
Metformin Is a Pyridoxal-5′-phosphate (PLP)-Competitive Inhibitor of SHMT2

 ,
,  , ,
, ,  , , ,
, , ,  ,
,  , ,
, ,  ,
, 
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Enzyme Purification
2.3. Inhibition Experiments with Purified Recombinant SHMT Isoforms
2.4. Differential Scanning Fluorimetry (DSF)
2.5. Inhibition Experiments in Cells
2.6. Molecular Docking Simulations
2.7. Molecular Dynamics Simulations
2.8. Cell Lines
2.9. Generation of NDI1- and SHMT2-Overexpressing Cell Lines
2.10. Immunoblotting
2.11. Stable Isotope Labeling
2.12. Cell Viability Assays
2.13. Real-Time Cell Growth
2.14. Extracellular Flux Assay
2.15. Mitochondrial Function Phenotyping
2.16. Statistical Analysis
3. Results
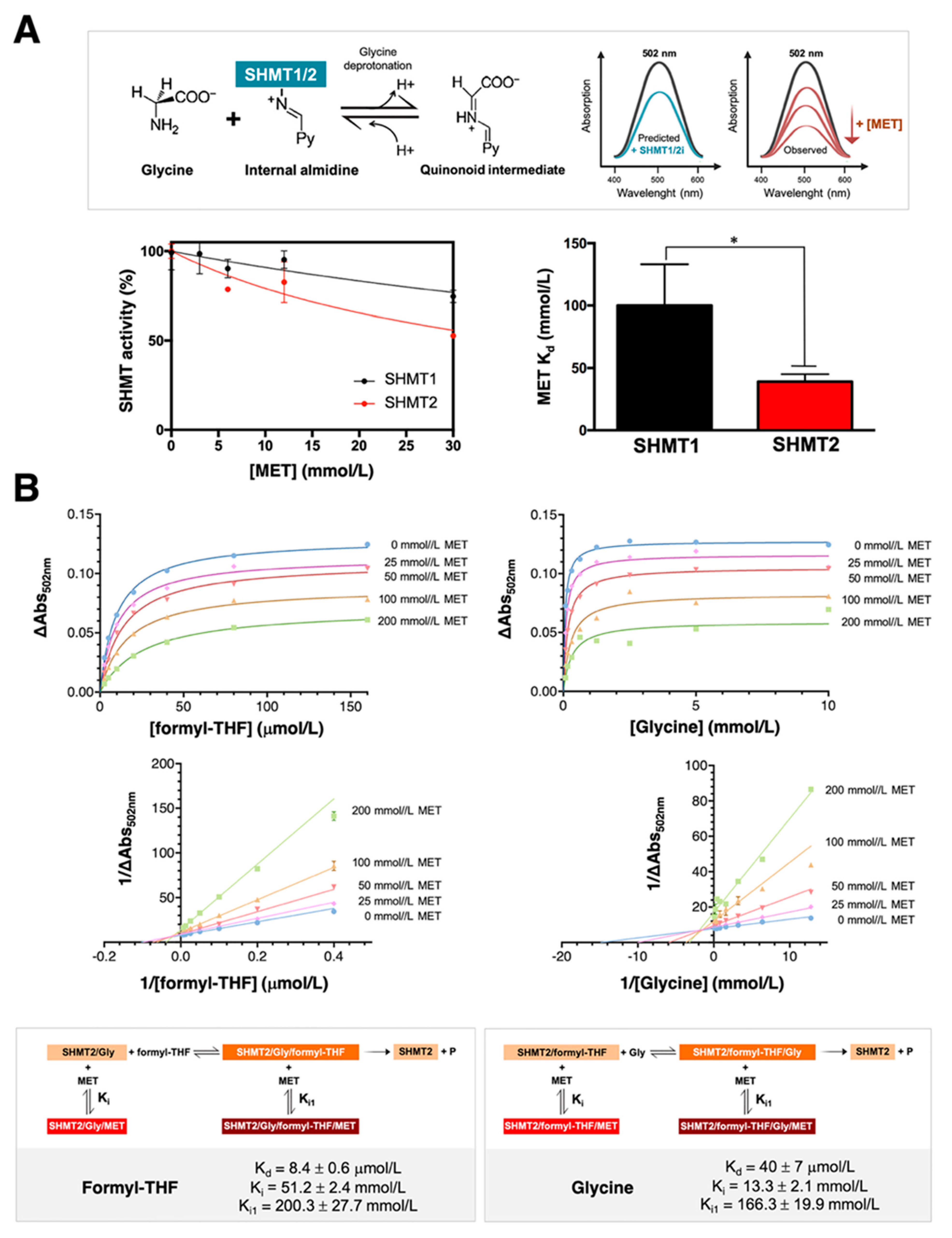
3.1. Metformin Directly Targets SHMT Enzymatic Activity in an Isoenzyme-Selective Manner
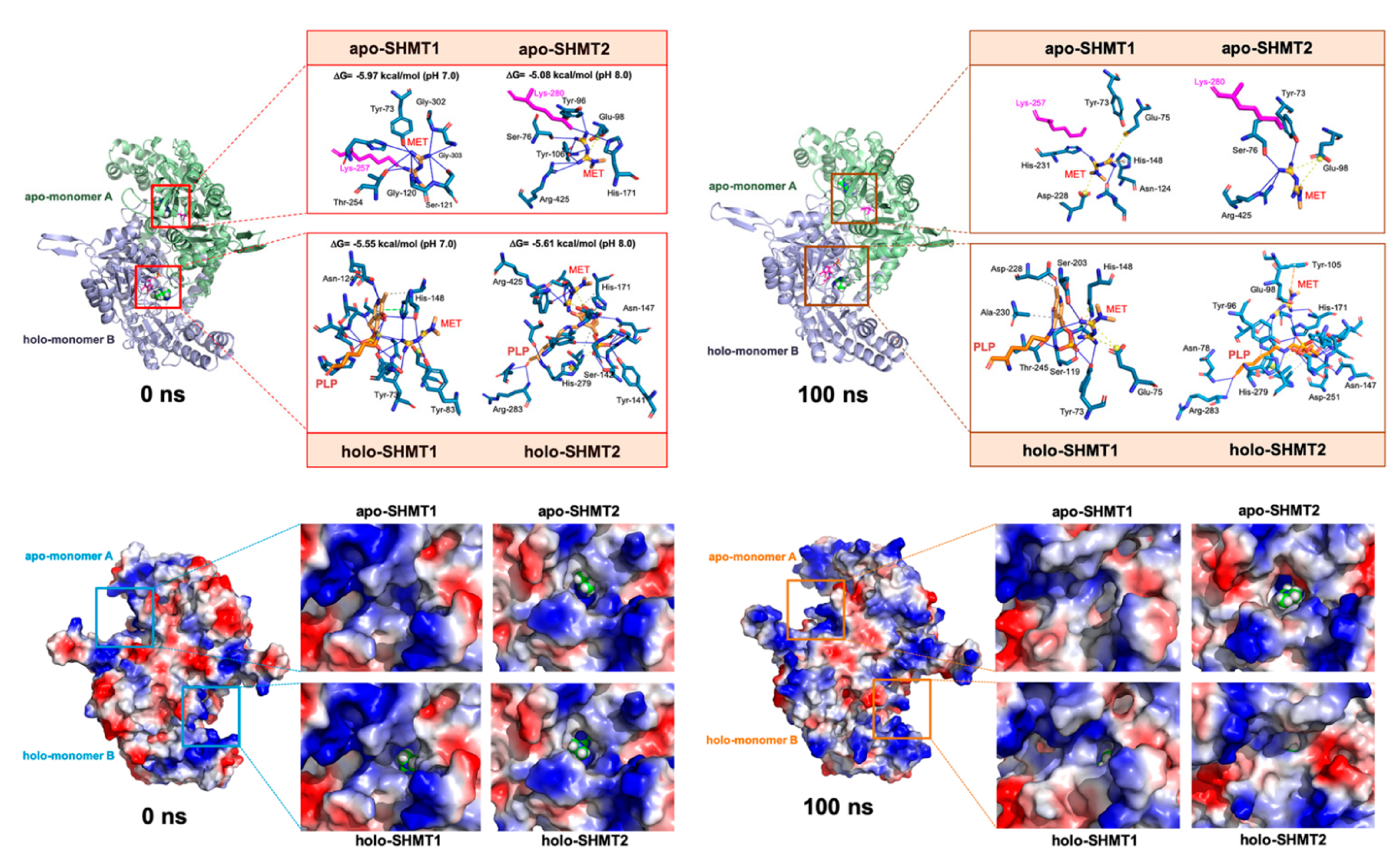
3.2. Binding of Metformin to SHMT2 Does Not Involve the SHMT2 Ligand-Binding Sites
3.3. Metformin Is Computationally Predicted to Occupy the Cofactor Pyridoxal 5′-phosphate Cavity of SHMTs
3.4. Metformin Is Computationally Predicted to Destabilize the Oligomerization of a Catalytically Active SHMT2 Conformation
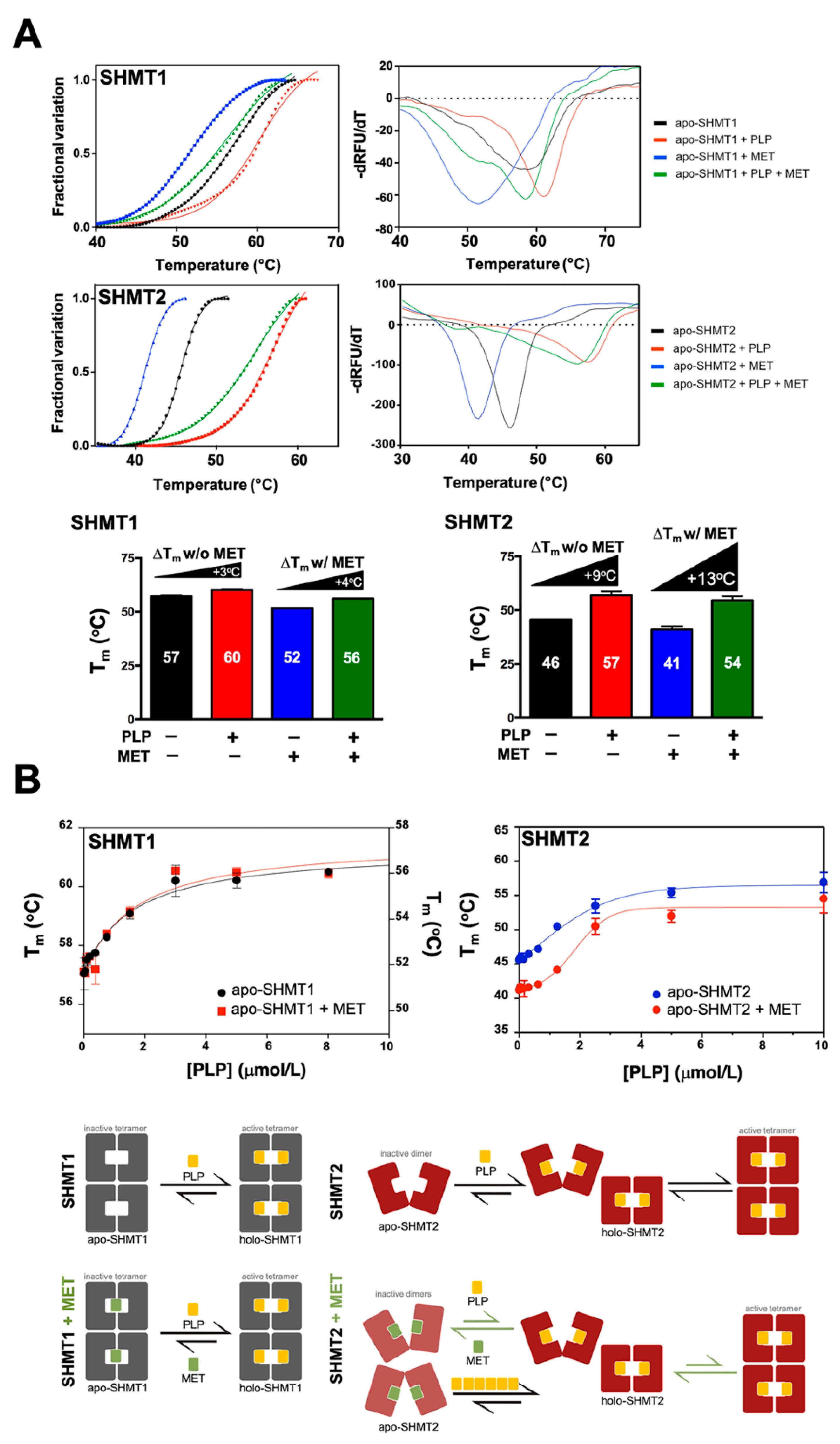
3.5. Metformin Impairs the Capacity of PLP to Promote SHMT2 Dimer-to-Tetramer Transition
3.6. Metformin Blocks SHMT2 Activity in Cancer Cells
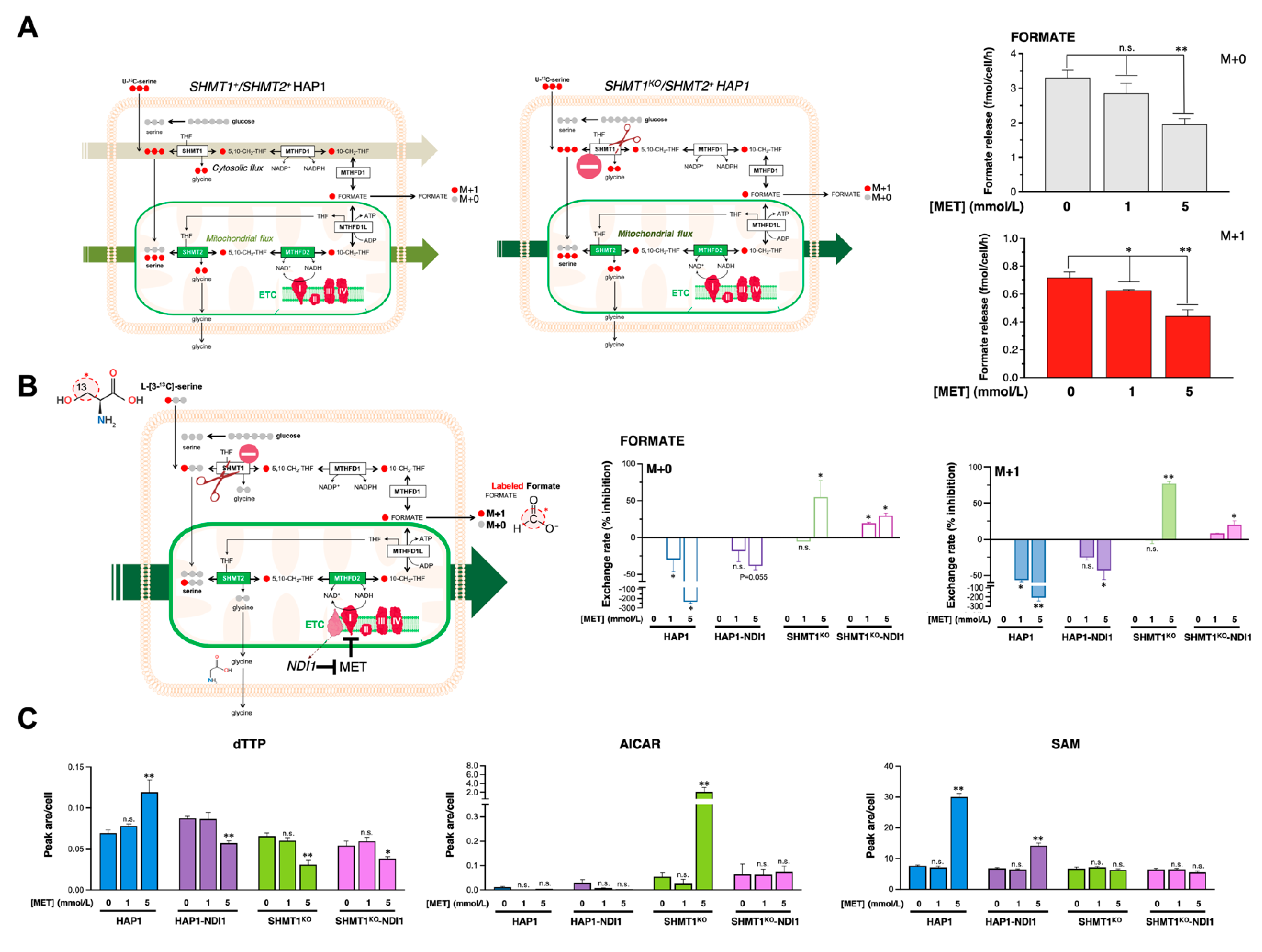
3.7. Metformin Reduces the SHMT2-Channeled Serine-to-Formate Flux
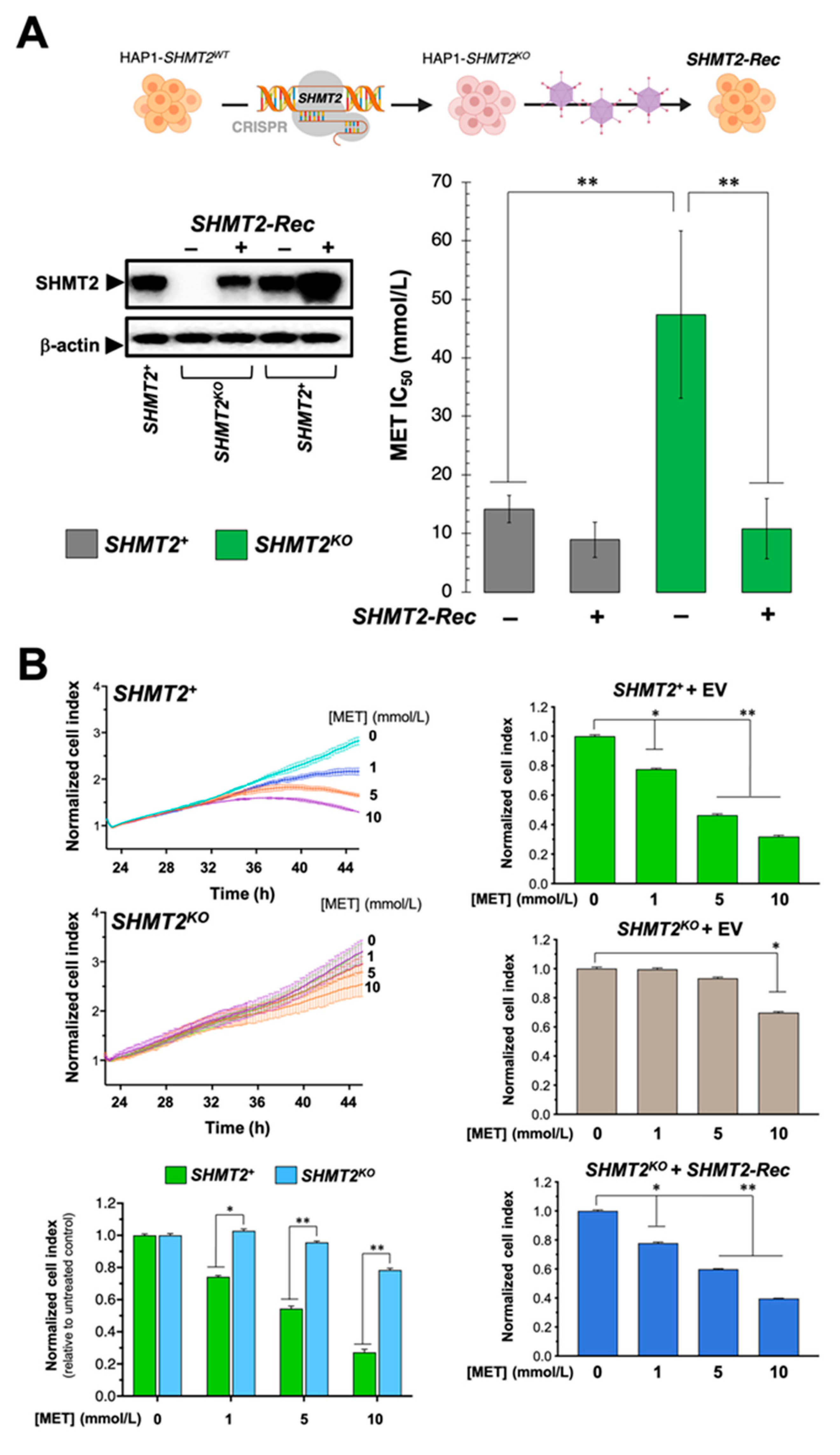
3.8. Loss of SHMT2 Prevents the Cytotoxic and Anti-Proliferative Effects of Metformin
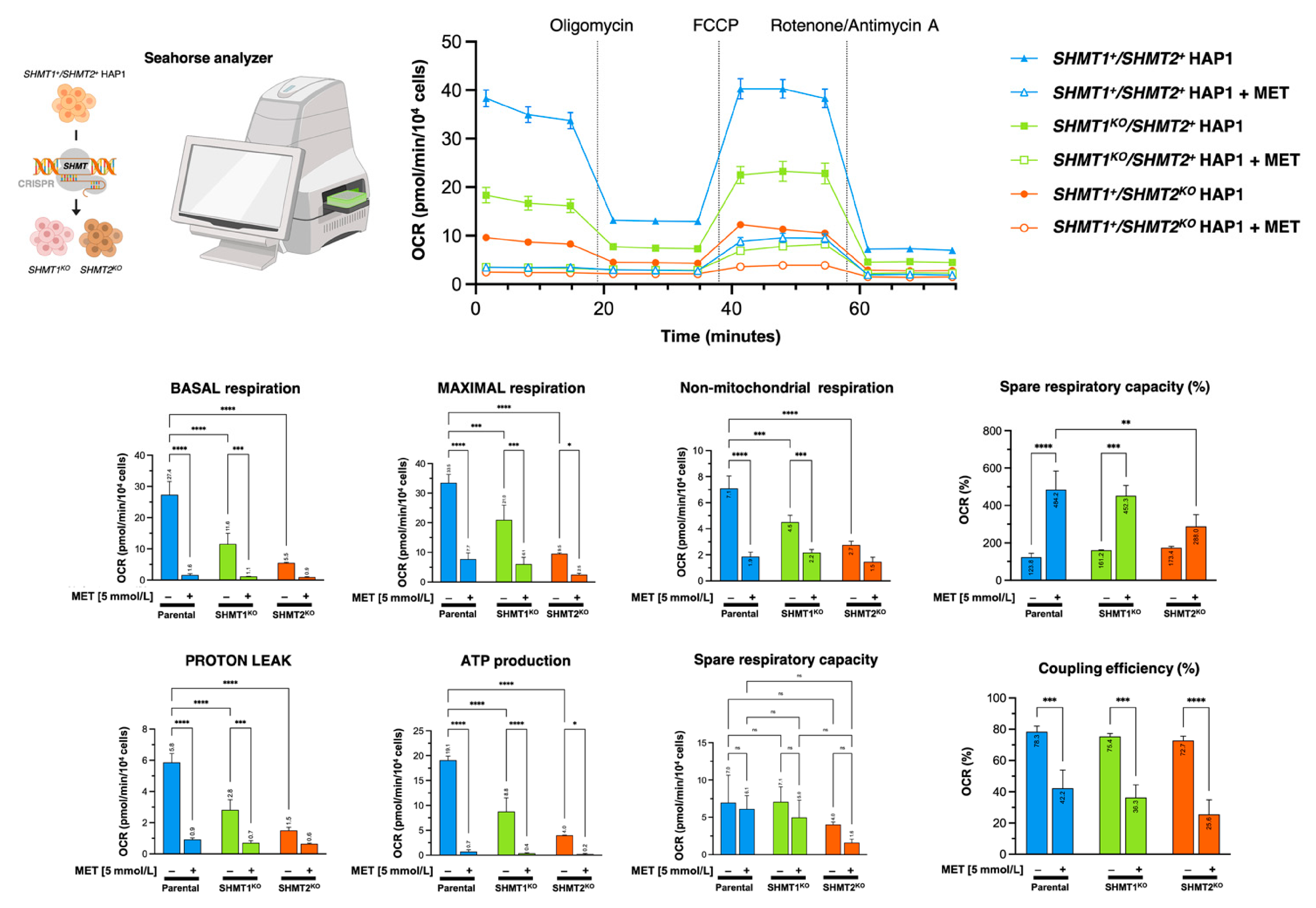
3.9. Mitochondrial Serine Catabolism Is a Determinant for the Anti-Cancer Activity of Metformin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Geeraerts, S.L.; Heylen, E.; De Keersmaecker, K.; Kampen, K.R. The ins and outs of serine and glycine metabolism in cancer. Nat. Metab. 2021, 3, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Reina-Campos, M.; Diaz-Meco, M.T.; Moscat, J. The complexity of the serine glycine one-carbon pathway in cancer. J. Cell Biol. 2020, 219, e201907022. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Ghergurovich, J.M.; Mainolfi, N.; Suri, V.; Jeong, S.K.; Hsin-Jung Li, S.; Friedman, A.; Manfredi, M.G.; Gitai, Z.; Kim, H.; et al. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2017, 114, 11404–11409. [Google Scholar] [CrossRef]
- Cuyàs, E.; Fernández-Arroyo, S.; Buxó, M.; Pernas, S.; Dorca, J.; Álvarez, I.; Martínez, S.; Pérez-Garcia, J.M.; Batista-López, N.; Rodríguez-Sánchez, C.A.; et al. Metformininduces a fasting- and antifolate-mimicking modification of systemic host metabolism in breast cancer patients. Aging 2019, 11, 2874–2888. [Google Scholar] [CrossRef]
