
Risks and Function of Breast Cancer Susceptibility Alleles

Abstract
:Simple Summary
Abstract
1. Introduction
2. Genetic Predisposition to Breast Cancer
2.1. High- and Moderate-Penetrance Susceptibility Genes
2.2. Common Low Penetrance Risk Alleles for Breast Cancer
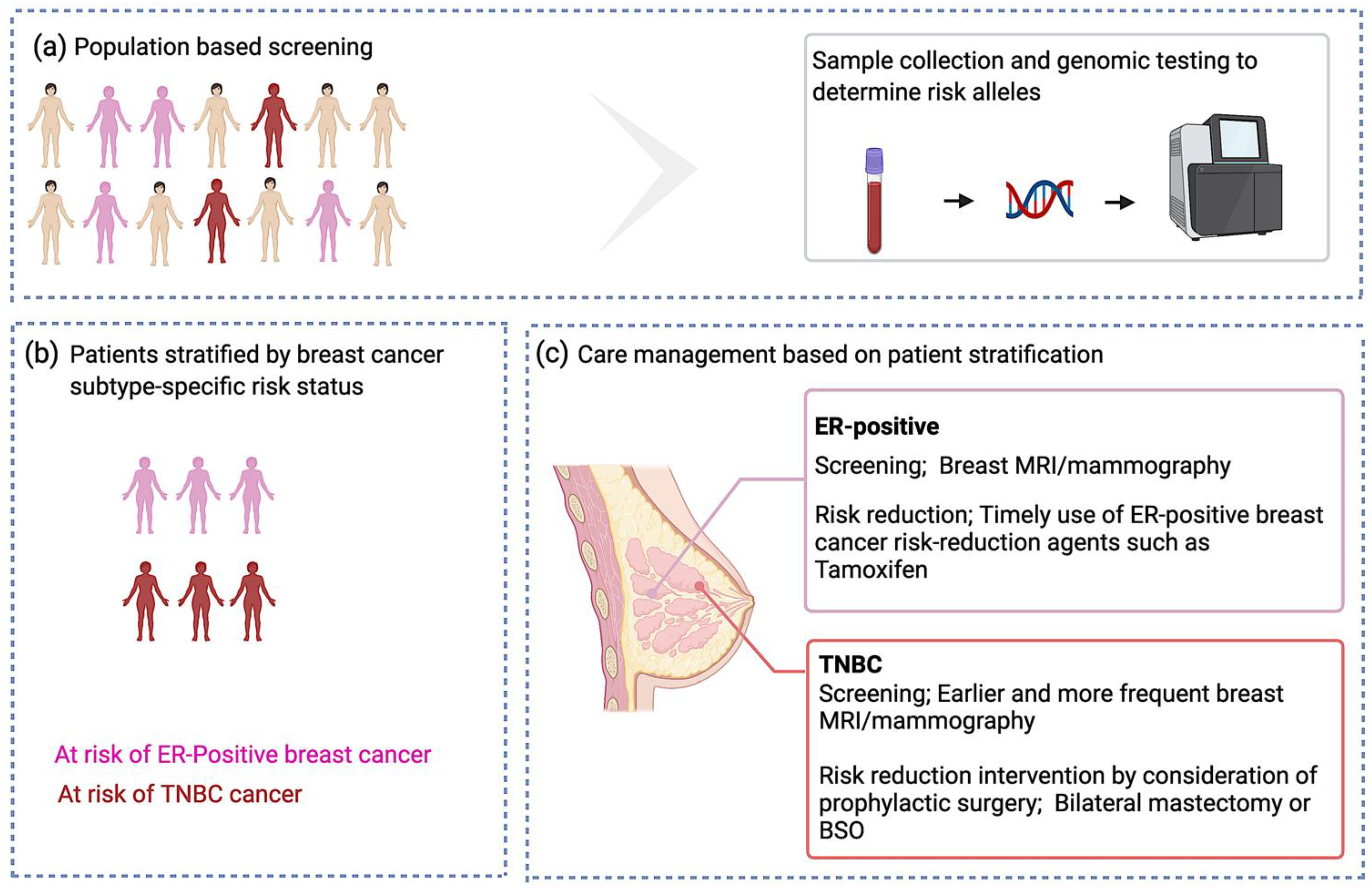
Clinical Utility of Identifying Common Risk Variants for Breast Cancer
2.3. Risk Associations with Breast Cancer Subtypes
2.3.1. Subtype Stratification for Coding Pathogenic Variants in High- and Moderate-Breast Cancer Susceptibility Genes
2.3.2. Subtype Stratification for Risk Variants Identified by Breast Cancer GWAS
3. The Functional Consequences of Coding and Non-Coding Risk Variants for Breast Cancer
3.1. Interpretation of Coding Risk Variants in High- to Moderate-Penetrance Susceptibility Genes
3.2. Functional Characterization of Common Low-Penetrance-Risk Alleles
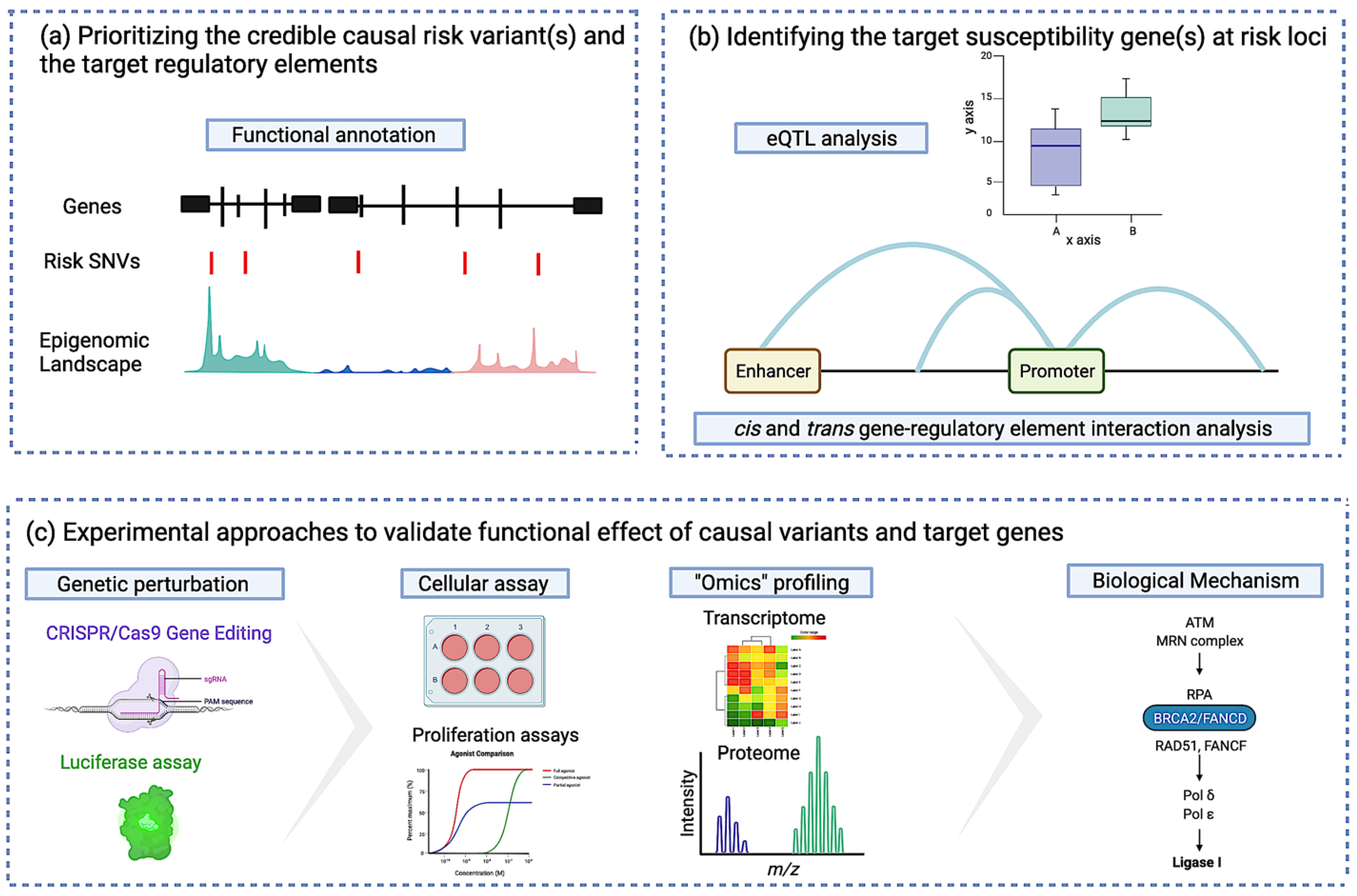
3.2.1. Identifying the Credible Causal Risk Variant and the Target Regulatory Element
3.2.2. Identifying the Target Susceptibility Gene(s) at Risk Loci
3.2.3. Identifying Susceptibility Gene-Regulatory Interactions at Risk Loci
3.2.4. Experimental Approaches to Validate the Functional Effect of Causal Variants and Target Genes
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Cancer Facts & Figures 2021. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2021.html (accessed on 6 July 2021).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Apostolou, P.; Fostira, F. Hereditary breast cancer: The era of new susceptibility genes. Biomed. Res. Int. 2013, 2013, 747318. [Google Scholar] [CrossRef] [Green Version]
- Shiovitz, S.; Korde, L.A. Genetics of breast cancer: A topic in evolution. Ann. Oncol. 2015, 26, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Mavaddat, N.; Peock, S.; Frost, D.; Ellis, S.; Platte, R.; Fineberg, E.; Evans, D.G.; Izatt, L.; Eeles, R.A.; Adlard, J.; et al. EMBRACE Cancer risks for BRCA1 and BRCA2 mutation carriers: Results from prospective analysis of EMBRACE. J. Natl. Cancer Inst. 2013, 105, 812–822. [Google Scholar] [CrossRef] [Green Version]
- Easton, D.F.; Ford, D.; Bishop, D.T. Breast and ovarian cancer incidence in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Am. J. Hum. Genet. 1995, 56, 265–271. [Google Scholar] [PubMed]
- Silvestri, V.; Leslie, G.; Barnes, D.R.; CIMBA Group; Agnarsson, B.A.; Aittomäki, K.; Alducci, E.; Andrulis, I.L.; Barkardottir, R.B.; Barroso, A.; et al. Characterization of the cancer spectrum in men with germline BRCA1 and BRCA2 pathogenic variants: Results from the consortium of investigators of modifiers of BRCA1/2 (CIMBA). JAMA Oncol. 2020, 6, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E. The cancer connection: BRCA1 and BRCA2 tumor suppression in mice and humans. Oncogene 2002, 21, 8994–9007. [Google Scholar] [CrossRef] [Green Version]
- Savage, K.I.; Harkin, D.P. BRCA1, a “complex” protein involved in the maintenance of genomic stability. FEBS J. 2015, 282, 630–646. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Sadeghi, F.; Asgari, M.; Matloubi, M.; Ranjbar, M.; Karkhaneh Yousefi, N.; Azari, T.; Zaki-Dizaji, M. Molecular contribution of BRCA1 and BRCA2 to genome instability in breast cancer patients: Review of radiosensitivity assays. Biol. Proced. Online 2020, 22, 23. [Google Scholar] [CrossRef]
- Lee, J.-K.; Choi, Y.-L.; Kwon, M.; Park, P.J. Mechanisms and Consequences of Cancer Genome Instability: Lessons from Genome Sequencing Studies. Annu. Rev. Pathol. 2016, 11, 283–312. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-Deegan, R.; Coffin, A.; et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752. [Google Scholar] [CrossRef] [Green Version]
- Rebbeck, T.R.; Friebel, T.M.; Friedman, E.; Hamann, U.; Huo, D.; Kwong, A.; Olah, E.; Olopade, O.I.; Solano, A.R.; Teo, S.-H.; et al. Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum. Mutat. 2018, 39, 593–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratton, M.R.; Rahman, N. The emerging landscape of breast cancer susceptibility. Nat. Genet. 2008, 40, 17–22. [Google Scholar] [CrossRef]
