
The Emerging Roles of Protein Interactions with O-GlcNAc Cycling Enzymes in Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
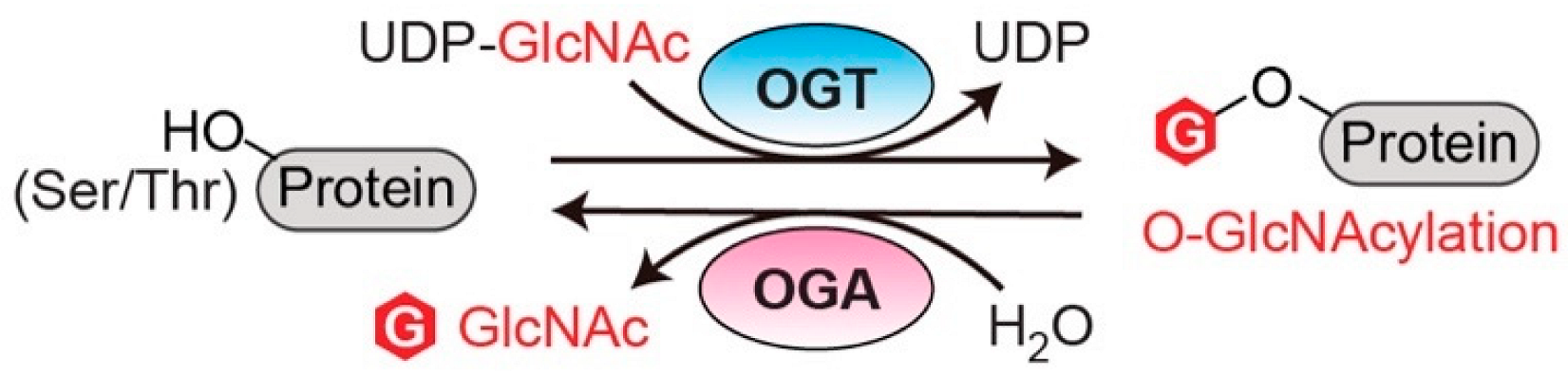
1. Introduction
2. Structural Insights of O-GlcNAc Cycling Enzymes as Potential Multi-Interface Hubs for Regulating Complex PPI Networks
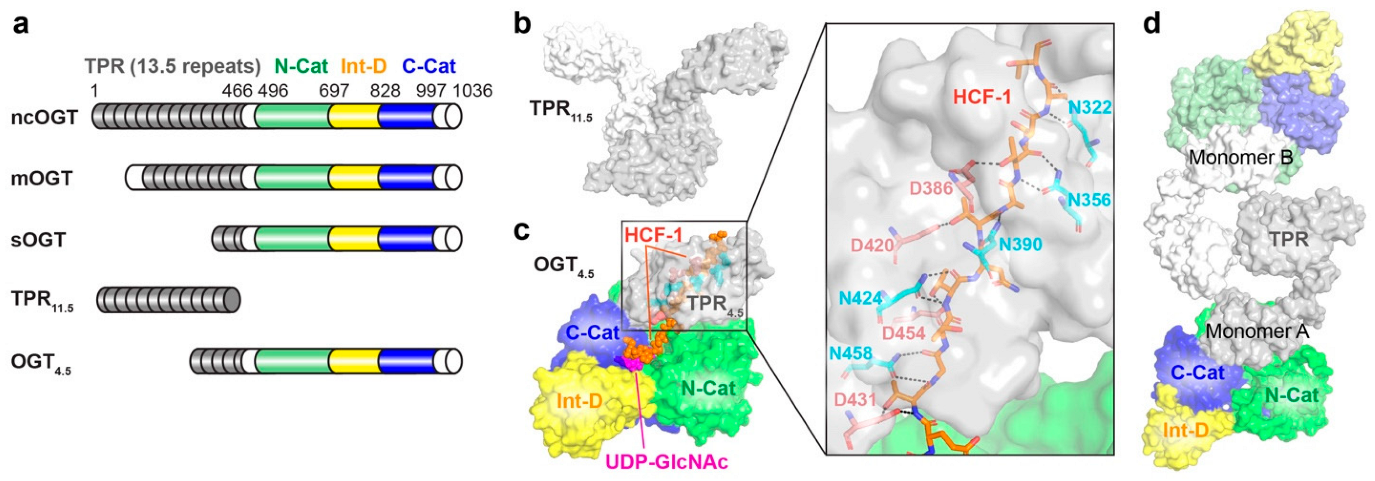
2.1. O-GlcNAc Transferase (OGT)
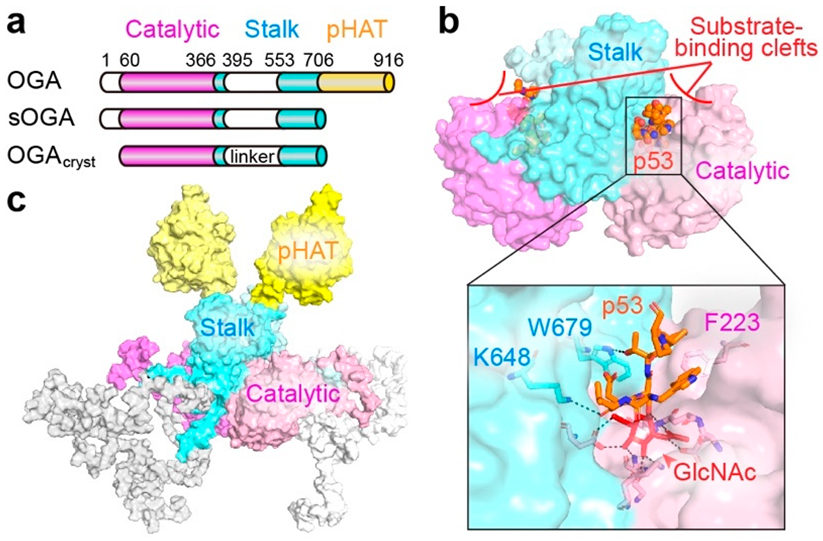
2.2. O-GlcNAcase (OGA)
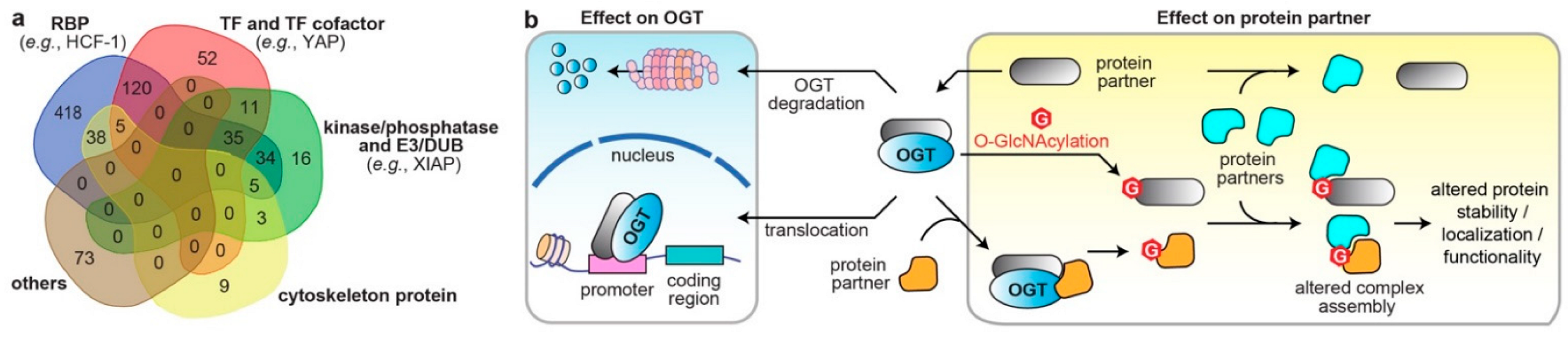
3. Systematic Analyses of OGT/OGA Associated PPI Networks in Cancer
4. Dysregulated Protein Functions by Rewired OGT/OGA Protein Networks in Cancer
4.1. Altered Cellular Localization and Protein Stability of O-GlcNAc Cycling Enzymes
4.2. Effects on the Direct Binding Partners/Substrates of O-GlcNAc Cycling Enzymes
4.3. Modulations through Binding Adaptors
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hart, G.W. Nutrient Regulation of Signaling and Transcription. J. Biol. Chem. 2019, 294, 2211–2231. [Google Scholar] [CrossRef] [Green Version]
- Haltiwanger, R.S.; Blomberg, M.A.; Hart, G.W. Glycosylation of Nuclear and Cytoplasmic Proteins. Purification and Characterization of a Uridine Diphospho-N-Acetylglucosamine:Polypeptide Beta-N-Acetylglucosaminyltransferase. J. Biol. Chem. 1992, 267, 9005–9013. [Google Scholar] [CrossRef]
- Lubas, W.A.; Frank, D.W.; Krause, M.; Hanover, J.A. O-Linked GlcNAc Transferase Is a Conserved Nucleocytoplasmic Protein Containing Tetratricopeptide Repeats. J. Biol. Chem. 1997, 272, 9316–9324. [Google Scholar] [CrossRef] [Green Version]
- Heckel, D.; Comtesse, N.; Brass, N.; Blin, N.; Zang, K.D.; Meese, E. Novel Immunogenic Antigen Homologous to Hyaluronidase in Meningioma. Hum. Mol. Genet. 1998, 7, 1859–1872. [Google Scholar] [CrossRef] [Green Version]
- Comtesse, N.; Maldener, E.; Meese, E. Identification of a Nuclear Variant of MGEA5, a Cytoplasmic Hyaluronidase and a β-N-Acetylglucosaminidase. Biochem. Biophys. Res. Commun. 2001, 283, 634–640. [Google Scholar] [CrossRef]
- Gao, Y.; Wells, L.; Comer, F.I.; Parker, G.J.; Hart, G.W. Dynamic O-Glycosylation of Nuclear and Cytosolic Proteins:Cloning and Characterization of A Neutral, Cytosolic Beta-N-Acetylglucosaminidase From Human Brain. J. Biol. Chem. 2001, 276, 9838–9845. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging Mechanisms and Functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef] [Green Version]
- Laarse, S.A.M.; Leney, A.C.; Heck, A.J.R. Crosstalk between Phosphorylation and O-GlcNAcylation: Friend or Foe. FEBS J. 2018, 285, 3152–3167. [Google Scholar] [CrossRef] [Green Version]