- Labuschagne, C.F.; van den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef]
- Kanarek, N.; Keys, H.R.; Cantor, J.R.; Lewis, C.A.; Chan, S.H.; Kunchok, T.; Abu-Remaileh, M.; Freinkman, E.; Schweitzer, L.D.; Sabatini, D.M. Histidinecatabolism is a major determinant of methotrexate sensitivity. Nature 2018, 559, 632–636. [Google Scholar] [CrossRef]
- Kim, D.; Fiske, B.P.; Birsoy, K.; Freinkman, E.; Kami, K.; Possemato, R.L.; Chudnovsky, Y.; Pacold, M.E.; Chen, W.W.; Cantor, J.R.; et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 2015, 520, 363–367. [Google Scholar] [CrossRef]
- Corominas-Faja, B.; Quirantes-Piné, R.; Oliveras-Ferraros, C.; Vazquez-Martin, A.; Cufí, S.; Martin-Castillo, B.; Micol, V.; Joven, J.; Segura-Carretero, A.; Menendez, J.A. Metabolomic fingerprint reveals thatmetforminimpairsone-carbonmetabolismin a manner similar to the antifolate class of chemotherapy drugs. Aging 2012, 4, 480–498. [Google Scholar] [CrossRef]
- Cuyàs, E.; Fernández-Arroyo, S.; Verdura, S.; García, R.Á.; Stursa, J.; Werner, L.; Blanco-González, E.; Montes-Bayón, M.; Joven, J.; Viollet, B.; et al. Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene 2018, 37, 963–970. [Google Scholar] [CrossRef]
- Gravel, S.P.; Hulea, L.; Toban, N.; Birman, E.; Blouin, M.J.; Zakikhani, M.; Zhao, Y.; Topisirovic, I.; St-Pierre, J.; Pollak, M. Serine deprivation enhances antineoplastic activity of biguanides. Cancer Res. 2014, 74, 7521–7533. [Google Scholar] [CrossRef]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; van den Broek, N.J.F.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef]
- Bridges, H.R.; Sirviö, V.A.; Agip, A.N.; Hirst, J. Molecular features ofbiguanidesrequired for targeting of mitochondrial respiratory complex I and activation of AMP-kinase. BMC Biol. 2016, 14, 65. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex Iof cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Meiser, J.; Schuster, A.; Pietzke, M.; Vande Voorde, J.; Athineos, D.; Oizel, K.; Burgos-Barragan, G.; Wit, N.; Dhayade, S.; Morton, J.P.; et al. Increased formate overflow is a hallmark of oxidative cancer. Nat. Commun. 2018, 9, 1368. [Google Scholar] [CrossRef]
- Kruschwitz, H.; Ren, S.; Di Salvo, M.; Schirch, V. Expression, purification, and characterization of human cytosolic serine hydroxymethyltransferase. Protein Expr. Purif. 1995, 6, 411–416. [Google Scholar] [CrossRef]