- Rahman, N.; Seal, S.; Thompson, D.; Kelly, P.; Renwick, A.; Elliott, A.; Reid, S.; Spanova, K.; Barfoot, R.; Chagtai, T.; et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 2007, 39, 165–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, M.R.; Low, C.E.; Chong, S.T.; Chan, S.H.; Ishak, N.D.B.; Courtney, E.; Kolinjivadi, A.M.; Rodrigue, A.; Masson, J.-Y.; Ngeow, J. Missense PALB2 germline variant disrupts nuclear localization of PALB2 in a patient with breast cancer. Fam Cancer 2020, 19, 123–131. [Google Scholar] [CrossRef]
- Decker, B.; Allen, J.; Luccarini, C.; Pooley, K.A.; Shah, M.; Bolla, M.K.; Wang, Q.; Ahmed, S.; Baynes, C.; Conroy, D.M.; et al. Rare, protein-truncating variants in ATM, CHEK2 and PALB2, but not XRCC2, are associated with increased breast cancer risks. J. Med. Genet. 2017, 54, 732–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renwick, A.; Thompson, D.; Seal, S.; Kelly, P.; Chagtai, T.; Ahmed, M.; North, B.; Jayatilake, H.; Barfoot, R.; Spanova, K.; et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 873–875. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Wokołorczyk, D.; Jakubowska, A.; Huzarski, T.; Byrski, T.; Gronwald, J.; Masojć, B.; Deebniak, T.; Górski, B.; Blecharz, P.; et al. Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J. Clin. Oncol. 2011, 29, 3747–3752. [Google Scholar] [CrossRef]
- Pharoah, P.D.; Guilford, P.; Caldas, C. International Gastric Cancer Linkage Consortium Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001, 121, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Weinstein, J.N.; Aladjem, M.I.; Kohn, K.W. Chk2 molecular interaction map and rationale for Chk2 inhibitors. Clin. Cancer Res. 2006, 12, 2657–2661. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.M.; Li, L.S.; Laquet, C.; Penault-Llorca, F.; Uhrhammer, N.; Xie, X.M.; Bignon, Y.J. Germline mutations of the E-cadherin gene in families with inherited invasive lobular breast carcinoma but no diffuse gastric cancer. Cancer 2011, 117, 3112–3117. [Google Scholar] [CrossRef]
- Masciari, S.; Larsson, N.; Senz, J.; Boyd, N.; Kaurah, P.; Kandel, M.J.; Harris, L.N.; Pinheiro, H.C.; Troussard, A.; Miron, P.; et al. Germline E-cadherin mutations in familial lobular breast cancer. J. Med. Genet. 2007, 44, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [Green Version]
- Sidransky, D.; Tokino, T.; Helzlsouer, K.; Zehnbauer, B.; Rausch, G.; Shelton, B.; Prestigiacomo, L.; Vogelstein, B.; Davidson, N. Inherited p53 gene mutations in breast cancer. Cancer Res. 1992, 52, 2984–2986. [Google Scholar] [PubMed]
- Pradella, L.M.; Evangelisti, C.; Ligorio, C.; Ceccarelli, C.; Neri, I.; Zuntini, R.; Amato, L.B.; Ferrari, S.; Martelli, A.M.; Gasparre, G.; et al. A novel deleterious PTEN mutation in a patient with early-onset bilateral breast cancer. BMC Cancer 2014, 14, 70. [Google Scholar] [CrossRef] [Green Version]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacocke, M.; et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Moyer, C.L.; Ivanovich, J.; Gillespie, J.L.; Doberstein, R.; Radke, M.R.; Richardson, M.E.; Kaufmann, S.H.; Swisher, E.M.; Goodfellow, P.J. Rare BRIP1 missense alleles confer risk for ovarian and breast cancer. Cancer Res. 2020, 80, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Singh, T.R.; Nassar, N.; Zhang, F.; Freund, M.; Hanenberg, H.; Meetei, A.R.; Andreassen, P.R. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene 2014, 33, 4803–4812. [Google Scholar] [CrossRef] [Green Version]
- Uzunoglu, H.; Korak, T.; Ergul, E.; Uren, N.; Sazci, A.; Utkan, N.Z.; Kargi, E.; Triyaki, Ç.; Yirmibesoglu, O. Association of the nibrin gene (NBN) variants with breast cancer. Biomed. Rep. 2016, 4, 369–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, W.; Hearle, N.; Shah, B.; Murday, V.; Hodgson, S.V.; Lucassen, A.; Eccles, D.; Talbot, I.; Neale, K.; Lim, A.G.; et al. Further observations on LKB1/STK11 status and cancer risk in Peutz-Jeghers syndrome. Br. J. Cancer 2003, 89, 308–313. [Google Scholar] [CrossRef]
- Banerjee, T.; Brosh, R.M. RECQL: A new breast cancer susceptibility gene. Cell Cycle 2015, 14, 3540–3543. [Google Scholar] [CrossRef] [Green Version]