- Bond, M.R.; Hanover, J.A. A Little Sugar Goes a Long Way: The Cell Biology of O-GlcNAc. J. Cell Biol. 2015, 208, 869–880. [Google Scholar] [CrossRef] [Green Version]
- Xue, Q.; Yan, R.; Ji, S.; Yu, S. Regulation of Mitochondrial Network Homeostasis by O-GlcNAcylation. Mitochondrion 2022, 65, 45–55. [Google Scholar] [CrossRef]
- de Queiroz, R.M.; Carvalho, Ã.; Dias, W.B. O-GlcNAcylation: The Sweet Side of the Cancer. Front. Oncol. 2014, 4, 132. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.B.; Pyo, K.-H.; Kim, H.R. Role and Function of O-GlcNAcylation in Cancer. Cancers 2021, 13, 5365. [Google Scholar] [CrossRef]
- Fardini, Y.; Dehennaut, V.; Lefebvre, T.; Issad, T. O-GlcNAcylation: A New Cancer Hallmark? Front. Endocrinol. 2013, 4, 99. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Vosseller, K. O-GlcNAc in Cancer Biology. Amino Acids 2013, 45, 719–733. [Google Scholar] [CrossRef]
- Itkonen, H.M.; Loda, M.; Mills, I.G. O-GlcNAc Transferase—An Auxiliary Factor or a Full-Blown Oncogene? Mol. Cancer Res. 2021, 19, 555–564. [Google Scholar] [CrossRef]
- Very, N.; El Yazidi-Belkoura, I. Targeting O-GlcNAcylation to Overcome Resistance to Anti-Cancer Therapies. Front. Oncol. 2022, 12, 960312. [Google Scholar] [CrossRef]
- Lu, Q.; Zhang, X.; Liang, T.; Bai, X. O-GlcNAcylation: An Important Post-Translational Modification and a Potential Therapeutic Target for Cancer Therapy. Mol. Med. 2022, 28, 115. [Google Scholar] [CrossRef]
- Singh, J.P.; Qian, K.; Lee, J.-S.; Zhou, J.; Han, X.; Zhang, B.; Ong, Q.; Ni, W.; Jiang, M.; Ruan, H.-B.; et al. O-GlcNAcase Targets Pyruvate Kinase M2 to Regulate Tumor Growth. Oncogene 2020, 39, 560–573. [Google Scholar] [CrossRef]
- Krześlak, A.; Forma, E.; Bernaciak, M.; Romanowicz, H.; Bryś, M. Gene Expression of O-GlcNAc Cycling Enzymes in Human Breast Cancers. Clin. Exp. Med. 2012, 12, 61–65. [Google Scholar] [CrossRef] [Green Version]
- Qian, K.; Wang, S.; Fu, M.; Zhou, J.; Singh, J.P.; Li, M.-D.; Yang, Y.; Zhang, K.; Wu, J.; Nie, Y.; et al. Transcriptional Regulation of O-GlcNAc Homeostasis Is Disrupted in Pancreatic Cancer. J. Biol. Chem. 2018, 293, 13989–14000. [Google Scholar] [CrossRef]
- Krzeslak, A.; Pomorski, L.; Lipinska, A. Elevation of Nucleocytoplasmic β-N-Acetylglucosaminidase (O-GlcNAcase) Activity in Thyroid Cancers. Int. Mol. Med. 2010, 25, 643–648. [Google Scholar] [CrossRef] [Green Version]
- Ding, N.; Ping, L.; Shi, Y.; Feng, L.; Zheng, X.; Song, Y.; Zhu, J. Thiamet-G-Mediated Inhibition of O-GlcNAcase Sensitizes Human Leukemia Cells to Microtubule-Stabilizing Agent Paclitaxel. Biochem. Biophys. Res. Commun. 2014, 453, 392–397. [Google Scholar] [CrossRef]