- Giardina, G.; Brunotti, P.; Fiascarelli, A.; Cicalini, A.; Costa, M.G.; Buckle, A.M.; di Salvo, M.L.; Giorgi, A.; Marani, M.; Paone, A.; et al. How pyridoxal 5’-phosphate differentially regulates human cytosolic anmitochondrial serine hydroxymethyltransferase oligomeric state. FEBS J. 2015, 282, 1225–1241. [Google Scholar] [CrossRef]
- Daidone, F.; Florio, R.; Rinaldo, S.; Contestabile, R.; di Salvo, M.L.; Cutruzzolà, F.; Bossa, F.; Paiardini, A. In silico and in vitro validation of serine hydroxymethyltransferase as a chemotherapeutic target of the antifolate drug pemetrexed. Eur. J. Med. Chem. 2011, 46, 1616–1621. [Google Scholar] [CrossRef]
- Marani, M.; Paone, A.; Fiascarelli, A.; Macone, A.; Gargano, M.; Rinaldo, S.; Giardina, G.; Pontecorvi, V.; Koes, D.; McDermott, L.; et al. A pyrazolopyran derivative preferentially inhibits the activity of human cytosolic serine hydroxymethyltransferase and induces cell death in lung cancer cells. Oncotarget 2016, 7, 4570–4583. [Google Scholar] [CrossRef]
- Schirch, L.; Ropp, M. Serine transhydroxymethylase. Affinity of tetrahydrofolate compounds for the enzyme and enzyme-glycine complex. Biochemistry. 1967, 6, 253–257. [Google Scholar] [CrossRef]
- Stover, P.J.; Chen, L.H.; Suh, J.R.; Stover, D.M.; Keyomarsi, K.; Shane, B. Molecular cloning, characterization, and regulation of the human mitochondrial serine hydroxymethyltransferase gene. J. Biol. Chem. 1997, 272, 1842–1848. [Google Scholar] [CrossRef]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins 2004, 57, 678–683. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinf. 2008, 8, 8–14. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Cuyàs, E.; Verdura, S.; Lozano-Sánchez, J.; Viciano, I.; Llorach-Parés, L.; Nonell-Canals, A.; Bosch-Barrera, J.; Brunet, J.; Segura-Carretero, A.; Sanchez-Martinez, M.; et al. The extra virgin olive oil phenolic oleacein is a dual substrate-inhibitor of catechol-O-methyltransferase. Food Chem. Toxicol. 2019, 128, 35–45. [Google Scholar] [CrossRef]
- Verdura, S.; Cuyàs, E.; Cortada, E.; Brunet, J.; Lopez-Bonet, E.; Martin-Castillo, B.; Bosch-Barrera, J.; Encinar, J.A.; Menendez, J.A. Resveratrol targets PD-L1 glycosylation and dimerization to enhance antitumor T-cell immunity. Aging 2020, 12, 8–34. [Google Scholar] [CrossRef]
- Encinar, J.A.; Menendez, J.A. Potential Drugs Targeting Early Innate Immune Evasion of SARS-Coronavirus 2 via 2′-O-Methylation of Viral RNA. Viruses 2020, 12, 525. [Google Scholar] [CrossRef]
