
Context-Specific Efficacy of Apalutamide Therapy in Preclinical Models of Pten-Deficient Prostate Cancer

 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
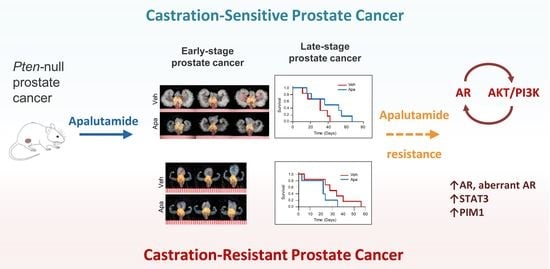
1. Introduction
2. Results
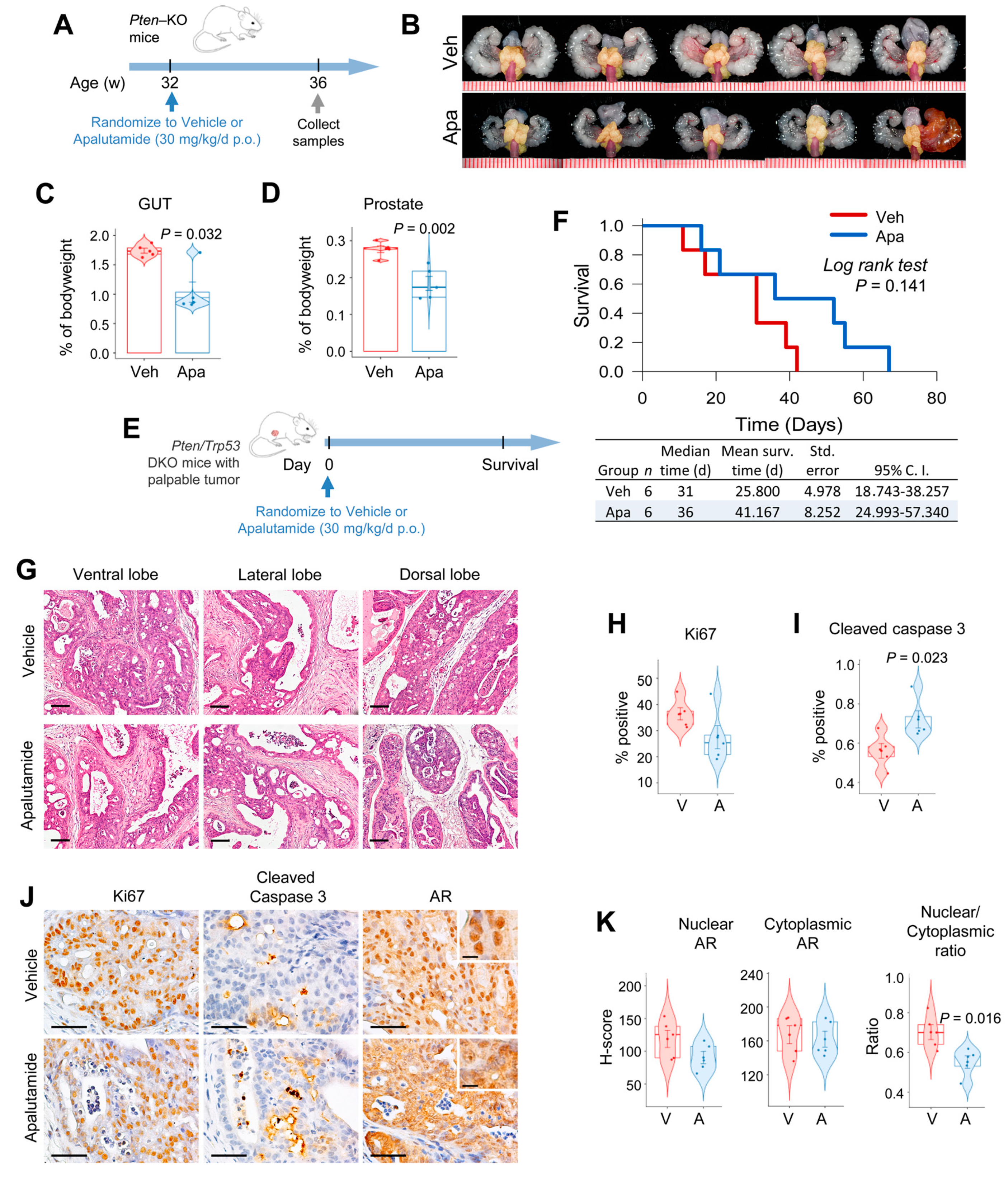
2.1. Apalutamide Shows Antitumor Efficacy in Mouse Castration-Naïve Pten-Deficient Prostate Cancer
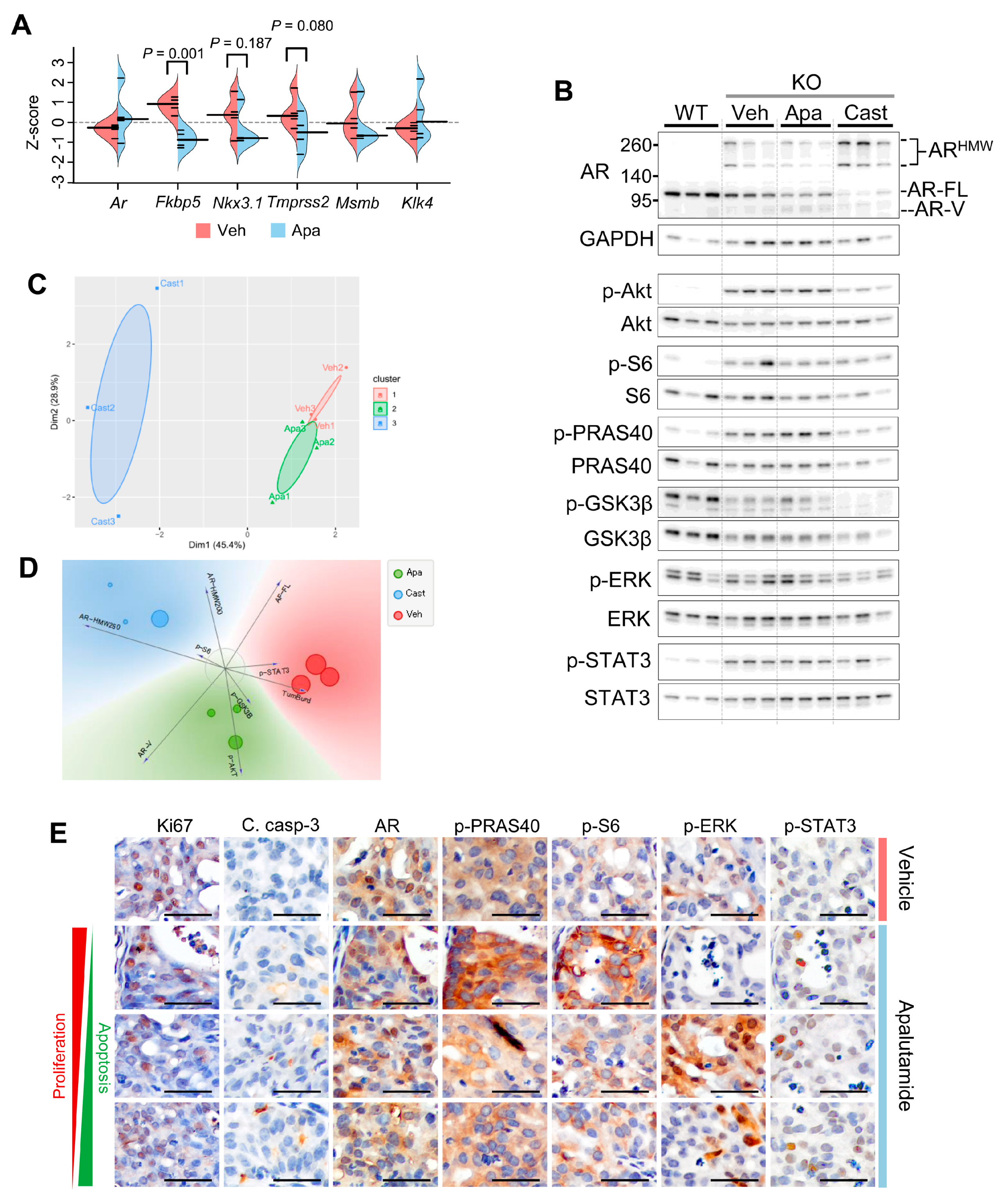
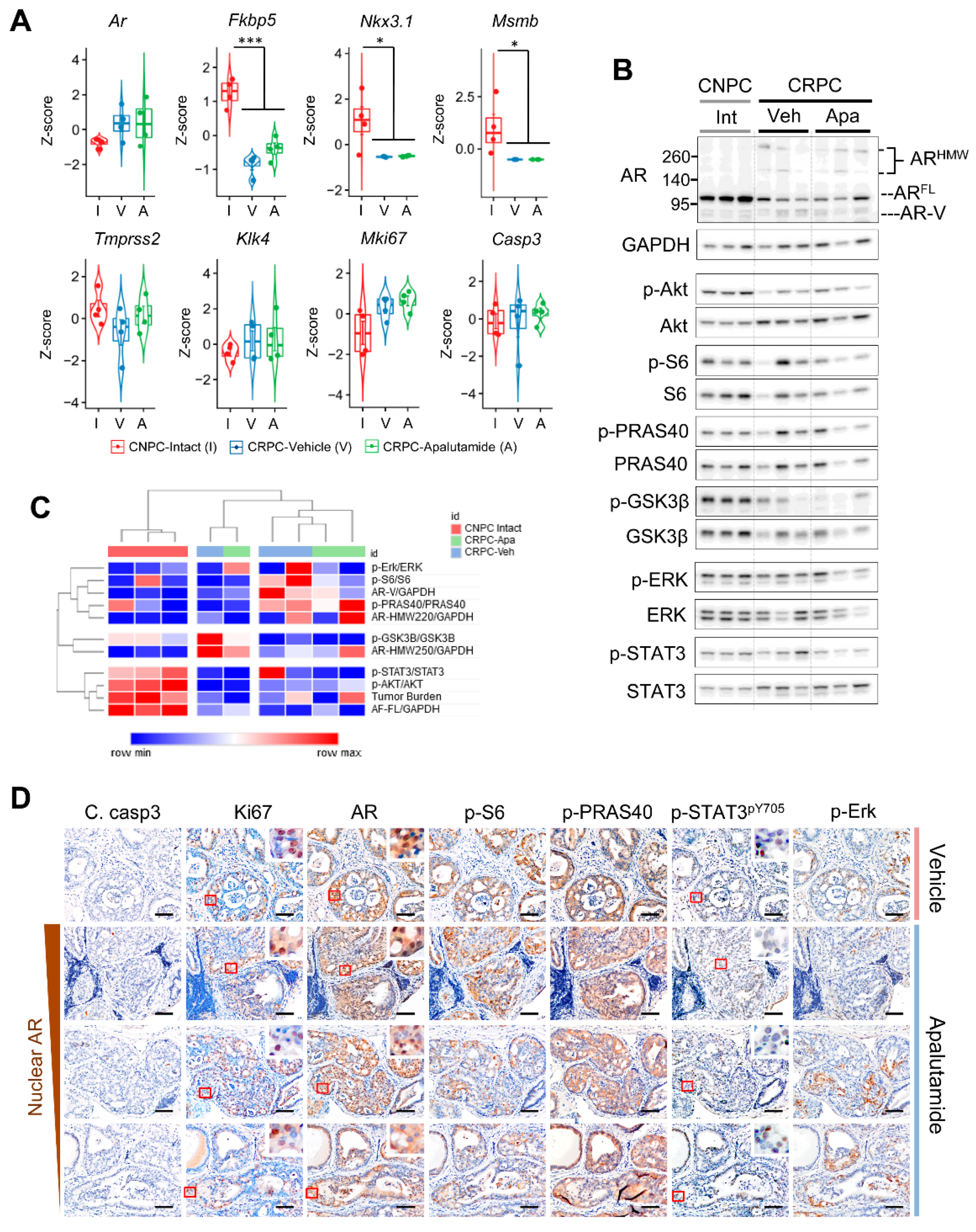
2.2. Molecular Characterization of Apalutamide in Pten-Deficient CNPC
2.3. Antitumor Activity of Apalutamide Monotherapy in Pten-Deficient CRPC
2.4. Molecular Characterization of Apalutamide Pten-Deficient CRPC
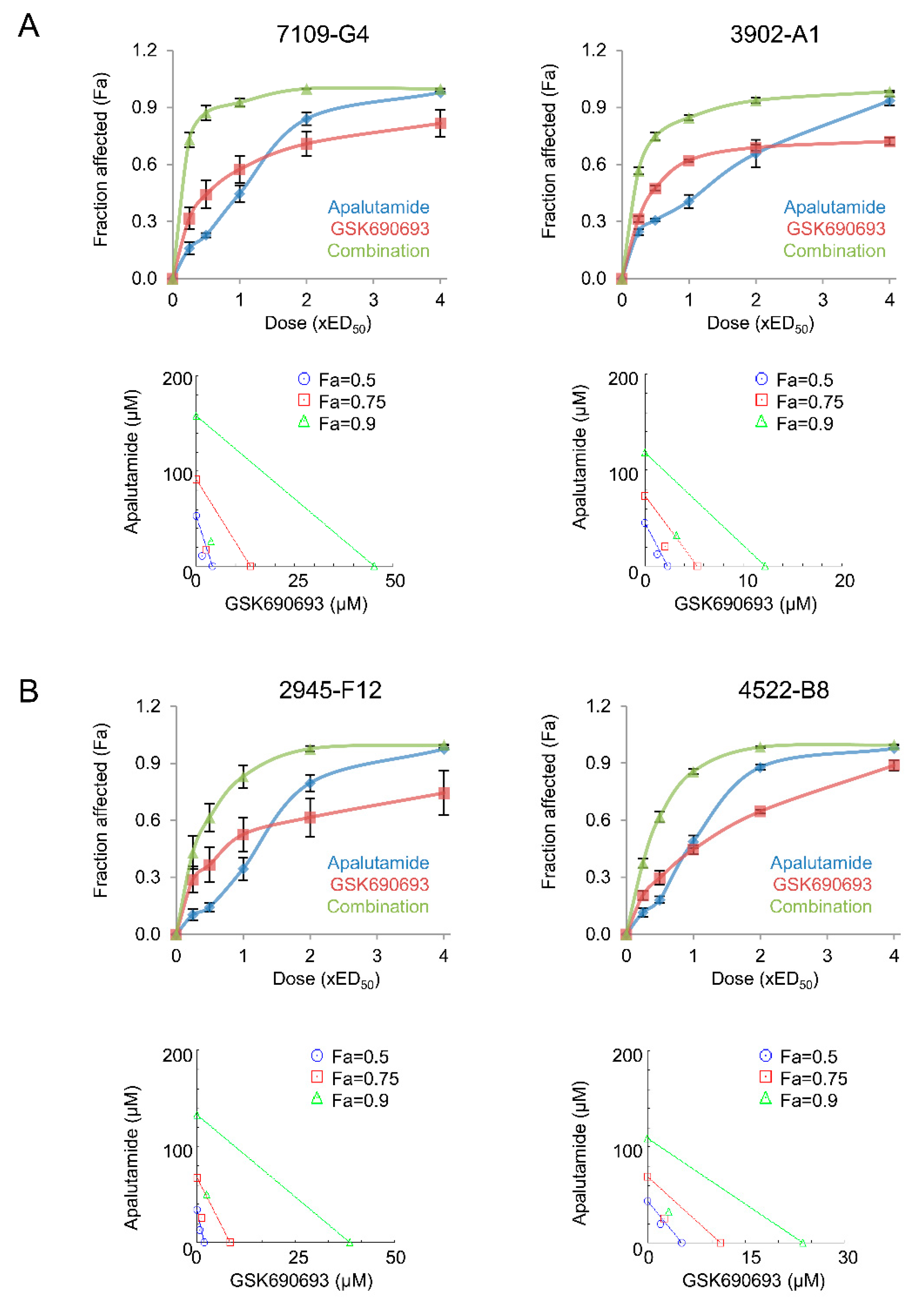
2.5. Apalutamide and AKT Inhibition Synergize to Suppress Prostate Cancer Cell Growth In Vitro
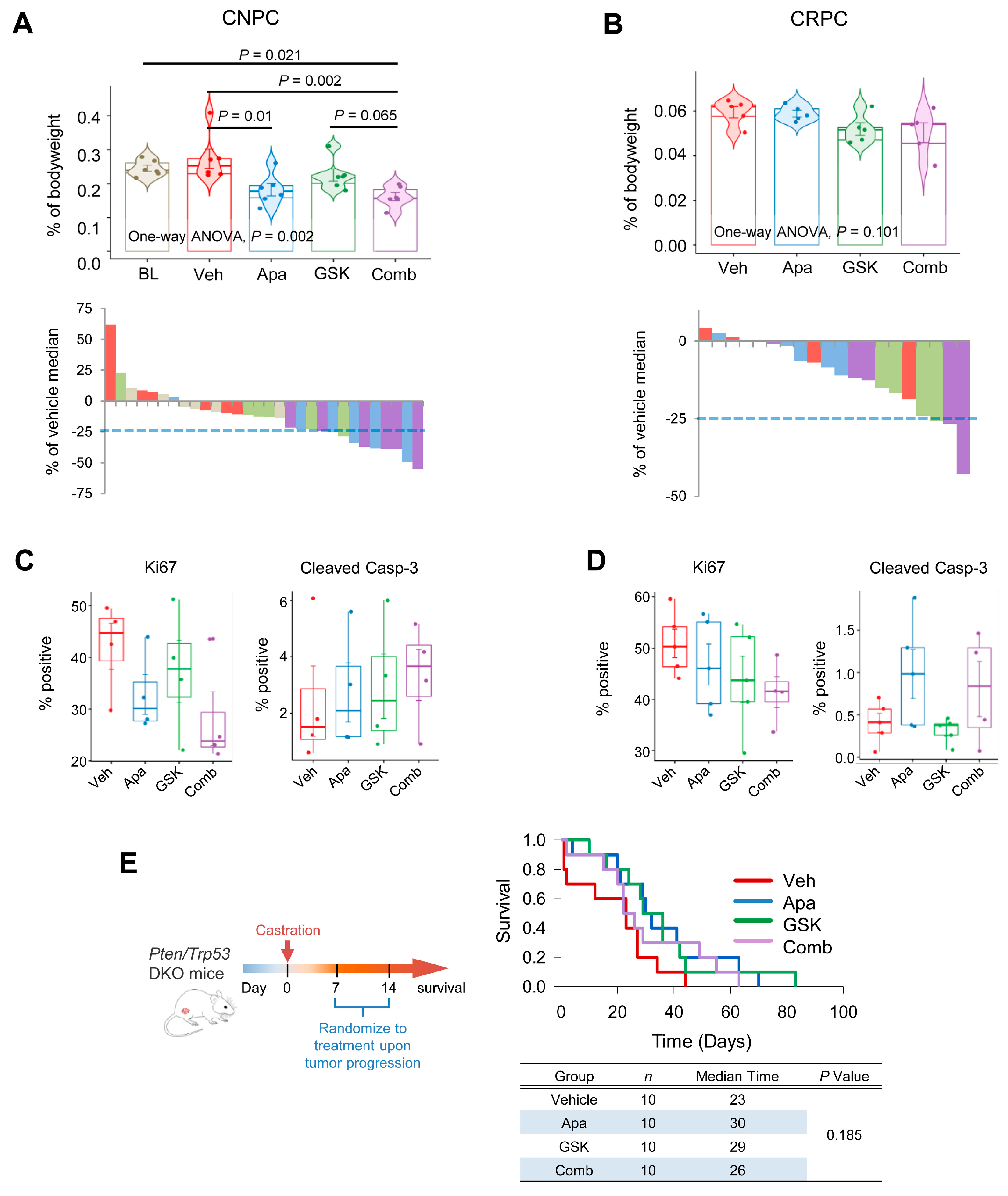
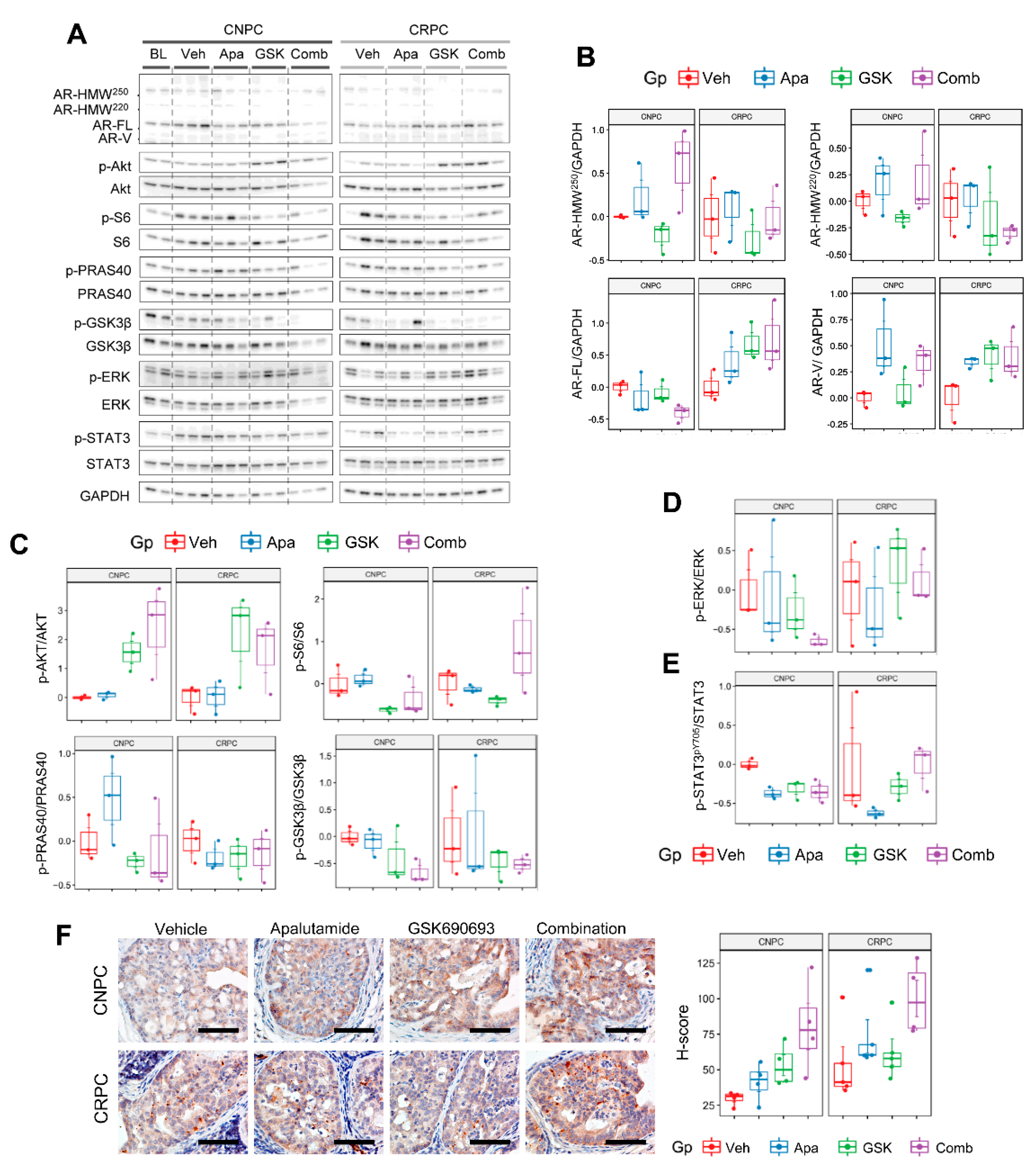
2.6. Combinatorial Effects of Apalutamide and AKT Inhibition In Vivo
3. Discussion
4. Materials and Methods
4.1. Treatment Compounds
4.2. Animals
4.3. Efficacy Studies
4.4. Histology and Immunohistochemistry (IHC)
4.5. Western Blot Analysis
4.6. RNA Extraction and qRT-PCR Analysis
4.7. In Vitro Studies