- Luanpitpong, S.; Chanthra, N.; Janan, M.; Poohadsuan, J.; Samart, P.; U-Pratya, Y.; Rojanasakul, Y.; Issaragrisil, S. Inhibition of O -GlcNAcase Sensitizes Apoptosis and Reverses Bortezomib Resistance in Mantle Cell Lymphoma through Modification of Truncated Bid. Mol. Cancer Ther. 2018, 17, 484–496. [Google Scholar] [CrossRef] [Green Version]
- Very, N.; Hardivillé, S.; Decourcelle, A.; Thévenet, J.; Djouina, M.; Page, A.; Vergoten, G.; Schulz, C.; Kerr-Conte, J.; Lefebvre, T.; et al. Thymidylate Synthase O-GlcNAcylation: A Molecular Mechanism of 5-FU Sensitization in Colorectal Cancer. Oncogene 2022, 41, 745–756. [Google Scholar] [CrossRef]
- Starska, K.; Forma, E.; Brzezińska-Błaszczyk, E.; Lewy-Trenda, I.; Bryś, M.; Jóźwiak, P.; Krześlak, A. Gene and Protein Expression of O-GlcNAc-Cycling Enzymes in Human Laryngeal Cancer. Clin. Exp. Med. 2015, 15, 455–468. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.R.; Jang, H.-J.; Yoon, S.; Lee, Y.H.; Nam, D.; Kim, I.S.; Lee, H.; Kim, H.; Choi, J.H.; Kang, B.H.; et al. OGA Heterozygosity Suppresses Intestinal Tumorigenesis in Apcmin/+ Mice. Oncogenesis 2014, 3, e109. [Google Scholar] [CrossRef] [Green Version]
- Jóźwiak, P.; Forma, E.; Bryś, M.; Anna Krześlak, A. O-GlcNAcylation and Metabolic Reprograming in Cancer. Front. Endocrinol. 2014, 5, 145. [Google Scholar] [CrossRef] [Green Version]
- Olivier-Van Stichelen, S.; Hanover, J.A. You Are What You Eat: O-Linked N-Acetylglucosamine in Disease, Development and Epigenetics. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 339–345. [Google Scholar] [CrossRef]
- Parker, M.P.; Peterson, K.R.; Slawson, C. O-GlcNAcylation and O-GlcNAc Cycling Regulate Gene Transcription: Emerging Roles in Cancer. Cancers 2021, 13, 1666. [Google Scholar] [CrossRef]
- Ouyang, M.; Yu, C.; Deng, X.; Zhang, Y.; Zhang, X.; Duan, F. O-GlcNAcylation and Its Role in Cancer-Associated Inflammation. Front. Immunol. 2022, 13, 861559. [Google Scholar] [CrossRef]
- Slawson, C.; Hart, G.W. O-GlcNAc Signalling: Implications for Cancer Cell Biology. Nat. Rev. Cancer 2011, 11, 678–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.R.; Song, M.; Lee, H.; Jeon, Y.; Choi, E.-J.; Jang, H.-J.; Moon, H.Y.; Byun, H.-Y.; Kim, E.-K.; Kim, D.H.; et al. O-GlcNAcase Is Essential for Embryonic Development and Maintenance of Genomic Stability: Genomic Instability by Dysregulation O-GlcNAcylation. Aging Cell 2012, 11, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivier-Van Stichelen, S.; Abramowitz, L.K.; Hanover, J.A. X Marks the Spot: Does It Matter That O-GlcNAc Transferase Is an X-Linked Gene? Biochem. Biophys. Res. Commun. 