- Burgos-Barragan, G.; Wit, N.; Meiser, J.; Dingler, F.A.; Pietzke, M.; Mulderrig, L.; Pontel, L.B.; Rosado, I.V.; Brewer, T.F.; Cordell, R.L.; et al. Mammals divert endogenous genotoxic formaldehyde into one-carbon metabolism. Nature 2017, 548, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Meiser, J.; Tumanov, S.; Maddocks, O.; Labuschagne, C.F.; Athineos, D.; Van Den Broek, N.; Mackay, G.M.; Gottlieb, E.; Blyth, K.; Vousden, K.; et al. Serine one-carbon catabolism with formate overflow. Sci. Adv. 2016, 2, e1601273. [Google Scholar] [CrossRef] [PubMed]
- Tramonti, A.; Paiardini, A.; Paone, A.; Bouzidi, A.; Giardina, G.; Guiducci, G.; Magnifico, M.C.; Rinaldo, S.; McDermott, L.; Menendez, J.A.; et al. Differential inhibitory effect of a pyrazolopyran compound on human serine hydroxymethyltransferase-amino acid complexes. Arch. Biochem. Biophys. 2018, 653, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Tramonti, A.; Nardella, C.; di Salvo, M.L.; Barile, A.; Cutruzzolà, F.; Contestabile, R. Human Cytosolic and Mitochondrial Serine Hydroxymethyltransferase Isoforms in Comparison: Full Kinetic Characterization and Substrate Inhibition Properties. Biochemistry 2018, 57, 6984–6996. [Google Scholar] [CrossRef]
- Renwick, S.B.; Snell, K.; Baumann, U. The crystal structure of human cytosolic serine hydroxymethyltransferase: A target for cancer chemotherapy. Structure 1998, 6, 1105–1116. [Google Scholar] [CrossRef]
- Scaletti, E.; Jemth, A.S.; Helleday, T.; Stenmark, P. Structural basis of inhibition of the human serine hydroxymethyltransferase SHMT2 by antifolate drugs. FEBS Lett. 2019, 593, 1863–1873. [Google Scholar] [CrossRef]
- Rabl, J.; Bunker, R.D.; Schenk, A.D.; Cavadini, S.; Gill, M.E.; Abdulrahman, W.; Andrés-Pons, A.; Luijsterburg, M.S.; Ibrahim, A.F.M.; Branigan, E.; et al. Structural Basis of BRCC36 Function in DNA Repair and Immune Regulation. Mol. Cell 2019, 75, 483–497. [Google Scholar] [CrossRef]
- Walden, M.; Tian, L.; Ross, R.L.; Sykora, U.M.; Byrne, D.P.; Hesketh, E.L.; Masandi, S.K.; Cassel, J.; George, R.; Ault, J.R.; et al. Metabolic control of BRISC-SHMT2 assembly regulates immune signalling. Nature 2019, 570, 194–199. [Google Scholar] [CrossRef]
- Delgado, J.; Radusky, L.G.; Cianferoni, D.; Serrano, L. FoldX 5.0: Working with RNA, small molecules and a new graphical interface. Bioinformatics 2019, 35, 4168–4169. [Google Scholar] [CrossRef]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef]
- Chilton, M.; Clennell, B.; Edfeldt, F.; Geschwindner, S. Hot-Spotting with Thermal Scanning: A Ligand- and Structure-Independent Assessment of Target Ligandability. J. Med. Chem. 2017, 60, 4923–4931. [Google Scholar] [CrossRef]