4.8. Plotting and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Joseph, J.D.; Lu, N.; Qian, J.; Sensintaffar, J.; Shao, G.; Brigham, D.; Moon, M.; Maneval, E.C.; Chen, I.; Darimont, B.; et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013, 3, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Fedor, H.L. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Eng. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Ferraldeschi, R.; Welti, J.; Luo, J.; Attard, G.; De Bono, J.S. Targeting the androgen receptor pathway in castration-resistant prostate cancer: Progresses and prospects. Oncogene 2015, 34, 1745–1757. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sircar, K.; Yoshimoto, M.; Monzon, F.A.; Koumakpayi, I.H.; Katz, R.L.; Khanna, A.; Alvarez, K.; Chen, G.; Darnel, A.D.; Aprikian, A.G.; et al. PTEN genomic deletion is associated with p-Akt and AR signalling in poorer outcome, hormone refractory prostate cancer. J. Pathol. 2009, 218, 505–513. [Google Scholar] [CrossRef]
- Ferraldeschi, R.; Rodrigues, D.N.; Riisnaes, R.; Miranda, S.; Figueiredo, I.; Rescigno, P.; Ravi, P.; Pezaro, C.; Omlin, A.; Lorente, D.; et al. PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur. Urol. 2015, 67, 795–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krohn, A.; Diedler, T.; Burkhardt, L.; Mayer, P.