam, M.M.; Gelderblom, H.; Melief, C.J.; Hogendoorn, P.C.; Egeler, R.M.; van Tol, M.J.; et al. NK cells recognize and lyse Ewing sarcoma cells through NKG2D and DNAM-1 receptor dependent pathways. Mol. Immunol. 2008, 45, 3917–3925. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.; Shook, D.R.; Shimasaki, N.; Chang, Y.H.; Fujisaki, H.; Campana, D. Cytotoxicity of activated natural killer cells against pediatric solid tumors. Clin. Cancer Res. 2010, 16, 3901–3909. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, M.; Seitz, G.; Ruck, P.; Mueller, C.; Steinle, A.; Lang, P.; Handgretinger, R.; Fuchs, J.; Warmann, S. CD155 is involved in NK-cell mediated lysis of human hepatoblastoma in vitro. Front. Biosci. 2011, 3, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Tarek, N.; Le Luduec, J.B.; Gallagher, M.M.; Zheng, J.; Venstrom, J.M.; Chamberlain, E.; Modak, S.; Heller, G.; Dupont, B.; Cheung, N.K.; et al. Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J. Clin. Investig. 2012, 122, 3260–3270. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Edberg, J.C.; Redecha, P.B.; Bansal, V.; Guyre, P.M.; Coleman, K.; Salmon, J.E.; Kimberly, R.P. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J. Clin. Investig. 1997, 100, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Koene, H.R.; Kleijer, M.; Algra, J.; Roos, D.; von dem Borne, A.E.; de Haas, M. FcγRIIIa-158V/F Polymorphism Influences the Binding of IgG by Natural Killer Cell FcγRIIIa, Independently of the FcγRIIIa-48L/R/H Phenotype. Blood 1997, 90, 1109–1114. [Google Scholar] [CrossRef] [Green Version]
- Weng, W.-K.; Negrin, R.S.; Lavori, P.; Horning, S.J. Immunoglobulin G Fc receptor FcgammaRIIIa 158 V/F polymorphism correlates with rituxi-mab-induced neutropenia after autologous trans-plantation in patients with non-Hodgkin’s lym-phoma. J. Clin. Oncol. 2010, 28, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Weng, W.K.; Levy, R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. 2003, 21, 3940–3947. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, P.; Krailo, M.D.; Buxton, A.; Hutson, P.R.; Davis, J.; Janeway, K.A.; Gorlick, R.G.; Isakoff, M. Phase II study of antidisialoganglioside antibody, dinutuximab, in combination with GM-CSF in patients with recurrent osteosarcoma (AOST1421): A report from the Children’s Oncology Group. JCO 2020, 38, 10508. [Google Scholar] [CrossRef]
- Saletta, F.; Vilain, R.E.; Gupta, A.K.; Nagabushan, S.; Yuksel, A.; Catchpoole, D.; Scolyer, R.A.; Byrne, J.A.; McCowage, G. Programmed Death-Ligand 1 Expression in a Large Cohort of Pediatric Patients with Solid Tumor and Association with Clinicopathologic Features in Neuroblastoma. JCO Precis. Oncol. 2017. [Google Scholar] [CrossRef]
- Dondero, A.; Pastorino, F.; Della Chiesa, M.; Corrias, M.V.; Morandi, F.; Pistoia, V.; Olive, D.; Bellora, F.; Locatelli, F.; Castellano, A.; et al. PD-L1 expression in metastatic neuroblastoma as an additional mechanism for limiting immune surveillance. Oncoimmunology 2016, 5, e1064578. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.M.; Quamine, A.E.; Olsen, M.R.; Capitini, C.M. Programmed cell death protein 1 on natural killer cells: Fact or fiction? J. Clin. Investig. 2020, 130, 2816–2819. [Google Scholar] [CrossRef]
- Lussier, D.M.; Johnson, J.L.; Hingorani, P.; Blattman, J.N. Combination immunotherapy with α-CTLA-4 and α-PD-L1 antibody blockade prevents immune escape and leads to complete control of metastatic osteosarcoma. J. Immunother. Cancer 2015, 3, 21. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, D.; Yang, Q.; Lv, X.; Huang, W.; Zhou, Z.; Wang, Y.; Zhang, Z.; Yuan, T.; Ding, X.; et al. Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat. Commun. 2020, 11, 6322. [Google Scholar] [CrossRef]
- Kendsersky, N.M.; Lindsay, J.; Kolb, E.A.; Smith, M.A.; Teicher, B.A.; Erickson, S.W.; Earley, E.J.; Mosse, Y.P.; Martinez, D.; Pogoriler, J.; et al. The B7-H3-Targeting Antibody-Drug Conjugate m276-SL-PBD Is Potently Effective Against Pediatric Cancer Preclinical Solid Tumor Models. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Siebert, N.; Zumpe, M.; Jüttner, M.; Troschke-Meurer, S.; Lode, H.N. PD-1 blockade augments anti-neuroblastoma immune response induced by anti-GD(2) antibody ch14.18/CHO. Oncoimmunology 2017, 6, e1343775. [Google Scholar] [CrossRef] [Green Version]
- Ehlert, K.; Hansjuergens, I.; Zinke, A.; Otto, S.; Siebert, N.; Henze, G.; Lode, H. Nivolumab and dinutuximab beta in two patients with refractory neuroblastoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, T.; Seow, S.V.; Wong, D.; Robinson, M.; Campana, D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J. Clin. Investig. 2019, 129, 2094–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Arooj, S.; Wang, H. NK Cell-Based Immune Checkpoint Inhibition. Front. Immunol. 2020, 11, 167. [Google Scholar] [CrossRef] [PubMed]