- Kraus, C.; Hoyer, J.; Vasileiou, G.; Wunderle, M.; Lux, M.P.; Fasching, P.A.; Krumbiegel, M.; Uebe, S.; Reuter, M.; Beckmann, M.W.; et al. Gene panel sequencing in familial breast/ovarian cancer patients identifies multiple novel mutations also in genes others than BRCA1/2. Int. J. Cancer 2017, 140, 95–102. [Google Scholar] [CrossRef]
- Desmond, A.; Kurian, A.W.; Gabree, M.; Mills, M.A.; Anderson, M.J.; Kobayashi, Y.; Horick, N.; Yang, S.; Shannon, K.M.; Tung, N.; et al. Clinical actionability of multigene panel testing for hereditary breast and ovarian cancer risk assessment. JAMA Oncol. 2015, 1, 943–951. [Google Scholar] [CrossRef]
- Daly, M.B.; Pilarski, R.; Yurgelun, M.B.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Garber, J.E.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020. J. Natl. Compr. Cancer Netw. 2020, 18, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Satagopan, J.M.; Offit, K.; Foulkes, W.; Robson, M.E.; Wacholder, S.; Eng, C.M.; Karp, S.E.; Begg, C.B. The lifetime risks of breast cancer in Ashkenazi Jewish carriers of BRCA1 and BRCA2 mutations. Cancer Epidemiol. Biomark. Prev. 2001, 10, 467–473. [Google Scholar]
- Scott, S.P.; Bendix, R.; Chen, P.; Clark, R.; Dork, T.; Lavin, M.F. Missense mutations but not allelic variants alter the function of ATM by dominant interference in patients with breast cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 925–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA 2015, 313, 1347–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, D.; Easton, D. Breast Cancer Linkage Consortium Variation in cancer risks, by mutation position, in BRCA2 mutation carriers. Am. J. Hum. Genet. 2001, 68, 410–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J. Clin. Oncol. 2015, 33, 304–311. [Google Scholar] [CrossRef]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.-R.; Winer, E.P.; Garber, J.E. Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churpek, J.E.; Walsh, T.; Zheng, Y.; Moton, Z.; Thornton, A.M.; Lee, M.K.; Casadei, S.; Watts, A.; Neistadt, B.; Churpek, M.M.; et al. Inherited predisposition to breast cancer among African American women. Breast Cancer Res. Treat. 2015, 149, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Breast Cancer Association Consortium; Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; et al. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef]
- Antoniou, A.C.; Easton, D.F. Models of genetic susceptibility to breast cancer. Oncogene 2006, 25, 5898–5905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavaddat, N.; Antoniou, A.C.; Easton, D.F.; Garcia-Closas, M. Genetic susceptibility to breast cancer. Mol. Oncol. 2010, 4, 174–191. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.; Dunning, A.M.; Garcia-Closas, M.; Balasubramanian, S.; Reed, M.W.R.; Pooley, K.A.; Scollen, S.; Baynes, C.; Ponder, B.A.J.; Chanock, S.; et al. Breast Cancer Association Consortium A common coding variant in CASP8 is associated with breast cancer risk. Nat. Genet. 2007, 39, 352–358. [Google Scholar] [CrossRef]
- Neuman, R.J.; Sung, Y.J. Multistage analysis strategies for genome-wide association studies: Summary of group 3 contributions to Genetic Analysis Workshop 16. Genet. Epidemiol. 2009, 33 (Suppl. S1), S19–S23. [Google Scholar] [CrossRef] [Green Version]
- Easton, D.F.; Pooley, K.A.; Dunning, A.M.; Pharoah, P.D.P.; Thompson, D.; Ballinger, D.G.; Struewing, J.P.; Morrison, J.; Field, H.; Luben, R.; et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007, 447, 1087–1093. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Thomas, G.; Ghoussaini, M.; Healey, C.S.; Humphreys, M.K.; Platte, R.; Morrison, J.; Maranian, M.; Pooley, K.A.; Luben, R.; et al. Newly discovered breast cancer susceptibility loci on 3p24 and 17q23.2. Nat. Genet. 2009, 41, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Long, J.; Gao, Y.-T.; Li, C.; Zheng, Y.; Xiang, Y.-B.; Wen, W.; Levy, S.; Deming, S.L.; Haines, J.L.; et al. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat. Genet. 2009, 41, 324–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacey, S.N.; Manolescu, A.; Sulem, P.; Thorlacius, S.; Gudjonsson, S.A.; Jonsson, G.F.; Jakobsdottir, M.; Bergthorsson, J.T.; Gudmundsson, J.; Aben, K.K.; et al. Common variants on chromosome 5p12 confer susceptibility to estrogen receptor-positive breast cancer. Nat. Genet. 2008, 40, 703–706. [Google Scholar] [CrossRef]