-S.; De Silva, C.; Meyer-Kornblum, M.; Kötschau, D.; Tennstedt, P.; Huang, J.; Gerhauser, C.; et al. Genomic deletion of PTEN is associated with tumor progression and early PSA recurrence in ERG fusion-positive and fusion-negative prostate cancer. Am. J. Pathol. 2012, 181, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Nan, B.; Snabboon, T.; Unni, E.; Yuan, X.J.; Whang, Y.; Marcelli, M. The PTEN tumor suppressor is a negative modulator of AR transcriptional activity. J. Mol. Endocrinol. 2003, 31, 169–183. [Google Scholar] [CrossRef] [Green Version]
- Watson, P.A.; Chen, Y.F.; Balbas, M.D.; Wongvipat, J.; Socci, N.D.; Viale, A.; Kim, K.; Sawyers, C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. USA 2010, 107, 16759–16765. [Google Scholar] [CrossRef] [Green Version]
- Liang, M.; Adisetiyo, H.; Liu, X.; Liu, R.; Gill, P.; Roy-Burman, P.; Jones, J.O.; Mulholland, D.J. Identification of androgen receptor splice variants in the pten deficient murine prostate cancer model. PLoS ONE 2015, 10, e0131232. [Google Scholar] [CrossRef]
- De Velasco, M.A.; Kura, Y.; Sakai, K.; Hatanaka, Y.; Davies, B.R.; Campbell, H.; Klein, S.; Kim, Y.; MacLeod, A.R.; Sugimoto, K.; et al. Targeting castration-resistant prostate cancer with androgen receptor antisense oligonucleotide therapy. JCI Insight 2019, 4, e122688. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choucair, K.; Ejdelman, J.; Brimo, F.; Aprikian, A.; Chevalier, S.; Lapointe, J. PTEN genomic deletion predicts prostate cancer recurrence and is associated with low AR expression and transcriptional activity. BMC Cancer 2012, 12, 543. [Google Scholar] [CrossRef] [Green Version]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Velasco, M.A.; Tanaka, M.; Yamamoto, Y.; Hatanaka, Y.; Koike, H.; Nishio, K.; Yoshikawa, K.; Uemura, H. Androgen deprivation induces phenotypic plasticity and promotes resistance to molecular targeted therapy in a PTEN-deficient mouse model of prostate cancer. Carcinogenesis 2014, 35, 2142–2153. [Google Scholar] [CrossRef] [Green Version]
- De Velasco, M.A.; Kura, Y.; Yoshikawa, K.; Nishio, K.; Davies, B.R.; Uemura, H. Efficacy of targeted AKT inhibition in genetically engineered mouse models of PTEN-deficient prostate cancer. Oncotarget 2016, 7, 15959–15976. [Google Scholar] [CrossRef] [Green Version]
- Rybak, A.P.; Bristow, R.G.; Kapoor, A. Prostate cancer stem cells: Deciphering the origins and pathways involved in prostate tumorigenesis and aggression. Oncotarget 2015, 6, 1900–1919. [Google Scholar] [CrossRef] [PubMed]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR pathway and prostate cancer: At the crossroads of AR, MAPK, and WNT signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Liu, C.; Zhu, Y.; Lou, W.; Cui, Y.; Evans, C.P.; Gao, A.C. Inhibition