- Borst, L.; van der Burg, S.H.; van Hall, T. The NKG2A-HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 5549–5556. [Google Scholar] [CrossRef] [PubMed]
- Haworth, K.B.; Arnold, M.A.; Pierson, C.R.; Leddon, J.L.; Kurmashev, D.K.; Swain, H.M.; Hutzen, B.J.; Roberts, R.D.; Cripe, T.P. Characterization of MHC Class I and β-2-Microglobulin Expression in Pediatric Solid Malignancies to Guide Selection of Immune-Based Therapeutic Trials. Pediatr. Blood Cancer 2016, 63, 618–626. [Google Scholar] [CrossRef]
- Ruggeri, L.; Urbani, E.; André, P.; Mancusi, A.; Tosti, A.; Topini, F.; Bléry, M.; Animobono, L.; Romagné, F.; Wagtmann, N.; et al. Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica 2016, 101, 626–633. [Google Scholar] [CrossRef] [Green Version]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef] [Green Version]
- Chauvin, J.M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013, 20, 456–464. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology 2018, 7, e1466769. [Google Scholar] [CrossRef]
- Yuan, H.; Liu, J.; Zhang, J. The Current Landscape of Immune Checkpoint Blockade in Metastatic Lung Squamous Cell Carcinoma. Molecules 2021, 26, 1392. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Lenvik, T.R.; McCullar, V.; Felices, M.; O’Brien, M.S.; Cooley, S.A.; Verneris, M.R.; Cichocki, F.; Holman, C.J.; Panoskaltsis-Mortari, A.; et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood 2012, 119, 3064–3072. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, L.C.; Lopez-Vergès, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther. 2018, 11, 7005–7009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, T.; Dierich, A.; Benoist, C.; Mathis, D. Independent modes of natural killing distinguished in mice lacking Lag3. Science 1996, 272, 405–408. [Google Scholar] [CrossRef]
- Huard, B.; Mastrangeli, R.; Prigent, P.; Bruniquel, D.; Donini, S.; El-Tayar, N.; Maigret, B.; Dréano, M.; Triebel, F. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc. Natl. Acad. Sci. USA 1997, 94, 5744–5749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemke, D.; Pfenning, P.N.; Sahm, F.; Klein, A.C.; Kempf, T.; Warnken, U.; Schnölzer, M.; Tudoran, R.; Weller, M.; Platten, M.; et al. Costimulatory protein 4IgB7H3 drives the malignant phenotype of glioblastoma by mediating immune escape and invasiveness. Clin. Cancer Res. 2012, 18, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Martin-Orozco, N.; Zheng, P.; Li, J.; Zhang, P.; Tan, H.; Park, H.J.; Jeong, M.; Chang, S.H.; Kim, B.S.; et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. 2017, 27, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Janakiram, M.; Shah, U.A.; Liu, W.; Zhao, A.; Schoenberg, M.P.; Zang, X. The third group of the B7-CD28 immune checkpoint family: HHLA2, TMIGD2, B7x, and B7-H3. Immunol. Rev. 2017, 276, 26–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 Expression in Solid Tumors and Role as a Target for Cancer Therapy. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Vey, N.; Karlin, L.; Sadot-Lebouvier, S.; Broussais, F.; Berton-Rigaud, D.; Rey, J.; Charbonnier, A.; Marie, D.; André, P.; Paturel, C.; et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D.; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget 2018, 9, 17675–17688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, X.; Li, K.; Zhang, T. FcγR-Binding Is an Important Functional Attribute for Immune Checkpoint Antibodies in Cancer Immunotherapy. Front. Immunol. 2019, 10, 292. [Google Scholar] [CrossRef] [PubMed]
- Hosseinkhani, N.; Derakhshani, A.; Kooshkaki, O.; Abdoli Shadbad, M.; Hajiasgharzadeh, K.; Baghbanzadeh, A.; Safarpour, H.; Mokhtarzadeh, A.; Brunetti, O.; Yue, S.C.; et al. Immune Checkpoints and CAR-T Cells: The Pioneers in Future Cancer Therapies? Int. J. Mol. Sci. 2020, 21, 8305. [Google Scholar] [CrossRef]
- Ruffo, E.; Wu, R.C.; Bruno, T.C.; Workman, C.J.; Vignali, D.A.A. Lymphocyte-activation gene 3 (LAG3): The next immune checkpoint receptor. Semin Immunol. 2019, 42, 101305. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef] [Green Version]
- Lecocq, Q.; Keyaerts, M.; Devoogdt, N.; Breckpot, K. The Next-Generation Immune Checkpoint LAG-3 and Its Therapeutic Potential in Oncology: Third Time’s a Charm. Int. J. Mol. Sci. 2020, 22, 75. [Google Scholar] [CrossRef]