- Stacey, S.N.; Manolescu, A.; Sulem, P.; Rafnar, T.; Gudmundsson, J.; Gudjonsson, S.A.; Masson, G.; Jakobsdottir, M.; Thorlacius, S.; Helgason, A.; et al. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat. Genet. 2007, 39, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K.; et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013, 45, 353–361.e1. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Beesley, J.; Lindstrom, S.; Canisius, S.; Dennis, J.; Lush, M.J.; Maranian, M.J.; Bolla, M.K.; Wang, Q.; Shah, M.; et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 2015, 47, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Michailidou, K.; Lindström, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemaçon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavaddat, N.; Michailidou, K.; Dennis, J.; Lush, M.; Fachal, L.; Lee, A.; Tyrer, J.P.; Chen, T.-H.; Wang, Q.; Bolla, M.K.; et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am. J. Hum. Genet. 2019, 104, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.P.; Beesley, J.; Amin Al Olama, A.; Michailidou, K.; Tyrer, J.; Kote-Jarai, Z.; Lawrenson, K.; Lindstrom, S.; Ramus, S.J.; Thompson, D.J.; et al. Genome-Wide Meta-Analyses of Breast, Ovarian, and Prostate Cancer Association Studies Identify Multiple New Susceptibility Loci Shared by at Least Two Cancer Types. Cancer Discov. 2016, 6, 1052–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bomba, L.; Walter, K.; Soranzo, N. The impact of rare and low-frequency genetic variants in common disease. Genome Biol. 2017, 18, 77. [Google Scholar] [CrossRef]
- Pharoah, P.D.P.; Antoniou, A.C.; Easton, D.F.; Ponder, B.A.J. Polygenes, risk prediction, and targeted prevention of breast cancer. N. Engl. J. Med. 2008, 358, 2796–2803. [Google Scholar] [CrossRef]
- Mavaddat, N.; Pharoah, P.D.; Michailidou, K.; Tyrer, J.; Brook, M.N.; Bolla, M.K.; Wang, Q.; Dennis, J.; Dunning, A.M.; Shah, M.; et al. Prediction of breast cancer risk based on profiling with common genetic variants. J. Natl. Cancer Inst. 2015, 107, djv036. [Google Scholar] [CrossRef]
- Barnes, D.R.; Rookus, M.A.; McGuffog, L.; Leslie, G.; Mooij, T.M.; Dennis, J.; Mavaddat, N.; Adlard, J.; Ahmed, M.; Aittomäki, K.; et al. Consortium of Investigators of Modifiers of BRCA and BRCA2 Polygenic risk scores and breast and epithelial ovarian cancer risks for carriers of BRCA1 and BRCA2 pathogenic variants. Genet. Med. 2020, 22, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.C.; Pharoah, P.P.D.; Smith, P.; Easton, D.F. The BOADICEA model of genetic susceptibility to breast and ovarian cancer. Br. J. Cancer 2004, 91, 1580–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.; Mavaddat, N.; Wilcox, A.N.; Cunningham, A.P.; Carver, T.; Hartley, S.; Babb de Villiers, C.; Izquierdo, A.; Simard, J.; Schmidt, M.K.; et al. BOADICEA: A comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet. Med. 2019, 21, 1708–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakeman, I.M.M.; Rodríguez-Girondo, M.; Lee, A.; Ruiter, R.; Stricker, B.H.; Wijnant, S.R.A.; Kavousi, M.; Antoniou, A.C.; Schmidt, M.K.; Uitterlinden, A.G.; et al. Validation of the BOADICEA model and a 313-variant polygenic risk score for breast cancer risk prediction in a Dutch prospective cohort. Genet. Med. 2020, 22, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Fang, H.; ULTRA-DD Consortium; De Wolf, H.; Knezevic, B.; Burnham, K.L.; Osgood, J.; Sanniti, A.; Lledó Lara, A.; Kasela, S.; De Cesco, S.; et al. A genetics-led approach defines the drug target landscape of 30 immune-related traits. Nat. Genet. 2019, 51, 1082–1091. [Google Scholar] [CrossRef]
- Bahcall, O. Functional annotation of susceptibility loci identified by COGS. Nat. Genet. 2013. [Google Scholar] [CrossRef]
- Johnson, K.S.; Conant, E.F.; Soo, M.S. Molecular subtypes of breast cancer: A review for breast radiologists. J. Breast Imaging 2021, 3, 12–24. [Google Scholar] [CrossRef]