2014, 453, 201–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkovskaya, A.; Seip, K.; Hilmarsdottir, B.; Maelandsmo, G.M.; Moestue, S.A.; Itkonen, H.M. O-GlcNAc Transferase Inhibition Differentially Affects Breast Cancer Subtypes. Sci. Rep. 2019, 9, 5670. [Google Scholar] [CrossRef] [Green Version]
- Trapannone, R.; Rafie, K.; van Aalten, D.M.F. O-GlcNAc Transferase Inhibitors: Current Tools and Future Challenges. Biohem. Soc. Trans. 2016, 44, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Muha, V.; Authier, F.; Szoke-Kovacs, Z.; Johnson, S.; Gallagher, J.; McNeilly, A.; McCrimmon, R.J.; Teboul, L.; van Aalten, D.M.F. Loss of O-GlcNAcase Catalytic Activity Leads to Defects in Mouse Embryogenesis. J. Biol. Chem. 2021, 296, 100439. [Google Scholar] [CrossRef]
- Huynh, V.N.; Wang, S.; Ouyang, X.; Wani, W.Y.; Johnson, M.S.; Chacko, B.K.; Jegga, A.G.; Qian, W.-J.; Chatham, J.C.; Darley-Usmar, V.M.; et al. Defining the Dynamic Regulation of O-GlcNAc Proteome in the Mouse Cortex—The O-GlcNAcylation of Synaptic and Trafficking Proteins Related to Neurodegenerative Diseases. Front. Aging 2021, 2, 757801. [Google Scholar] [CrossRef]
- Levine, Z.G.; Potter, S.C.; Joiner, C.M.; Fei, G.Q.; Nabet, B.; Sonnett, M.; Zachara, N.E.; Gray, N.S.; Paulo, J.A.; Walker, S. Mammalian Cell Proliferation Requires Noncatalytic Functions of O-GlcNAc Transferase. Proc. Natl. Acad. Sci. USA 2021, 118, e2016778118. [Google Scholar] [CrossRef]
- Gulati, S.; Cheng, T.M.K.; Bates, P.A. Cancer Networks and beyond: Interpreting Mutations Using the Human Interactome and Protein Structure. Semin. Cancer Biol. 2013, 23, 219–226. [Google Scholar] [CrossRef]
- Yoon, T.-Y.; Lee, H.-W. Shedding Light on Complexity of Protein–Protein Interactions in Cancer. Curr. Opin. Chem. Biol. 2019, 53, 75–81. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent Advances in the Development of Protein–Protein Interactions Modulators: Mechanisms and Clinical Trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Worth, M.; Li, H.; Jiang, J. Chemical and Biochemical Strategies To Explore the Substrate Recognition of O-GlcNAc-Cycling Enzymes. ChemBioChem 2019, 20, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Cai, M.; Xing, X.; Ji, J.; Yang, E.; Wu, J. PINA 3.0: Mining Cancer Interactome. Nucleic Acids Res. 2021, 49, D1351–D1357. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Chen, K.; Zhong, C.; Zhu, S.; Ma, X. Network-Based Protein-Protein Interaction Prediction Method Maps Perturbations of Cancer Interactome. PLoS Genet. 2021, 17, e1009869. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Davé, V.; Iakoucheva, L.M.; Malaney, P.; Metallo, S.J.; Pathak, R.R.; Joerger, A.C. Pathological Unfoldomics of Uncontrolled Chaos: Intrinsically Disordered Proteins and Human Diseases. Chem. Rev. 2014, 114, 6844–6879. [Google Scholar] [CrossRef] [Green Version]
- Kar, G.; Gursoy, A.; Keskin, O. Human Cancer Protein-Protein Interaction Network: A Structural Perspective. PLoS Comput. Biol. 2009, 5, e1000601. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, I.; Nakajima, Y.; Ito, M.; Fukuchi, S.; Homma, K.; Nishikawa, K. Computational Prediction of O-Linked Glycosylation Sites That Preferentially Map on Intrinsically Disordered Regions of Extracellular Proteins. IJMS 2010, 11, 4991–5008. [Google Scholar] [CrossRef]
- Hanover, J.A.; Yu, S.; Lubas, W.B.; Shin, S.H.; Ragano-Caracciola, M.; Kochran, J.; Love, D.C. Mitochondrial and Nucleocytoplasmic Isoforms of O-Linked GlcNAc Transferase Encoded by a Single Mammalian Gene. Arch. Biochem. Biophys. 2003, 409, 287–297. [Google Scholar] [CrossRef]
- Love, D.C.; Kochan, J.; Cathey, R.L.; Shin, S.-H.; Hanover, J.A.; Kochran, J. Mitochondrial and Nucleocytoplasmic Targeting of O-Linked GlcNAc Transferase. J. Cell Sci. 2003, 116, 647–654. [Google Scholar] [CrossRef] [Green Version]
- Zeytuni, N.; Zarivach, R. Structural and Functional Discussion of the Tetra-Trico-Peptide Repeat, a Protein Interaction Module. Structure 2012, 20, 397–405. [Google Scholar] [CrossRef]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, Functions, and Mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jínek, M.; Rehwinkel, J.; Lazarus, B.D.; Izaurralde, E.; Hanover, J.A.; Conti, E. The Superhelical TPR-Repeat Domain of O-Linked GlcNAc Transferase Exhibits Structural Similarities to Importin α. Nat. Struct. Mol. Biol. 2004, 11, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Rafie, K.; Raimi, O.; Ferenbach, A.T.; Borodkin, V.S.; Kapuria, V.; van Aalten, D.M.F. Recognition of a Glycosylation Substrate by the O-GlcNAc Transferase TPR Repeats. Open Biol. 2017, 7, 170078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, Z.G.; Walker, S. The Biochemistry of O-GlcNAc Transferase: Which Functions Make It Essential in Mammalian Cells? Annu. Rev. Biochem. 2016, 85, 631–657. [Google Scholar] [CrossRef]
- Nagel, A.K.; Ball, L.E. O-GlcNAc Transferase and O-GlcNAcase: Achieving Target Substrate Specificity. Amino Acids 2014, 46, 2305–2316. [Google Scholar] [CrossRef] [Green Version]