- Gao, K.; Oerlemans, R.; Groves, M.R. Theory and applications of differential scanning fluorimetry in early-stage drug discovery. Biophys. Rev. 2020, 12, 85–104. [Google Scholar] [CrossRef]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef]
- Zhang, R.; Monsma, F. Fluorescence-based thermal shift assays. Curr. Opin. Drug Discov. Dev. 2010, 13, 389–402. [Google Scholar]
- Paone, A.; Marani, M.; Fiascarelli, A.; Rinaldo, S.; Giardina, G.; Contestabile, R.; Paiardini, A.; Cutruzzolà, F. SHMT1 knockdown induces apoptosis in lung cancer cells by causing uracil misincorporation. Cell Death Dis. 2014, 5, e1525. [Google Scholar] [CrossRef]
- Ducker, G.S.; Chen, L.; Morscher, R.J.; Ghergurovich, J.M.; Esposito, M.; Teng, X.; Kang, Y.; Rabinowitz, J.D. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab. 2016, 23, 1140–1153. [Google Scholar] [CrossRef]
- Chandel, N. Four key questions about metformin and cancer. BMC Biol. 2014, 12, 85. [Google Scholar] [CrossRef]
- Chandel, N.S.; Avizonis, D.; Reczek, C.R.; Weinberg, S.E.; Menz, S.; Neuhaus, R.; Christian, S.; Haegebarth, A.; Algire, C.; Pollak, M. Are Metformin Doses Used in Murine Cancer Models Clinically Relevant? Cell Metab. 2016, 23, 569–570. [Google Scholar] [CrossRef]
- Tan, Y.L.; Sou, N.L.; Tang, F.Y.; Ko, H.A.; Yeh, W.T.; Peng, J.H.; Chiang, E.I. Tracing Metabolic Fate of Mitochondrial Glycine Cleavage System Derived Formate In Vitro and In Vivo. Int. J. Mol. Sci. 2020, 21, 8808. [Google Scholar] [CrossRef]
- Sou, N.L.; Huang, Y.H.; Chen, D.Y.; Chen, Y.M.; Tang, F.Y.; Ko, H.A.; Fan, Y.H.; Lin, Y.Y.; Wang, Y.C.; Chih, H.M.; et al. Folinate Supplementation Ameliorates Methotrexate Induced Mitochondrial Formate Depletion In Vitro and In Vivo. Int. J. Mol. Sci. 2021, 22, 1350. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, L.F.; Appling, D.R. Characterization of the folate-dependent mitochondrial oxidation of carbon 3 of serine. Biochemistry 1993, 32, 4671–4676. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.B.; Kitajima-Ihara, T.; Chan, E.K.; Scheffler, I.E.; Matsuno-Yagi, A.; Yagi, T. Molecular remedy of complex I defects: Rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 1998, 95, 9167–9171. [Google Scholar] [CrossRef]
- Nishimura, T.; Nakata, A.; Chen, X.; Nishi, K.; Meguro-Horike, M.; Sasaki, S.; Kita, K.; Horike, S.I.; Saitoh, K.; Kato, K.; et al. Cancer stem-like properties and gefitinib resistance are dependent on purine synthetic metabolism mediated by the mitochondrial enzyme MTHFD2. Oncogene 2019, 38, 2464–2481. [Google Scholar] [CrossRef]