- Barton, V.N.; D’Amato, N.C.; Gordon, M.A.; Christenson, J.L.; Elias, A.; Richer, J.K. Androgen receptor biology in triple negative breast cancer: A case for classification as AR+ or quadruple negative disease. Horm. Cancer 2015, 6, 206–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buys, S.S.; Sandbach, J.F.; Gammon, A.; Patel, G.; Kidd, J.; Brown, K.L.; Sharma, L.; Saam, J.; Lancaster, J.; Daly, M.B. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 2017, 123, 1721–1730. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.R.F.; Bateman, A.C.; Hanson, H.; An, Q.; Evans, G.; Rahman, N.; Jones, J.L.; Eccles, D.M. A novel HER2-positive breast cancer phenotype arising from germline TP53 mutations. J. Med. Genet. 2010, 47, 771–774. [Google Scholar] [CrossRef] [Green Version]
- Melhem-Bertrandt, A.; Bojadzieva, J.; Ready, K.J.; Obeid, E.; Liu, D.D.; Gutierrez-Barrera, A.M.; Litton, J.K.; Olopade, O.I.; Hortobagyi, G.N.; Strong, L.C.; et al. Early onset HER2-positive breast cancer is associated with germline TP53 mutations. Cancer 2012, 118, 908–913. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Closas, M.; Couch, F.J.; Lindstrom, S.; Michailidou, K.; Schmidt, M.K.; Brook, M.N.; Orr, N.; Rhie, S.K.; Riboli, E.; Feigelson, H.S.; et al. Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat. Genet. 2013, 45, 392–398.e1. [Google Scholar] [CrossRef] [Green Version]
- Purrington, K.S.; Slager, S.; Eccles, D.; Yannoukakos, D.; Fasching, P.A.; Miron, P.; Carpenter, J.; Chang-Claude, J.; Martin, N.G.; Montgomery, G.W.; et al. Genome-wide association study identifies 25 known breast cancer susceptibility loci as risk factors for triple-negative breast cancer. Carcinogenesis 2014, 35, 1012–1019. [Google Scholar] [CrossRef] [Green Version]
- Milne, R.L.; Kuchenbaecker, K.B.; Michailidou, K.; Beesley, J.; Kar, S.; Lindström, S.; Hui, S.; Lemaçon, A.; Soucy, P.; Dennis, J.; et al. Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat. Genet. 2017, 49, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Fachal, L.; Aschard, H.; Beesley, J.; Barnes, D.R.; Allen, J.; Kar, S.; Pooley, K.A.; Dennis, J.; Michailidou, K.; Turman, C.; et al. Fine-mapping of 150 breast cancer risk regions identifies 191 likely target genes. Nat. Genet. 2020, 52, 56–73. [Google Scholar] [CrossRef]
- Holm, J.; Li, J.; Darabi, H.; Eklund, M.; Eriksson, M.; Humphreys, K.; Hall, P.; Czene, K. Associations of breast cancer risk prediction tools with tumor characteristics and metastasis. J. Clin. Oncol. 2016, 34, 251–258. [Google Scholar] [CrossRef]
- Li, J.; Holm, J.; Bergh, J.; Eriksson, M.; Darabi, H.; Lindström, L.S.; Törnberg, S.; Hall, P.; Czene, K. Breast cancer genetic risk profile is differentially associated with interval and screen-detected breast cancers. Ann. Oncol. 2015, 26, 517–522. [Google Scholar] [CrossRef]
- Wu, K.; Hinson, S.R.; Ohashi, A.; Farrugia, D.; Wendt, P.; Tavtigian, S.V.; Deffenbaugh, A.; Goldgar, D.; Couch, F.J. Functional evaluation and cancer risk assessment of BRCA2 unclassified variants. Cancer Res. 2005, 65, 417–426. [Google Scholar]
- Mesman, R.L.S.; Calléja, F.M.G.R.; Hendriks, G.; Morolli, B.; Misovic, B.; Devilee, P.; van Asperen, C.J.; Vrieling, H.; Vreeswijk, M.P.G. The functional impact of variants of uncertain significance in BRCA2. Genet. Med. 2019, 21, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Fortuno, C.; Lee, K.; Olivier, M.; Pesaran, T.; Mai, P.L.; de Andrade, K.C.; Attardi, L.D.; Crowley, S.; Evans, D.G.; Feng, B.-J.; et al. ClinGen TP53 Variant Curation Expert Panel Specifications of the ACMG/AMP variant interpretation guidelines for germline TP53 variants. Hum. Mutat. 2021, 42, 223–236. [Google Scholar] [CrossRef]
- Mester, J.L.; Ghosh, R.; Pesaran, T.; Huether, R.; Karam, R.; Hruska, K.S.; Costa, H.A.; Lachlan, K.; Ngeow, J.; Barnholtz-Sloan, J.; et al. Gene-specific criteria for PTEN variant curation: Recommendations from the ClinGen PTEN Expert Panel. Hum. Mutat. 2018, 39, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Krempely, K.; Roberts, M.E.; Anderson, M.J.; Carneiro, F.; Chao, E.; Dixon, K.; Figueiredo, J.; Ghosh, R.; Huntsman, D.; et al. Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum. Mutat. 2018, 39, 1553–1568. [Google Scholar] [CrossRef] [PubMed]