- Johnston, J.A.; Bacon, C.M.; Finbloom, D.S.; Rees, R.C.; Kaplan, D.; Shibuya, K.; Ortaldo, J.R.; Gupta, S.; Chen, Y.Q.; Giri, J.D.; et al. Tyrosine phosphorylation and activation of STAT5, STAT3, and Janus kinases by interleukins 2 and 15. Proc. Natl. Acad. Sci. USA 1995, 92, 8705–8709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buddingh, E.P.; Schilham, M.W.; Ruslan, S.E.N.; Berghuis, D.; Szuhai, K.; Suurmond, J.; Taminiau, A.H.M.; Gelderblom, H.; Egeler, R.M.; Serra, M.; et al. Chemotherapy-resistant osteosarcoma is highly susceptible to IL-15-activated allogeneic and autologous NK cells. Cancer Immunol. Immunother. 2011, 60, 575–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hüber, C.; Doisne, J.-M.; Colucci, F. IL-12/15/18-preactivated NK cells suppress GvHD in a mouse model of mismatched hematopoietic cell transplantation. Eur. J. Immunol. 2015, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vela, M.; Corral, D.; Carrasco, P.; Fernández, L.; Valentín, J.; González, B.; Escudero, A.; Balas, A.; de Paz, R.; Torres, J.; et al. Haploidentical IL-15/41BBL activated and expanded natural killer cell infusion therapy after salvage chemotherapy in children with relapsed and refractory leukemia. Cancer Lett. 2018, 422, 107–117. [Google Scholar] [CrossRef]
- Lee, S.H.; Shin, D.J.; Kim, Y.; Kim, C.J.; Lee, J.J.; Yoon, M.S.; Uong, T.N.T.; Yu, D.; Jung, J.Y.; Cho, D.; et al. Comparison of Phenotypic and Functional Characteristics Between Canine Non-B, Non-T Natural Killer Lymphocytes and CD3(+)CD5(dim)CD21(-) Cytotoxic Large Granular Lymphocytes. Front. Immunol. 2018, 9, 841. [Google Scholar] [CrossRef] [Green Version]
- Michael, H.T.; Ito, D.; McCullar, V.; Zhang, B.; Miller, J.S.; Modiano, J.F. Isolation and characterization of canine natural killer cells. Vet. Immunol. Immunopathol. 2013, 155, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canter, R.J.; Grossenbacher, S.K.; Foltz, J.A.; Sturgill, I.R.; Park, J.S.; Luna, J.I.; Kent, M.S.; Culp, W.T.N.; Chen, M.; Modiano, J.F.; et al. Radiotherapy enhances natural killer cell cytotoxicity and localization in pre-clinical canine sarcomas and first-in-dog clinical trial. J. Immunother. Cancer 2017, 5, 98. [Google Scholar] [CrossRef] [Green Version]
- Judge, S.J.; Yanagisawa, M.; Sturgill, I.R.; Bateni, S.B.; Gingrich, A.A.; Foltz, J.A.; Lee, D.A.; Modiano, J.F.; Monjazeb, A.M.; Culp, W.T.N.; et al. Blood and tissue biomarker analysis in dogs with osteosarcoma treated with palliative radiation and intra-tumoral autologous natural killer cell transfer. PLoS ONE 2020, 15, e0224775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Khan, A.A.; Gunn, H.J.; Day, M.J.; Tayebi, M.; Ryan, S.D.; Kuntz, C.A.; Saad, E.S.; Richardson, S.J.; Danks, J.A. Immunohistochemical Validation of Spontaneously Arising Canine Osteosarcoma as a Model for Human Osteosarcoma. J. Comp. Pathol. 2017, 157, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Fenger, J.M.; London, C.A.; Kisseberth, W.C. Canine Osteosarcoma: A Naturally Occurring Disease to Inform Pediatric Oncology. ILAR J. 2014, 55, 69–85. [Google Scholar] [CrossRef] [Green Version]
- Meazza, C.; Cefalo, G.; Massimino, M.; Daolio, P.; Pastorino, U.; Scanagatta, P.; Morosi, C.; Podda, M.; Ferrari, A.; Terenziani, M.; et al. Primary metastatic osteosarcoma: Results of a prospective study in children given chemotherapy and interleukin-2. Med. Oncol. 2017, 34, 191. [Google Scholar] [CrossRef]
- Roberts, R.D.; Lizardo, M.M.; Reed, D.R.; Hingorani, P.; Glover, J.; Allen-Rhoades, W.; Fan, T.; Khanna, C.; Sweet-Cordero, E.A.; Cash, T.; et al. Provocative questions in osteosarcoma basic and translational biology: A report from the Children’s Oncology Group. Cancer 2019, 125, 3514–3525. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, S.-H.; Kim, C.-J.; Lee, J.-J.; Yu, D.; Ahn, S.; Shin, D.-J.; Kim, S.-K. Canine non-B, non-T NK lymphocytes have a potential antibody-dependent cellular cytotoxicity function against antibody-coated tumor cells. BMC Vet. Res. 2019, 15, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addissie, S.; Klingemann, H. Cellular Immunotherapy of Canine Cancer. Vet. Sci. 2018, 5, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grøndahl-Rosado, C.; Boysen, P.; Johansen, G.M.; Brun-Hansen, H.; Storset, A.K. NCR1 is an activating receptor expressed on a subset of canine NK cells. Vet. Immunol. Immunopathol. 2016, 177, 7–15. [Google Scholar] [CrossRef]
- Dobrenkov, K.; Ostrovnaya, I.; Gu, J.; Cheung, I.Y.; Cheung, N.-K.V. Oncotargets GD2 and GD3 are highly expressed in sarcomas of children, adolescents, and young adults. Pediatr. Blood Cancer 2016, 63, 1780–1785. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.L.; Chen, L.; Li, Y.J.; Kong, D.L. PD-L1/PD-1 axis serves an important role in natural killer cell-induced cytotoxicity in osteosarcoma. Oncol. Rep. 2019, 42, 2049–2056. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.; Pfeiffer, M.; Müller, I.; Schumm, M.; Ebinger, M.; Koscielniak, E.; Feuchtinger, T.; Föll, J.; Martin, D.; Handgretinger, R. Haploidentical stem cell transplantation in patients with pediatric solid tumors: Preliminary results of a pilot study and analysis of graft versus tumor effects. Klin Padiatr. 2006, 218, 321–326. [Google Scholar] [CrossRef]