- Stephen, H.M.; Adams, T.M.; Wells, L. Regulating the Regulators: Mechanisms of Substrate Selection of the O-GlcNAc Cycling Enzymes OGT and OGA. Glycobiology 2021, 31, 724–733. [Google Scholar] [CrossRef]
- Lazarus, M.B.; Nam, Y.; Jiang, J.; Sliz, P.; Walker, S. Structure of Human O-GlcNAc Transferase and Its Complex with a Peptide Substrate. Nature 2011, 469, 564–567. [Google Scholar] [CrossRef]
- Lazarus, M.B.; Jiang, J.; Kapuria, V.; Bhuiyan, T.; Janetzko, J.; Zandberg, W.F.; Vocadlo, D.J.; Herr, W.; Walker, S. HCF-1 Is Cleaved in the Active Site of O-GlcNAc Transferase. Science 2013, 342, 1235–1239. [Google Scholar] [CrossRef] [Green Version]
- Levine, Z.G.; Fan, C.; Melicher, M.S.; Orman, M.; Benjamin, T.; Walker, S. O-GlcNAc Transferase Recognizes Protein Substrates Using an Asparagine Ladder in the Tetratricopeptide Repeat (TPR) Superhelix. J. Am. Chem. Soc. 2018, 140, 3510–3513. [Google Scholar] [CrossRef]
- Joiner, C.M.; Levine, Z.G.; Aonbangkhen, C.; Woo, C.M.; Walker, S. Aspartate Residues Far from the Active Site Drive O-GlcNAc Transferase Substrate Selection. J. Am. Chem. Soc. 2019, 141, 12974–12978. [Google Scholar] [CrossRef]
- Joiner, C.M.; Hammel, F.A.; Janetzko, J.; Walker, S. Protein Substrates Engage the Lumen of O-GlcNAc Transferase’s Tetratricopeptide Repeat Domain in Different Ways. Biochemistry 2021, 60, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Kositzke, A.; Fan, D.; Wang, A.; Li, H.; Worth, M.; Jiang, J. Elucidating the Protein Substrate Recognition of O-GlcNAc Transferase (OGT) toward O-GlcNAcase (OGA) Using a GlcNAc Electrophilic Probe. Int. J. Biol. Macromol. 2021, 169, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Kreppel, L.K.; Blomberg, M.A.; Hart, G.W. Dynamic Glycosylation of Nuclear and Cytosolic Proteins. J. Biol. Chem. 1997, 272, 9308–9315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Ongusaha, P.P.; Miles, P.D.; Havstad, J.C.; Zhang, F.; So, W.V.; Kudlow, J.E.; Michell, R.H.; Olefsky, J.M.; Field, S.J.; et al. Phosphoinositide Signalling Links O-GlcNAc Transferase to Insulin Resistance. Nature 2008, 451, 964–969. [Google Scholar] [CrossRef] [PubMed]
- Meek, R.W.; Blaza, J.N.; Busmann, J.A.; Alteen, M.G.; Vocadlo, D.J.; Davies, G.J. Cryo-EM Structure Provides Insights into the Dimer Arrangement of the O-Linked β-N-Acetylglucosamine Transferase OGT. Nat. Commun. 2021, 12, 6508. [Google Scholar] [CrossRef]