- Golubeva, V.A.; Nepomuceno, T.C.; Monteiro, A.N.A. Germline missense variants in BRCA1: New trends and challenges for clinical annotation. Cancers 2019, 11, 522. [Google Scholar] [CrossRef] [Green Version]
- Meulemans, L.; Mesman, R.L.S.; Caputo, S.M.; Krieger, S.; Guillaud-Bataille, M.; Caux-Moncoutier, V.; Léone, M.; Boutry-Kryza, N.; Sokolowska, J.; Révillion, F.; et al. Skipping nonsense to maintain function: The paradigm of BRCA2 exon 12. Cancer Res. 2020, 80, 1374–1386. [Google Scholar] [CrossRef]
- Mazoyer, S.; Dunning, A.M.; Serova, O.; Dearden, J.; Puget, N.; Healey, C.S.; Gayther, S.A.; Mangion, J.; Stratton, M.R.; Lynch, H.T.; et al. A polymorphic stop codon in BRCA2. Nat. Genet. 1996, 14, 253–254. [Google Scholar] [CrossRef] [PubMed]
- Meeks, H.D.; Song, H.; Michailidou, K.; Bolla, M.K.; Dennis, J.; Wang, Q.; Barrowdale, D.; Frost, D.; EMBRACE; McGuffog, L.; et al. BRCA2 polymorphic stop codon K3326X and the risk of breast, prostate, and ovarian cancers. J. Natl. Cancer Inst. 2016, 108, djv315. [Google Scholar] [CrossRef] [PubMed]
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 25 June 2021).
- Hansen, T.V.O.; Steffensen, A.Y.; Jønson, L.; Andersen, M.K.; Ejlertsen, B.; Nielsen, F.C. The silent mutation nucleotide 744 G--> A, Lys172Lys, in exon 6 of BRCA2 results in exon skipping. Breast Cancer Res. Treat. 2010, 119, 547–550. [Google Scholar] [CrossRef] [Green Version]
- Findlay, G.M.; Daza, R.M.; Martin, B.; Zhang, M.D.; Leith, A.P.; Gasperini, M.; Janizek, J.D.; Huang, X.; Starita, L.M.; Shendure, J. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018, 562, 217–222. [Google Scholar] [CrossRef]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [Green Version]
- Cooper, G.M.; Shendure, J. Needles in stacks of needles: Finding disease-causal variants in a wealth of genomic data. Nat. Rev. Genet. 2011, 12, 628–640. [Google Scholar] [CrossRef]
- Meyer, K.B.; O’Reilly, M.; Michailidou, K.; Carlebur, S.; Edwards, S.L.; French, J.D.; Prathalingham, R.; Dennis, J.; Bolla, M.K.; Wang, Q.; et al. Fine-scale mapping of the FGFR2 breast cancer risk locus: Putative functional variants differentially bind FOXA1 and E2F1. Am. J. Hum. Genet. 2013, 93, 1046–1060. [Google Scholar] [CrossRef] [Green Version]
- French, J.D.; Ghoussaini, M.; Edwards, S.L.; Meyer, K.B.; Michailidou, K.; Ahmed, S.; Khan, S.; Maranian, M.J.; O’Reilly, M.; Hillman, K.M.; et al. Functional variants at the 11q13 risk locus for breast cancer regulate cyclin D1 expression through long-range enhancers. Am. J. Hum. Genet. 2013, 92, 489–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoussaini, M.; Edwards, S.L.; Michailidou, K.; Nord, S.; Cowper-Sal Lari, R.; Desai, K.; Kar, S.; Hillman, K.M.; Kaufmann, S.; Glubb, D.M.; et al. Evidence that breast cancer risk at the 2q35 locus is mediated through IGFBP5 regulation. Nat. Commun. 2014, 4, 4999. [Google Scholar] [CrossRef] [Green Version]
- Lawrenson, K.; Kar, S.; McCue, K.; Kuchenbaeker, K.; Michailidou, K.; Tyrer, J.; Beesley, J.; Ramus, S.J.; Li, Q.; Delgado, M.K.; et al. Functional mechanisms underlying pleiotropic risk alleles at the 19p13.1 breast-ovarian cancer susceptibility locus. Nat. Commun. 2016, 7, 12675. [Google Scholar] [CrossRef] [Green Version]
- Gasperskaja, E.; Kučinskas, V. The most common technologies and tools for functional genome analysis. Acta Med. Litu. 2017, 24, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Udler, M.S.; Tyrer, J.; Easton, D.F. Evaluating the power to discriminate between highly correlated SNPs in genetic association studies. Genet. Epidemiol. 2010, 34, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Sur, I.; Taipale, J. The role of enhancers in cancer. Nat. Rev. Cancer 2016, 16, 483–493. [Google Scholar] [CrossRef]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FANTOM Consortium and the RIKEN PMI and CLST (DGT); Forrest, A.R.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; de Hoon, M.J.L.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Walavalkar, N.M.; Dozmorov, M.G.; Rich, S.S.; Civelek, M.; Guertin, M.J. Identification of breast cancer associated variants that modulate transcription factor binding. PLoS Genet. 2017, 13, e1006761. [Google Scholar] [CrossRef]