- Pende, D.; Marcenaro, S.; Falco, M.; Martini, S.; Bernardo, M.E.; Montagna, D.; Romeo, E.; Cognet, C.; Martinetti, M.; Maccario, R.; et al. Anti-leukemia activity of alloreactive NK cells in KIR ligand-mismatched haploidentical HSCT for pediatric patients: Evaluation of the functional role of activating KIR and redefinition of inhibitory KIR specificity. Blood 2009, 113, 3119–3129. [Google Scholar] [CrossRef] [Green Version]
- Rutella, S.; Rumi, C.; Laurenti, L.; Pierelli, L.; Sora, F.; Sica, S.; Leone, G. Immune reconstitution after transplantation of autologous peripheral CD34+ cells: Analysis of predictive factors and comparison with unselected progenitor transplants. Br. J. Haematol. 2000, 108, 105–115. [Google Scholar] [CrossRef]
- Asai, O.; Longo, D.L.; Tian, Z.G.; Hornung, R.L.; Taub, D.D.; Ruscetti, F.W.; Murphy, W.J. Suppression of graft-versus-host disease and amplification of graft-versus-tumor effects by activated natural killer cells after allogeneic bone marrow transplantation. J. Clin. Investig. 1998, 101, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Jiang, J.; Wu, C. CAR-NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer Lett. 2020, 472, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Pende, D.; Falco, M.; Della Chiesa, M.; Moretta, A.; Moretta, L. NK Cells Mediate a Crucial Graft-versus-Leukemia Effect in Haploidentical-HSCT to Cure High-Risk Acute Leukemia. Trends Immunol. 2018, 39, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.K.; Kushner, B.H.; Yeh, S.D.; Larson, S.M. 3F8 monoclonal antibody treatment of patients with stage 4 neuroblastoma: A phase II study. Int. J. Oncol. 1998, 12, 1299–1306. [Google Scholar] [CrossRef]
- Yu, A.L.; Uttenreuther-Fischer, M.M.; Huang, C.S.; Tsui, C.C.; Gillies, S.D.; Reisfeld, R.A.; Kung, F.H. Phase I trial of a human-mouse chimeric anti-disialoganglioside monoclonal antibody ch14.18 in patients with refractory neuroblastoma and osteosarcoma. J. Clin. Oncol. 1998, 16, 2169–2180. [Google Scholar] [CrossRef]
- Simon, T.; Hero, B.; Faldum, A.; Handgretinger, R.; Schrappe, M.; Klingebiel, T.; Berthold, F. Long term outcome of high-risk neuroblastoma patients after immunotherapy with antibody ch14.18 or oral metronomic chemotherapy. BMC Cancer 2011, 11, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [Green Version]
- Kushner, B.H.; Cheung, I.Y.; Modak, S.; Basu, E.M.; Roberts, S.S.; Cheung, N.K. Humanized 3F8 Anti-GD2 Monoclonal Antibody Dosing with Granulocyte-Macrophage Colony-Stimulating Factor in Patients With Resistant Neuroblastoma: A Phase 1 Clinical Trial. JAMA Oncol. 2018, 4, 1729–1735. [Google Scholar] [CrossRef] [Green Version]
- Meazza, R.; Azzarone, B.; Orengo, A.; Ferrini, S. Role of Common-Gamma Chain Cytokines in NK Cell Development and Function: Perspectives for Immunotherapy. J. Biomed. Biotechnol. 2011, 2011, 861920. [Google Scholar] [CrossRef]
- Davila, M.L.; Brentjens, R.J. CD19-Targeted CAR T cells as novel cancer immunotherapy for relapsed or refractory B-cell acute lymphoblastic leukemia. Clin. Adv. Hematol. Oncol. 2016, 14, 802–808. [Google Scholar]
- Wu, Y.; Tian, Z.; Wei, H. Developmental and Functional Control of Natural Killer Cells by Cytokines. Front. Immunol. 2017, 8, 930. [Google Scholar] [CrossRef] [PubMed]
- Parihar, R.; Rivas, C.; Huynh, M.; Omer, B.; Lapteva, N.; Metelitsa, L.S.; Gottschalk, S.M.; Rooney, C.M. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors. Cancer Immunol. Res. 2019, 7, 363–375. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Heczey, A.; Courtney, A.N.; Montalbano, A.; Robinson, S.; Liu, K.; Li, M.; Ghatwai, N.; Dakhova, O.; Liu, B.; Raveh-Sadka, T.; et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: An interim analysis. Nat. Med. 2020, 26, 1686–1690. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, K.; Capitini, C.M.; Saha, K. Genome engineering of induced pluripotent stem cells to manufacture natural killer cell therapies. Stem Cell Res. 2020, 11, 234. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Huang, F.; Wang, Y.; Xu, Y.; Yang, T.; Fan, Z.; Lin, R.; Xu, N.; Xuan, L.; Ye, J.; et al. Haploidentical transplantation might have superior graft-versus-leukemia effect than HLA-matched sibling transplantation for high-risk acute myeloid leukemia in first complete remission: A prospective multicentre cohort study. Leukemia 2020, 34, 1433–1443. [Google Scholar] [CrossRef]
- Arai, S.; Vogelsang, G.B. Management of graft-versus-host disease. Blood Rev. 2000, 14, 190–204. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [Green Version]
- Bregni, M.; Herr, W.; Blaise, D. Allogeneic stem cell transplantation for renal cell carcinoma. Expert Rev. Anticancer 2011, 11, 901–911. [Google Scholar] [CrossRef]
- Tykodi, S.S.; Sandmaier, B.M.; Warren, E.H.; Thompson, J.A. Allogeneic hematopoietic cell transplantation for renal cell carcinoma: Ten years after. Expert Opin. Biol. 2011, 11, 763–773. [Google Scholar] [CrossRef] [Green Version]