- Oizel, K.; Tait-Mulder, J.; Fernandez-de-Cossio-Diaz, J.; Pietzke, M.; Brunton, H.; Lilla, S.; Dhayade, S.; Athineos, D.; Blanco, G.R.; Sumpton, D.; et al. Formate induces a metabolic switch in nucleotide and energy metabolism. Cell Death Dis. 2020, 11, 310. [Google Scholar] [CrossRef]
- Bao, X.R.; Ong, S.E.; Goldberger, O.; Peng, J.; Sharma, R.; Thompson, D.A.; Vafai, S.B.; Cox, A.G.; Marutani, E.; Ichinose, F.; et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife 2016, 5, e10575. [Google Scholar] [CrossRef]
- Lucas, S.; Chen, G.; Aras, S.; Wang, J. Serine catabolism is essential to maintain mitochondrial respiration in mammalian cell. Life Sci. Alliance 2018, 1, e201800036. [Google Scholar] [CrossRef]
- Minton, D.R.; Nam, M.; McLaughlin, D.J.; Shin, J.; Bayraktar, E.C.; Alvarez, S.W.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Sabatini, D.M.; Birsoy, K.; et al. Serine Catabolism by SHMT2 Is Required for Proper Mitochondrial Translation Initiation and Maintenance of Formylmethionyl-tRNAs. Mol. Cell 2018, 69, 610–621. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metforminas a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef]
- Visentin, M.; Zhao, R.; Goldman, I.D. The antifolates. Hematol. Oncol. Clin. N. Am. 2012, 26, 629–648. [Google Scholar] [CrossRef]
- Zhao, R.; Goldman, I.D. Resistance to antifolates. Oncogene 2003, 22, 7431–7457. [Google Scholar] [CrossRef]
- Cuthberston, C.R.; Arabzada, Z.; Bankhead, A., III; Kyani, A.; Neamati, N. A review of small-molecule inhibitors of one-carbon enzymes: SHMT2 and MTHFD2 in the spotlight. ACS Pharmacol. Transl. Sci. 2021, 4, 624–646. [Google Scholar] [CrossRef]
- Dekhne, A.S.; Hou, Z.; Gangjee, A.; Matherly, L.H. Therapeutic Targeting of Mitochondrial One-Carbon Metabolism in Cancer. Mol. Cancer Ther. 2020, 19, 2245–2255. [Google Scholar] [CrossRef]
- Zhao, L.N.; Björklund, M.; Caldez, M.J.; Zheng, J.; Kaldis, P. Therapeutic targeting of the mitochondrial one-carbon pathway: Perspectives, pitfalls, and potential. Oncogene 2021, 40, 2339–2354. [Google Scholar] [CrossRef]
- Geeraerts, S.L.; Kampen, K.R.; Rinaldi, G.; Gupta, P.; Planque, M.; Louros, N.; Heylen, E.; De Cremer, K.; De Brucker, K.M.; Vereecke, S.; et al. Repurposing the Antidepressant Sertraline as SHMT Inhibitor to Suppress Serine/Glycine Synthesis-Addicted Breast Tumor Growth. Mol. Cancer Ther. 2021, 20, 50–63. [Google Scholar] [CrossRef]
- Nonaka, H.; Nakanishi, Y.; Kuno, S.; Ota, T.; Mochidome, K.; Saito, Y.; Sugihara, F.; Takakusagi, Y.; Aoki, I.; Nagatoishi, S.; et al. Design strategy for serine hydroxymethyltransferase probes based on retro-aldol-type reaction. Nat. Commun. 2019, 10, 876. [Google Scholar] [CrossRef]