of constitutively active Stat3 reverses enzalutamide resistance in LNCaP derivative prostate cancer cells. Prostate 2014, 74, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Cen, B.; Mahajan, S.; Wang, W.; Kraft, A.S. Elevation of receptor tyrosine kinases by small molecule AKT inhibitors in prostate cancer is mediated by Pim-1. Cancer Res. 2013, 73, 3402–3411. [Google Scholar] [CrossRef] [Green Version]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 174, 758–769. [Google Scholar] [CrossRef] [Green Version]
- Navone, N.M.; Troncoso, P.; Pisters, L.L.; Goodrow, T.L.; Palmer, J.L.; Nichols, W.W.; Von Eschenbach, A.C.; Conti, C.J. P53 protein accumulation and gene mutation in the progression of human prostate carcinoma. JNCI 1993, 85, 1657–1669. [Google Scholar] [CrossRef]
- Cronauer, M.V.; Schulz, W.A.; Burchardt, T.; Ackermann, R.; Burchardt, M. Inhibition of p53 function diminishes androgen receptor-mediated signaling in prostate cancer cell lines. Oncogene 2004, 23, 3541–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alimirah, F.; Panchanathan, R.; Chen, J.; Zhang, X.; Ho, S.-M.; Choubey, D. Expression of androgen receptor is negatively regulated by p53. Neoplasia 2007, 9, 1152–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [Green Version]