- Llosa, N.J.; Cooke, K.R.; Chen, A.R.; Gamper, C.J.; Klein, O.R.; Zambidis, E.T.; Luber, B.; Rosner, G.; Siegel, N.; Holuba, M.J.; et al. Reduced-Intensity Haploidentical Bone Marrow Transplantation with Post-Transplant Cyclophosphamide for Solid Tumors in Pediatric and Young Adult Patients. Biol. Blood Marrow Transplant. 2017, 23, 2127–2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunzmann, V.; Bauer, E.; Wilhelm, M. Gamma/delta T-cell stimulation by pamidronate. N. Engl. J. Med. 1999, 340, 737–738. [Google Scholar] [CrossRef]
- Nussbaumer, O.; Gruenbacher, G.; Gander, H.; Thurnher, M. DC-like cell-dependent activation of human natural killer cells by the bisphosphonate zoledronic acid is regulated by γδ T lymphocytes. Blood 2011, 118, 2743–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pytel, N.; McDowell, K.; Desantes, K.; Capitini, C.; Hematti, P.; Leith, C.; Sondel, P.; Otto, M. 2019 ASPHO ABSTRACTS. Pediatr. Blood Cancer 2019, 66, e27713. [Google Scholar] [CrossRef]
- Buddingh, E.P.; Ruslan, S.E.N.; Berghuis, D.; Gelderblom, H.; Anninga, J.K.; Hogendoorn, P.C.W.; Egeler, R.M.; Schilham, M.W.; Lankester, A.C. Intact interferon signaling in peripheral blood leukocytes of high-grade osteosarcoma patients. Cancer Immunol. Immunother. 2012, 61, 941–947. [Google Scholar] [CrossRef] [Green Version]
- Markiewicz, K.; Zeman, K.; Kozar, A.; Gołębiowska-Wawrzyniak, M.; Woźniak, W. Evaluation of selected parameters of cellular immunity in children with osteosarcoma at diagnosis. Med. Wieku Rozw. 2012, 16, 212–221. [Google Scholar]
- Moore, C.; Eslin, D.; Levy, A.; Roberson, J.; Giusti, V.; Sutphin, R. Prognostic significance of early lymphocyte recovery in pediatric osteosarcoma. Pediatr. Blood Cancer 2010, 55, 1096–1102. [Google Scholar] [CrossRef]
- Erbe, A.K.; Wang, W.; Carmichael, L.; Kim, K.; Mendonça, E.A.; Song, Y.; Hess, D.; Reville, P.K.; London, W.B.; Naranjo, A.; et al. Neuroblastoma Patients’ KIR and KIR-Ligand Genotypes Influence Clinical Outcome for Dinutuximab-based Immunotherapy: A Report from the Children’s Oncology Group. Clin. Cancer Res. 2018, 24, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Federico, S.M.; McCarville, M.B.; Shulkin, B.L.; Sondel, P.M.; Hank, J.A.; Hutson, P.; Meagher, M.; Shafer, A.; Ng, C.Y.; Leung, W.; et al. A Pilot Trial of Humanized Anti-GD2 Monoclonal Antibody (hu14.18K322A) with Chemotherapy and Natural Killer Cells in Children with Recurrent/Refractory Neuroblastoma. Clin. Cancer Res. 2017, 23, 6441–6449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modak, S.; Le Luduec, J.B.; Cheung, I.Y.; Goldman, D.A.; Ostrovnaya, I.; Doubrovina, E.; Basu, E.; Kushner, B.H.; Kramer, K.; Roberts, S.S.; et al. Adoptive immunotherapy with haploidentical natural killer cells and Anti-GD2 monoclonal antibody m3F8 for resistant neuroblastoma: Results of a phase I study. Oncoimmunology 2018, 7, e1461305. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Target | Indications | Therapy | Drug Type | Status |
|---|---|---|---|---|
| GD2 [108] | Melanoma, neuroblastoma, osteosarcoma | 3F8 | Mouse IgG3 | Phase I, II |
| ch14.18 (Dinutuximab) | Mouse human chimeric IgG1 | FDA approved (neuroblastoma) | ||
| hu14.18-IL2 | Humanized 14.18 fused with IL-2 | Phase I, II | ||
| hu14.18K322A | Point mutation in hu14.18 | Phase I, II | ||
| hu3F8 (Naxitumab) | Humanized 3F8 | FDA approved (neuroblastoma) | ||
| NKG2A [91] | NSCLC, head and neck squamous cell carcinoma, colorectal cancer, gynecologic cancers, other advanced solid malignancies | IPH2201 (Monalizumab) | Humanized IgG4 | Phase I, II |
| KIR2D [109] | Breast, kidney, and ovarian carcinomas | IPH2102 (alirocumab) | Human IgG4 | Phase I |
| TIGIT [92,110,111] | Advanced solid tumors | OMP-313M32 (Etigilimab) | Human IgG1 | Phase I |
| NSCLC, SCLC, esophageal cancer, advanced solid tumors | MTIG7192A/RG-6058 (Tiragolumab) | Human IgG1 | FDA approved (NSCLC) | |
| Melanoma | MK-7684 (Vibostolimab) | Humanized IgG1 | Phase I, II | |
| Multiple myeloma | BMS-986207 | Human IgG1 | Phase I | |
| Advanced solid tumors | ASP-8374 | Human IgG4 | Phase I | |
| Metastatic solid tumors | BGB-A1217 | Humanized IgG1 | Phase I | |
| NSCLC, advanced solid tumors | AB-154 | Humanized IgG1 | Phase I | |
| TIM-3 [98,110] | Liver cancer, advanced solid tumors | TSR-022 | Human IgG4 | Phase I |
| Advanced malignancies, AML, MDS | MBG453 | Humanized IgG4 | Phase I, II | |
| Solid tumors and lymphomas | SYM023 | Human IgG1 | Phase I | |
| Solid Tumors | INCAGN2390 | Human IgG1 | Phase I | |
| Advanced solid tumors | LY3321367 | Human IgG1 | Phase I | |
| Advanced malignancies | BMS-986258 | Human IgG1 | Phase I | |
| Solid Tumors | BGBA425 | Humanized IgG1 | Phase I | |