- Tani, H.; Ohnishi, S.; Shitara, H.; Mito, T.; Yamaguchi, M.; Yonekawa, H.; Hashizume, O.; Ishikawa, K.; Nakada, K.; Hayashi, J.I. Mice deficient in the Shmt2 gene have mitochondrial respiration defects and are embryonic lethal. Sci. Rep. 2018, 8, 425. [Google Scholar] [CrossRef]
- Lee, W.D.; Pirona, A.C.; Sarvin, B.; Stern, A.; Nevo-Dinur, K.; Besser, E.; Sarvin, N.; Lagziel, S.; Mukha, D.; Raz, S.; et al. Tumor Reliance on Cytosolic versus Mitochondrial One-Carbon Flux Depends on Folate Availability. Cell Metab. 2021, 33, 190–198. [Google Scholar] [CrossRef]
- Peralta, S.; Pinto, M.; Arguello, T.; Garcia, S.; Diaz, F.; Moraes, C.T. Metformin delays neurological symptom onset in a mouse model of neuronal complex I deficiency. JCI Insight 2020, 5, e141183. [Google Scholar] [CrossRef]
- Yang, L.; Garcia Canaveras, J.C.; Chen, Z.; Wang, L.; Liang, L.; Jang, C.; Mayr, J.A.; Zhang, Z.; Ghergurovich, J.M.; Zhan, L.; et al. Serine Catabolism Feeds NADH when Respiration Is Impaired. Cell Metab. 2020, 31, 809–821. [Google Scholar] [CrossRef]
- Cabreiro, F.; Au, C.; Leung, K.Y.; Vergara-Irigaray, N.; Cochemé, H.M.; Noori, T.; Weinkove, D.; Schuster, E.; Greene, N.D.; Gems, D. Metformin retards aging in C elegans by altering microbial folate and methionine metabolism. Cell 2013, 153, 228–239. [Google Scholar] [CrossRef]
- Gabel, S.A.; Duff, M.R.; Pedersen, L.C.; DeRose, E.F.; Krahn, J.M.; Howell, E.E.; London, R.E. A Structural Basis for Biguanide Activity. Biochemistry 2017, 56, 4786–4798. [Google Scholar] [CrossRef]
- Adams, J.F.; Clark, J.S.; Ireland, J.T.; Kesson, C.M.; Watson, W.S. Malabsorption of vitamin B12 and intrinsic factor secretion during biguanide therapy. Diabetologia 1983, 24, 16–18. [Google Scholar] [CrossRef]
- Andrès, E.; Federici, L. Vitamin B12 deficiency in patients receiving metformin: Clinical data. Arch. Intern. Med. 2007, 167, 729. [Google Scholar]
- Herbert, V. Metformin and B-12 malabsorption. Ann. Intern. Med. 1972, 76, 140–141. [Google Scholar] [CrossRef]
- Stowers, J.M.; Smith, O.A. Vitamin B 12 and metformin. Br. Med. J. 1971, 3, 246–247. [Google Scholar] [CrossRef]
- Nijhout, H.F.; Reed, M.C.; Budu, P.; Ulrich, C.M. A mathematical model of the folate cycle: New insights into folate homeostasis. J. Biol. Chem. 2004, 279, 55008–55016. [Google Scholar] [CrossRef]
- Scott, J.M.; Weir, D.G. Themethylfolatetrap A physiological response in man to preventmethylgroup deficiency in kwashiorkor (methionine deficiency) and an explanation for folic-acid induced exacerbation of subacute combined degeneration in pernicious anaemia. Lancet 1981, 2, 337–340. [Google Scholar] [CrossRef]