- Small, E.J.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Final survival results from SPARTAN, a phase III study of apalutamide (APA) versus placebo (PBO) in patients (pts) with nonmetastatic castration-resistant prostate cancer (nmCRPC). J. Clin. Oncol. 2020, 38, 5516. [Google Scholar] [CrossRef]
- Chi, K.N.; Agarwal, N.; Bjartell, A.; Chung, B.H.; Gomes, A.J.P.D.S.; Given, R.; Given, R.; Soto, Á.J.; Merseburger, A.S.; Özgüroglu, M.; et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N. Eng. J. Med. 2019, 381, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Marques, R.B.; Aghai, A.; de Ridder, C.M.; Stuurman, D.; Hoeben, S.; Boer, A.; Ellston, R.; Barry, S.T.; Davies, B.R.; Trapman, J.; et al. High efficacy of combination therapy using PI3K/AKT inhibitors with androgen deprivation in prostate cancer preclinical models. Eur. Urol. 2015, 67, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Toren, P.; Kim, S.; Cordonnier, T.; Crafter, C.; Davies, B.R.; Fazli, L.; Gleave, M.E.; Zoubeidi, A. Combination AZD5363 with enzalutamide significantly delays enzalutamide-resistant prostate cancer in preclinical models. Eur. Urol. 2015, 67, 986–990. [Google Scholar] [CrossRef]