| Advanced solid tumors | SHR-1702 (Camrelizumab) | Humanized IgG4 | Phase I | |
| Melanoma, NSCLC, solid tumors | RO7121661 | Bi-specific antibody targeting PD-1 and TIM-3 | Phase I | |
| DNAM-1 [92] | Advanced solid tumors | LY3435151 | DNAM-1 agonist | Phase I |
| LAG-3 [112,113,114,115] | Breast carcinoma, melanoma, RCC, solid tumors | IMP321 (Eftilagimod alpha) | LAG-3 IgG1 Fc fusion protein | Phase I, II |
| Advanced solid tumors, melanoma, colon cancer, hematologic malignancies, glioblastoma, gliosarcoma, advanced gastric cancer, NSCLC, RCC | BMS-986016 (Relatlimab) | Human IgG4 | Phase II, III | |
| Advanced solid tumors, advanced hematologic malignancies | LAG525 | Humanized IgG4 | Phase I, II | |
| Advanced solid tumors, hematologic malignancies | MK-4280 | Humanized IgG4 | Phase I, II | |
| Malignancies | REGN3767 | Human IgG4 | Phase I | |
| Advanced solid tumors | TSR-033 | Humanized IgG4 | Phase I, II | |
| Advanced solid tumors, cholangiocarcinoma, liver cancer, gastric cancer, breast cancer, esophageal cancer, hematologic malignancies | MGD013 | Bi-specific DART® IgG4k antibody targeting PD-1 and Lag-3 | Phase I | |
| Advanced malignancies | FS118 | Bi-specific antibody targeting PD-L1 and LAG-3 | Phase I | |
| Advanced solid tumors, lymphomas | Sym022 | Human Fc-inert | Phase I | |
| Solid tumors | XmAb22841 | Bi-specific Fc-inert antibody | Phase I | |
| B7-H3 [110] | Solid tumors, pediatric B7-H3 expressing solid tumors, prostate cancer | MGA271 (Enblituzumab) | Humanized IgG1 | Phase I, II |
| Advanced solid tumors | MGD009 | Bi-specific DART® IgG1 antibody targeting B7-H3 and CD3 | Phase I | |
| DSRCT, advanced CNS or leptomeningeal cancer, neuroblastoma, gliomas | 131I-8H9 (Omburtamab) | Murine IgG1 | Phase I |
| Clinical Trial Number | Phase | Title | Sponsor Institution | Status | Condition Treated | Trial Goals | Treatment Notes |
|---|---|---|---|---|---|---|---|
| NCT00582816 | 1 | Haploidentical Transplant with NK Cell Infusion for Pediatric Acute Leukemia and Solid Tumors | University of Wisconsin, Madison | Terminated (toxicity) | Relapsed/refractory leukemia or solid tumors | Primary outcomes: GVHD, engraftment failure, number of days until engraftment criteria were met, mortality rate | Methylprednisolone, Equine ATG, Cyclosporine, Fludarabine, Melphalan, Thiotepa and Rituximab |
| NCT01875601 | 1 | NK White Blood Cells and Interleukin in Children and Young Adults with Advanced Solid Tumors | National Cancer Institute (NCI) | Completed | Relapsed/refractory solid tumors | Primary objectives are: (1) to assess the feasibility of harvesting and expanding activated NK cells to meet escalating dose goals in Cohort A, (2) to assess the toxicity of infusing escalating doses of activated NK cells following lymphodepleting chemotherapy without rhIL15 (cohort A), and (3) to assess the toxicity of infusing NK activated cells with escalating doses of rhIL15 (cohort B) in pediatric patients with refractory malignant solid tumors. | All patients receive pre-NK lymphodepleting chemotherapy with cyclophosphamide. Cohort A receives escalating doses of NK cells; Cohort B receives escalating doses of NK cells and rhIL15 |
| NCT00640796 | 1 | Pilot Study of Expanded, Donor Natural Killer Cell Infusions for Refractory Non-B Lineage Hematologic Malignancies and Solid Tumors | St. Jude Children’s Research Hospital | Completed | Relapsed/refractory hematologic malignancies, Ewing sarcoma family of tumors (ESFT) and rhabdomyosarcoma (RMS) | To determine the maximum tolerated dose of expanded NK cells in research participants with relapsed or refractory hematologic malignancies and sarcomas. | Haploidentical NK cells + Cyclophosphamide, Fludarabine, Interleukin-2, Mesna |
| NCT02130869 | 1 | A Pilot Study of Immunotherapy Including Haploidentical NK Cell Infusion Following CD133+ Positively-Selected Autologous Hematopoietic Stem Cells in Children with High-Risk Solid Tumors or Lymphomas | St. Jude Children’s Research Hospital | Completed | Relapsed/refractory neuroblastoma, lymphoma, high risk solid tumor | To evaluate day +35 absolute neutrophil count (and) engraftment in autologous stem cell transplantation for high-risk pediatric malignancies after stem cell selection and immunotherapy. | All participants first receive standard of care high-dose chemotherapy specific to their tumor type. Group C participants receive melphalan, etoposide (or etoposide phosphate), carboplatin, CD133+ selected autologous stem cell infusion, IL-2, haploidentical natural killer cell infusion, G-CSF, and GM-CSF. |