- Palmer, A.M.; Kamynina, E.; Field, M.S.; Stover, P.J. Folate rescues vitamin B12depletion-induced inhibition of nuclear thymidylate biosynthesis and genome instability. Proc. Natl. Acad. Sci. USA 2017, 114, E4095–E4102. [Google Scholar] [CrossRef]
- Locasale, J.W. Serine, glycine andone-carbonunits: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.C.; Maddocks, O.D.K. One-carbon metabolism in cancer. Br. J. Cancer. 2017, 116, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.C.; Maddocks, O.D.K. Serine and Functional Metabolites in Cancer. Trends Cell Biol. 2017, 27, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Gupta, V.; Patterson-Fortin, J.; Bhattacharya, S.; Katlinski, K.; Wu, J.; Varghese, B.; Carbone, C.J.; Aressy, B.; Fuchs, S.Y.; et al. ABRISC-SHMT complex deubiquitinates IFNAR1 and regulates interferon responses. Cell Rep. 2013, 5, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Li, A.M.; Ye, J. Reprogramming of serine, glycine and one-carbon metabolism in cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165841. [Google Scholar] [CrossRef]
- Gao, X.; Locasale, J.W. Serine Metabolism Links Tumor Suppression to the Epigenetic Landscape. Cell Metab. 2016, 24, 777–779. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer. 2016, 16, 650–662. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tramonti, A.; Cuyàs, E.; Encinar, J.A.; Pietzke, M.; Paone, A.; Verdura, S.; Arbusà, A.; Martin-Castillo, B.; Giardina, G.; Joven, J.; et al. Metformin Is a Pyridoxal-5′-phosphate (PLP)-Competitive Inhibitor of SHMT2. Cancers 2021, 13, 4009. https://doi.org/10.3390/cancers13164009
Tramonti A, Cuyàs E, Encinar JA, Pietzke M, Paone A, Verdura S, Arbusà A, Martin-Castillo B, Giardina G, Joven J, et al. Metformin Is a Pyridoxal-5′-phosphate (PLP)-Competitive Inhibitor of SHMT2. Cancers. 2021; 13(16):4009. https://doi.org/10.3390/cancers13164009
Chicago/Turabian StyleTramonti, Angela, Elisabet Cuyàs, José Antonio Encinar, Matthias Pietzke, Alessio Paone, Sara Verdura, Aina Arbusà, Begoña Martin-Castillo, Giorgio Giardina, Jorge Joven, and et al. 2021. "Metformin Is a Pyridoxal-5′-phosphate (PLP)-Competitive Inhibitor of SHMT2" Cancers 13, no. 16: 4009. https://doi.org/10.3390/cancers13164009
APA StyleTramonti, A., Cuyàs, E., Encinar, J. A., Pietzke, M., Paone, A., Verdura, S., Arbusà, A., Martin-Castillo, B., Giardina, G., Joven, J., Vazquez, A., Contestabile, R., Cutruzzolà, F., & Menendez, J. A. (2021). Metformin Is a Pyridoxal-5′-phosphate (PLP)-Competitive Inhibitor of SHMT2. Cancers, 13(16), 4009. https://doi.org/10.3390/cancers13164009