- Warfel, N.A.; Kraft, A.S. PIM kinase (and Akt) biology and signaling in tumors. Pharm. Ther. 2015, 151, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdman, A.; Fang, X.; Pang, S.-T.; Ekman, P.; Egevad, L. Pim-1 expression in prostatic intraepithelial neoplasia and human prostate cancer. Prostate 2004, 60, 367–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanasekaran, S.M.; Barrette, T.R.; Ghosh, D.; Shah, R.; Varambally, S.; Kurachi, K.; Pienta, K.; Rubin, M.; Chinnaiyan, A.M. Delineation of prognostic biomarkers in prostate cancer. Nature 2001, 412, 822–826. [Google Scholar] [CrossRef]
- Nowak, D.G.; Cho, H.; Herzka, T.; Watrud, K.; Demarco, D.V.; Wang, V.; Senturk, S.; Fellmann, C.; Ding, D.; Beinortas, T.; et al. MYC drives Pten/Trp53-deficient proliferation and metastasis due to IL6 secretion and AKT suppression via PHLPP2. Cancer Discov. 2015, 5, 636–651. [Google Scholar] [CrossRef] [Green Version]
- Ha, S.; Iqbal, N.; Mita, P.; Ruoff, R.; Gerald, W.L.; Lepor, H.; Taneja, S.S.; Lee, P.; Melamed, J.; Garabedian, M.J.; et al. Phosphorylation of the androgen receptor by PIM1 in hormone refractory prostate cancer. Oncogene 2013, 32, 3992–4000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open-source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]
- ImageJ Image Processing and Analysis in Java. Available online: https://imagej.nih.gov/ij/ (accessed on 17 May 2020).