- Marianayagam, N.J.; Sunde, M.; Matthews, J.M. The Power of Two: Protein Dimerization in Biology. Trends Biochem. Sci. 2004, 29, 618–625. [Google Scholar] [CrossRef]
- Joiner, C.M.; Li, H.; Jiang, J.; Walker, S. Structural Characterization of the O-GlcNAc Cycling Enzymes: Insights into Substrate Recognition and Catalytic Mechanisms. Curr. Opin. Struct. Biol. 2019, 56, 97–106. [Google Scholar] [CrossRef]
- Elsen, N.L.; Patel, S.B.; Ford, R.E.; Hall, D.L.; Hess, F.; Kandula, H.; Kornienko, M.; Reid, J.; Selnick, H.; Shipman, J.M.; et al. Insights into Activity and Inhibition from the Crystal Structure of Human O-GlcNAcase. Nat. Chem. Biol. 2017, 13, 613–615. [Google Scholar] [CrossRef]
- Li, B.; Li, H.; Lu, L.; Jiang, J. Structures of Human O-GlcNAcase and Its Complexes Reveal a New Substrate Recognition Mode. Nat. Struct. Mol. Biol. 2017, 24, 362–369. [Google Scholar] [CrossRef]
- Roth, C.; Chan, S.; Offen, W.A.; Hemsworth, G.R.; Willems, L.I.; King, D.T.; Varghese, V.; Britton, R.; Vocadlo, D.J.; Davies, G.J. Structural and Functional Insight into Human O-GlcNAcase. Nat. Chem. Biol. 2017, 13, 610–612. [Google Scholar] [CrossRef]
- Rao, F.V.; Dorfmueller, H.C.; Villa, F.; Allwood, M.; Eggleston, I.M.; van Aalten, D.M.F. Structural Insights into the Mechanism and Inhibition of Eukaryotic O-GlcNAc Hydrolysis. EMBO J. 2006, 25, 1569–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, R.J.; Taylor, E.J.; Macauley, M.S.; Stubbs, K.A.; Turkenburg, J.P.; Hart, S.J.; Black, G.N.; Vocadlo, D.J.; Davies, G.J. Structure and Mechanism of a Bacterial β-Glucosaminidase Having O-GlcNAcase Activity. Nat. Struct. Mol. Biol. 2006, 13, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Schimpl, M.; Schüttelkopf, A.W.; Borodkin, V.S.; van Aalten, D.M.F. Human OGA Binds Substrates in a Conserved Peptide Recognition Groove. Biochem. J. 2010, 432, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, A.; Gundogdu, M.; Ferenbach, A.T.; Lebedev, A.A.; van Aalten, D.M.F. Evidence for a Functional O-Linked N-Acetylglucosamine (O-GlcNAc) System in the Thermophilic Bacterium Thermobaculum Terrenum. J. Biol. Chem. 2015, 290, 30291–30305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Li, H.; Hu, C.-W.; Jiang, J. Structural Insights into the Substrate Binding Adaptability and Specificity of Human O-GlcNAcase. Nat. Commun. 2017, 8, 666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Tong, M.; Suttapitugsakul, S.; Wu, R. Spatial and Temporal Proteomics Reveals the Distinct Distributions and Dynamics of O-GlcNAcylated Proteins. Cell Rep. 2022, 39, 110946. [Google Scholar] [CrossRef]