- Cookson, W.; Liang, L.; Abecasis, G.; Moffatt, M.; Lathrop, M. Mapping complex disease traits with global gene expression. Nat. Rev. Genet. 2009, 10, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Gamazon, E.R.; Wheeler, H.E.; Shah, K.P.; Mozaffari, S.V.; Aquino-Michaels, K.; Carroll, R.J.; Eyler, A.E.; Denny, J.C.; GTEx Consortium; Nicolae, D.L.; et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015, 47, 1091–1098. [Google Scholar] [CrossRef] [Green Version]
- Gusev, A.; Ko, A.; Shi, H.; Bhatia, G.; Chung, W.; Penninx, B.W.J.H.; Jansen, R.; de Geus, E.J.C.; Boomsma, D.I.; Wright, F.A.; et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016, 48, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, M.A.; Gamazon, E.R.; Al-Ejeh, F.; Aittomäki, K.; Andrulis, I.L.; Anton-Culver, H.; Arason, A.; Arndt, V.; Aronson, K.J.; Arun, B.K.; et al. Genome-wide association and transcriptome studies identify target genes and risk loci for breast cancer. Nat. Commun. 2019, 10, 1741. [Google Scholar] [CrossRef] [Green Version]
- Neavin, D.; Nguyen, Q.; Daniszewski, M.S.; Liang, H.H.; Chiu, H.S.; Wee, Y.K.; Senabouth, A.; Lukowski, S.W.; Crombie, D.E.; Lidgerwood, G.E.; et al. Single cell eQTL analysis identifies cell type-specific genetic control of gene expression in fibroblasts and reprogrammed induced pluripotent stem cells. Genome Biol. 2021, 22, 76. [Google Scholar] [CrossRef]
- Sun, Y.; Ye, C.; Guo, X.; Wen, W.; Long, J.; Gao, Y.-T.; Shu, X.O.; Zheng, W.; Cai, Q. Evaluation of potential regulatory function of breast cancer risk locus at 6q25.1. Carcinogenesis 2016, 37, 163–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Zhang, Y.; Zheng, W.; Michailidou, K.; Ghoussaini, M.; Bolla, M.K.; Wang, Q.; Dennis, J.; Lush, M.; Milne, R.L.; et al. Fine-scale mapping of 8q24 locus identifies multiple independent risk variants for breast cancer. Int. J. Cancer 2016, 139, 1303–1317. [Google Scholar] [CrossRef] [Green Version]
- Fagny, M.; Platig, J.; Kuijjer, M.L.; Lin, X.; Quackenbush, J. Nongenic cancer-risk SNPs affect oncogenes, tumour-suppressor genes, and immune function. Br. J. Cancer 2020, 122, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitz, M.J.; Ay, F.; Heidmann, J.D.; Lerner, P.L.; Noble, W.S.; Steelman, B.N.; Hoffman, A.R. Genomic interaction profiles in breast cancer reveal altered chromatin architecture. PLoS ONE 2013, 8, e73974. [Google Scholar] [CrossRef] [Green Version]
- Nasser, J.; Bergman, D.T.; Fulco, C.P.; Guckelberger, P.; Doughty, B.R.; Patwardhan, T.A.; Jones, T.R.; Nguyen, T.H.; Ulirsch, J.C.; Lekschas, F.; et al. Genome-wide enhancer maps link risk variants to disease genes. Nature 2021, 593, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Chai, P.; Zhang, H.; Fan, X. Novel insights into chromosomal conformations in cancer. Mol. Cancer 2017, 16, 173. [Google Scholar] [CrossRef] [Green Version]
- Cai, M.; Kim, S.; Wang, K.; Farnham, P.J.; Coetzee, G.A.; Lu, W. 4C-seq revealed long-range interactions of a functional enhancer at the 8q24 prostate cancer risk locus. Sci. Rep. 2016, 6, 22462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomerantz, M.M.; Ahmadiyeh, N.; Jia, L.; Herman, P.; Verzi, M.P.; Doddapaneni, H.; Beckwith, C.A.; Chan, J.A.; Hills, A.; Davis, M.; et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat. Genet. 2009, 41, 882–884. [Google Scholar] [CrossRef] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Mumbach, M.R.; Rubin, A.J.; Flynn, R.A.; Dai, C.; Khavari, P.A.; Greenleaf, W.J.; Chang, H.Y. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 2016, 13, 919–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barutcu, A.R.; Hong, D.; Lajoie, B.R.; McCord, R.P.; van Wijnen, A.J.; Lian, J.B.; Stein, J.L.; Dekker, J.; Imbalzano, A.N.; Stein, G.S. RUNX1 contributes to higher-order chromatin organization and gene regulation in breast cancer cells. Biochim. Biophys. Acta 2016, 1859, 1389–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mourad, R.; Hsu, P.-Y.; Juan, L.; Shen, C.; Koneru, P.; Lin, H.; Liu, Y.; Nephew, K.; Huang, T.H.; Li, L. Estrogen induces global reorganization of chromatin structure in human breast cancer cells. PLoS ONE 2014, 9, e113354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodorou, V.; Stark, R.; Menon, S.; Carroll, J.S. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013, 23, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Hilton, I.B.; Gersbach, C.A. Enabling functional genomics with genome engineering. Genome Res. 2015, 25, 1442–1455. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Gasperini, M.; Hill, A.J.; McFaline-Figueroa, J.L.; Martin, B.; Kim, S.; Zhang, M.D.; Jackson, D.; Leith, A.; Schreiber, J.; Noble, W.S.; et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 2019, 176, 377–390.e19. [Google Scholar] [CrossRef] [Green Version]