- Takao, A.; Yoshikawa, K.; Karnan, S.; Ota, A.; Uemura, H.; De Velasco, M.A.; Kura, Y.; Suzuki, S.; Ueda, R.; Nishino, T.; et al. Generation of PTEN-knockout (−/−) murine prostate cancer cells using the CRISPR/Cas9 system and comprehensive gene expression profiling. Oncol. Rep. 2018, 40, 2455–2466. [Google Scholar] [CrossRef] [Green Version]
- Morpheus. Available online: https://software.broadinstitute.org/morpheus (accessed on 12 January 2021).
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 1 October 2020).
- Demšar, J.; Curk, T.; Erjavec, A.; Gorup, Č.; Hočevar, T.; Milutinovič, M.; Možina, M.; Polajnar, M.; Toplak, M.; Starič, A.; et al. Orange: Data Mining Toolbox in Python. J. Mach. Learn. Res. 2013, 14, 2349–2353. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Description | Apalutamide | GSK690693 | Combination Index | |||
|---|---|---|---|---|---|---|---|
| Mean ED50 | SE | Mean ED50 | SE | Mean CI | SE | ||
| (µM) | (µM) | ||||||
| 7109-G4 | CNPC Pten-deficient tumor | 28.70 | 8.16 | 9.18 | 2.57 | 0.585 | 0.053 |
| 2945-F12 | CRPC Pten-deficient tumor | 56.17 | 5.16 | 1.28 | 0.33 | 0.628 | 0.046 |
| 3902-A1 | CNPC Pten/Trp53-deficient tumor | 41.77 | 5.59 | 1.87 | 0.69 | 0.73 | 0.063 |
| 4522-B8 | CRPC Pten/Trp53-deficient tumor | 36.37 | 4.1 | 7.79 | 1.33 | 0.791 | 0.053 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Velasco, M.A.; Kura, Y.; Ando, N.; Sako, N.; Banno, E.; Fujita, K.; Nozawa, M.; Yoshimura, K.; Sakai, K.; Yoshikawa, K.; et al. Context-Specific Efficacy of Apalutamide Therapy in Preclinical Models of Pten-Deficient Prostate Cancer. Cancers 2021, 13, 3975. https://doi.org/10.3390/cancers13163975
De Velasco MA, Kura Y, Ando N, Sako N, Banno E, Fujita K, Nozawa M, Yoshimura K, Sakai K, Yoshikawa K, et al. Context-Specific Efficacy of Apalutamide Therapy in Preclinical Models of Pten-Deficient Prostate Cancer. Cancers. 2021; 13(16):3975. https://doi.org/10.3390/cancers13163975
Chicago/Turabian StyleDe Velasco, Marco A., Yurie Kura, Naomi Ando, Noriko Sako, Eri Banno, Kazutoshi Fujita, Masahiro Nozawa, Kazuhiro Yoshimura, Kazuko Sakai, Kazuhiro Yoshikawa, and et al. 2021. "Context-Specific Efficacy of Apalutamide Therapy in Preclinical Models of Pten-Deficient Prostate Cancer" Cancers 13, no. 16: 3975. https://doi.org/10.3390/cancers13163975
APA StyleDe Velasco, M. A., Kura, Y., Ando, N., Sako, N., Banno, E., Fujita, K., Nozawa, M., Yoshimura, K., Sakai, K., Yoshikawa, K., Nishio, K., & Uemura, H. (2021). Context-Specific Efficacy of Apalutamide Therapy in Preclinical Models of Pten-Deficient Prostate Cancer. Cancers, 13(16), 3975. https://doi.org/10.3390/cancers13163975