| NCT02100891 | 2 | Phase 2 STIR Trial: Haploidentical Transplant and Donor Natural Killer Cells for Solid Tumors (STIR) | Medical College of Wisconsin | Active, not recruiting | Relapsed/refractory neuroblastoma, Ewing sarcoma, rhabdomyosarcoma, osteosarcoma, CNS tumors | Disease-control rate | Patients will receive a reduced-intensity conditioning regimen for 6 days that consists of Fludarabine 150 mg/m2, Cyclophosphamide 29 mg/kg, and 3 Gy total body irradiation (TBI), followed by HLA-haploidentical marrow from a family member on Day 0. On Days +3 and +4, Cyclophosphamide 50 mg/kg will be infused for selective in vivo T cell depletion. Additional post-grafting immune suppression will consist of mycophenolate mofetil and either tacrolimus or sirolimus. PBMCs will be collected from donors on Day +6, from which NK cells will be selected and infused into patients on Day +7. |
| NCT02508038 | 1 | Alpha/Beta CD19+ Depleted Haploidentical Transplantation + Zometa for Pediatric Hematologic Malignancies and Solid Tumors | University of Wisconsin, Madison | Recruiting | Relapsed/refractory leukemia, lymphoma, neuroblastoma, Ewing sarcoma, rhabdomyosarcoma, osteosarcoma, PNET | Incidence of acute GVHD. Incidence of graft failure | Patients with high-risk leukemia will receive myeloablative conditioning with anti-thymocyte globulin intravenously (IV) over 4–6 h on days −12 through −9, Fludarabine IV over 30 min on days −8 through −5, Thiotepa IV every 12 h on day −4 and total body irradiation (TBI) on days −3 through −1. All other patients receive reduced intensity conditioning consisting of anti-thymocyte globulin intravenously (IV) over 4–6 h on days −12 through −9, fludarabine IV over 30 min on days −8 through −5, thiotepa IV over 4 h every 12 h on day −4, and melphalan IV on days −3 and −2. Patients undergo TCR-alpha/beta+ and CD19+ depleted KIR/KIR ligand-mismatched haploidentical donor PBMC transplantation on day 0. |
| NCT01576692 | 1 | Combination Chemotherapy, Monoclonal Antibody, and Natural Killer Cells in Treating Young Patients with Recurrent or Refractory Neuroblastoma | St. Jude Children’s Research Hospital | Completed | Relapsed/refractory neuroblastoma | To observe and describe the toxicities associated with humanized anti-GD2 antibody (hu14.18K322A) with and without allogeneic NK cells when given with repeated cycles of chemotherapy to children with refractory/relapsed neuroblastoma. | A maximum of 6 courses of therapy may be given on the following schedule: Courses 1, 3, and 5: Humanized anti-GD2 antibody + chemotherapy. Courses 2, 4, and 6: Humanized anti-GD2 antibody + chemotherapy, with or without natural killer (NK) cells (depending on availability of appropriate NK donor). NK Cell dosage: minimum of 0.1 × 106 cells/kg; maximum of 400 × 106 CD45+ cells/kg, given once |
| NCT03209869 | 1 | Treatment of Relapsed or Refractory Neuroblastoma and Osteosarcoma with Expanded Haploidentical NK Cells and Hu14.18-IL2 | University of Wisconsin, Madison | Suspended (COVID) | Relapsed/refractory neuroblastoma and osteosarcoma | Safety: Incidence of treatment-emergent adverse events of treatment with AENK cells and hu14.18-IL2 | All subjects will receive Ex vivo Expanded and Activated Haploidentical Donor NK Cells + hu14.18-IL2 |
| NCT00877110 | 1 | Anti-GD2 3F8 Antibody and Allogeneic Natural Killer Cells for High-Risk Neuroblastoma | Memorial Sloan Kettering Cancer Center | Completed | Relapsed/refractory neuroblastoma | Assess the feasibility and safety of administering allogeneic haploidentical NK infusions with 3F8 in patients with high-risk NB | Patients will receive combination chemotherapy with intravenous cyclophosphamide for two days, IV vincristine for one day, and IV topotecan 2.4 mg/m2/day for 3 days. |
| NCT02650648 | 1 | Humanized Anti-GD2 Antibody Hu3F8 and Allogeneic Natural Killer Cells for High-Risk Neuroblastoma | Memorial Sloan Kettering Cancer Center | Active, not recruiting | Relapsed/refractory neuroblastoma | The number patient responses observed at each dose level | Following chemotherapy, three dose levels of NK cells, starting at dose level 1, will be evaluated in this treatment protocol. Cyclophosphamide will be given for two days (days −6 and −5). On Days −1, +1, +5, +7, and +9 hu3F8 is administered. On day 0, daily from +2 through +4, day +6, and day +8, rIL-2 is administered subcutaneously. |
| NCT02573896 | 1 | Immunotherapy of Relapsed Refractory Neuroblastoma with Expanded NK Cells | New Approaches to Neuroblastoma Therapy Consortium | Recruiting | Relapsed/refractory neuroblastoma | Feasibility of expanding NK cells from neuroblastoma patients and cryopreserving, shipping, and infusing multiple doses of NK cells | NK cells on Day 5 and 17.5 mg/m2/dose of Ch14.18 on Day 1–4. Patients will also receive 25 mg/m2/dose of Lenalidomide during Day −6 through 14 of treatment. |
| NCT00698009 | 1 | Haploidentical Natural Killer (NK) Cells in Patients with Relapsed or Refractory Neuroblastoma | M.D. Anderson Cancer Center | Terminated (slow accrual) | Relapsed/refractory neuroblastoma | Participant Disease Response | Fludarabine 25 mg/m2 intravenous daily starting 6 days before the NK cell infusion (considered Day −6) and once a day through Day −2. Cyclophosphamide 60 mg/kg IV days −5 and −4. Natural Killer Cell Infusion on Day 0. |