- Inoue, F.; Ahituv, N. Decoding enhancers using massively parallel reporter assays. Genomics 2015, 106, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Bourges, C.; Groff, A.F.; Burren, O.S.; Gerhardinger, C.; Mattioli, K.; Hutchinson, A.; Hu, T.; Anand, T.; Epping, M.W.; Wallace, C.; et al. Resolving mechanisms of immune-mediated disease in primary CD4 T cells. EMBO Mol. Med. 2020, 12, e12112. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Zhang, Q.; Wu, J.; Liang, J.; Yu, S.; Wei, G.-H.; White, K.P.; Wang, X. Systematic identification of regulatory variants associated with cancer risk. Genome Biol. 2017, 18, 194. [Google Scholar] [CrossRef] [Green Version]
- Klein, J.C.; Keith, A.; Agarwal, V.; Durham, T.; Shendure, J. Functional characterization of enhancer evolution in the primate lineage. Genome Biol. 2018, 19, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Bennett, R.L.; Buchanan, A.; Pearlman, R.; Wiesner, G.L.; Guideline Development Group; American College of Medical Genetics and Genomics Professional Practice and Guidelines Committee and National Society of Genetic Counselors Practice Guidelines Committee. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet. Med. 2015, 17, 70–87. [Google Scholar] [CrossRef] [Green Version]
- Peshkin, B.N.; Isaacs, C.; Finch, C.; Kent, S.; Schwartz, M.D. Tamoxifen as chemoprevention in BRCA1 and BRCA2 mutation carriers with breast cancer: A pilot survey of physicians. J. Clin. Oncol. 2003, 21, 4322–4328. [Google Scholar] [CrossRef] [PubMed]
- Sestak, I. Preventative therapies for healthy women at high risk of breast cancer. Cancer Manag. Res. 2014, 6, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Turashvili, G.; Brogi, E. Tumor heterogeneity in breast cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Genomic Location | Odds Ratio * (95% CI) | Associated with Other Cancers # | Syndrome | Absolute Risk # |
|---|---|---|---|---|---|
| ATM | 11q22 | 2.1 (1.71–2.57) | Ovarian, Pancreatic | Ataxia-telangiectasia | 15–40% |
| BARD1 | 2q35 | 2.09 (1.35–3.23) | Insufficient data to define | ||
| BRCA1 | 17q21 | 10.57 (8.02–13.93) | Ovarian, Pancreatic, Prostate | >60% | |
| BRCA2 | 13q12 | 5.85 (4.85–7.06) | Ovarian, Pancreatic, Prostate, Melanoma | >60% | |
| CHEK2 | 22q12 | 2.54 (2.21–2.91) | Colon | 15–40% | |
| PALB2 | 16p12 | 5.02 | Ovarian, Pancreatic | Fanconi anemia | 41–60% |
| PTEN | 10q23 | 2.25 (0.85–6.00) | Thyroid, Colon, Endometrial | Cowden’s syndrome | 40–60% |
| RAD51C | 17q23 | 1.93 (1.20–3.11) | Ovarian | 15–40% | |
| RAD51D | 17q12 | 1.8 (1.11–2.93) | Ovarian | 15–40% | |
| TP53 | 17p13 | 3.06 (0.63–14.91) | Pancreatic, and cancers associated with Li-Fraumeni syndrome | Li-Fraumeni syndrome (expanded to heritable TP53-related cancer (hTP53rc) syndrome) | >60% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torabi Dalivandan, S.; Plummer, J.; Gayther, S.A. Risks and Function of Breast Cancer Susceptibility Alleles. Cancers 2021, 13, 3953. https://doi.org/10.3390/cancers13163953
Torabi Dalivandan S, Plummer J, Gayther SA. Risks and Function of Breast Cancer Susceptibility Alleles. Cancers. 2021; 13(16):3953. https://doi.org/10.3390/cancers13163953
Chicago/Turabian StyleTorabi Dalivandan, Saeideh, Jasmine Plummer, and Simon A. Gayther. 2021. "Risks and Function of Breast Cancer Susceptibility Alleles" Cancers 13, no. 16: 3953. https://doi.org/10.3390/cancers13163953
APA StyleTorabi Dalivandan, S., Plummer, J., & Gayther, S. A. (2021). Risks and Function of Breast Cancer Susceptibility Alleles. Cancers, 13(16), 3953. https://doi.org/10.3390/cancers13163953