| NCT03294954 | 1 | GD2 Specific CAR and Interleukin-15 Expressing Autologous NKT Cells to Treat Children with Neuroblastoma (GINAKIT2) | Texas Children’s Hospital | Recruiting | Relapsed/refractory neuroblastoma | Maximum tolerated dose of autologous NKTs expressing a 2nd generation GD2-specific chimeric antigen receptor administered to patients with relapsed or refractory neuroblastoma | GINAKIT cells + Cytoxan + fludara |
| NCT01287104 | 1 | A Phase I Study of NK Cell Infusion Following Allogeneic Peripheral Blood Stem Cell Transplantation from Related or Matched Unrelated Donors in Pediatric Patients with Solid Tumors and Leukemias | National Cancer Institute (NCI) | Completed | Relapsed/refractory leukemia or solid tumors | To assess the feasibility and toxicity of infusing escalating doses of donor-derived activated NK cell donor lymphocyte infusions (NK-DLI) on Days 7 plus or minus 2 days and 49 plus or minus 7 days following human leukocyte antigen (HLA)-matched T cell depleted (TCD) peripheral blood stem cell transplant (PBSCT) | A phase 1 cell dose escalation of donor derived NK-DLI will be performed using 3 dose levels infused on days 21 more or less 3 post-PBSCT and a second infusion on day 49 more or less 7 post-PBSCT. |
| NCT03420963 | 1 | Donor Natural Killer Cells, Cyclophosphamide, and Etoposide in Treating Children and Young Adults with Relapsed or Refractory Solid Tumors | MD Anderson | Recruiting | Relapsed/refractory solid tumors | Determine the safety, maximum tolerated dose and/or recommended phase II dose of cord blood-derived expanded allogeneic natural killer cells following chemotherapy | Patients receive cyclophosphamide IV QD over 30 min and etoposide IV QD over 60 min on days 1–5 in the absence of unacceptable toxicity. Patients then receive cord blood derived allogeneic NK cells IV on day 8. |
| NCT04211675 | 1/2 | A Phase I-II Study of Ex-Vivo Expanded Autologous NK Cells Infusions in Combination with Irinotecan, Temozolomide, and Dinutuximab in Patients with Relapsed or Refractory Neuroblastoma: The STING Trial | Nationwide Children’s Hospital | Not yet recruiting | Relapsed/refractory neuroblastoma | NK cells safety and tolerability: Number of participants with treatment-related adverse events and toxicities. Response to NK Cell treatment as determine by CT/MRI imaging, MIBG imaging, and bone marrow aspiration. | 6 cycles of 21 days each consisting of irinotecan, temozolomide, Dinutuximab, and Sargramostim (Cycle 1), or irinotecan, temozolomide, Dinutuximab, Sargramostim, and natural killer (NK) cells (Cycles 2–6). |
| NCT02409576 | 1/2 | Pilot Study of Expanded, Activated Haploidentical Natural Killer Cell Infusions for Sarcomas (NKEXPSARC) | National University Hospital, Singapore | Recruiting | Relapsed/refractory Ewing sarcoma, rhabdomyosarcoma | Disease response after expanded activated NK cell infusion | Day −7 Cyclophosphamide at 60 mg/kg. Day −6 Fludarabine at 25 mg/m2 daily for 5 days. Each patient will receive radiation within 48 h of NK cell infusion to make the tumor cells more sensitive to NK cell killing Radiation 2 Gy. Each patient will receive IL-2 to support NK cell activation and expansion in vivofor a total of 6 doses. Expanded activated haploidentical NK cells will be infused on day 0. |
| NCT04214730 | N/A | Study of Natural Killer Cell Combined with Chemotherapy for Advanced Solid Tumor | Yantai Yuhuangding Hospital | Recruiting | Relapsed/refractory solid tumors | Disease Control Rates | Patients in group A will receive 4 cycles of NK treatments within 8 months. Patients in group B will have no immunotherapy. Chemotherapy is available in both groups. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quamine, A.E.; Olsen, M.R.; Cho, M.M.; Capitini, C.M. Approaches to Enhance Natural Killer Cell-Based Immunotherapy for Pediatric Solid Tumors. Cancers 2021, 13, 2796. https://doi.org/10.3390/cancers13112796
Quamine AE, Olsen MR, Cho MM, Capitini CM. Approaches to Enhance Natural Killer Cell-Based Immunotherapy for Pediatric Solid Tumors. Cancers. 2021; 13(11):2796. https://doi.org/10.3390/cancers13112796
Chicago/Turabian StyleQuamine, Aicha E., Mallery R. Olsen, Monica M. Cho, and Christian M. Capitini. 2021. "Approaches to Enhance Natural Killer Cell-Based Immunotherapy for Pediatric Solid Tumors" Cancers 13, no. 11: 2796. https://doi.org/10.3390/cancers13112796
APA StyleQuamine, A. E., Olsen, M. R., Cho, M. M., & Capitini, C. M. (2021). Approaches to Enhance Natural Killer Cell-Based Immunotherapy for Pediatric Solid Tumors. Cancers, 13(11), 2796. https://doi.org/10.3390/cancers13112796