- Cortese, M.S.; Uversky, V.N.; Keith Dunker, A. Intrinsic Disorder in Scaffold Proteins: Getting More from Less. Prog. Biophys. Mol. Biol. 2008, 98, 85–106. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible Nets. The Roles of Intrinsic Disorder in Protein Interaction Networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef]
- Butkinaree, C.; Cheung, W.D.; Park, S.; Park, K.; Barber, M.; Hart, G.W. Characterization of β-N-Acetylglucosaminidase Cleavage by Caspase-3 during Apoptosis. J. Biol. Chem. 2008, 283, 23557–23566. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. 20 Years of the SMART Protein Domain Annotation Resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- Rao, F.V.; Schüttelkopf, A.W.; Dorfmueller, H.C.; Ferenbach, A.T.; Navratilova, I.; van Aalten, D.M.F. Structure of a Bacterial Putative Acetyltransferase Defines the Fold of the Human O-GlcNAcase C-Terminal Domain. Open Biol. 2013, 3, 130021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Roth, C.; Turkenburg, J.P.; Davies, G.J. Three-Dimensional Structure of a Streptomyces Sviceus GNAT Acetyltransferase with Similarity to the C-Terminal Domain of the Human GH84 O-GlcNAcase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 186–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macauley, M.S.; Vocadlo, D.J. Enzymatic Characterization and Inhibition of the Nuclear Variant of Human O-GlcNAcase. Carbohydr. Res. 2009, 344, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Keembiyehetty, C.N.; Krzeslak, A.; Love, D.C.; Hanover, J.A. A Lipid-Droplet-Targeted O-GlcNAcase Isoform Is a Key Regulator of the Proteasome. J. Cell Sci. 2011, 124, 2851–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grechkin, M.; Logsdon, B.A.; Gentles, A.J.; Lee, S.-I. Identifying Network Perturbation in Cancer. PLoS Comput. Biol. 2016, 12, e1004888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, R.-P.; He, X.; Guo, S.-J.; Liu, W.-F.; Tao, Y.; Tao, S.-C. Global Identification of O-GlcNAc Transferase (OGT) Interactors by a Human Proteome Microarray and the Construction of an OGT Interactome. Proteomics 2014, 14, 1020–1030. [Google Scholar] [CrossRef]
- Martinez, M.; Renuse, S.; Kreimer, S.; O’Meally, R.; Natov, P.; Madugundu, A.K.; Nirujogi, R.S.; Tahir, R.; Cole, R.; Pandey, A.; et al. Quantitative Proteomics Reveals That the OGT Interactome Is Remodeled in Response to Oxidative Stress. Mol. Cell. Proteom. 2021, 20, 100069. [Google Scholar] [CrossRef]
- Ma, J.; Hou, C.; Li, Y.; Chen, S.; Wu, C. OGT Protein Interaction Network (OGT-PIN): A Curated Database of Experimentally Identified Interaction Proteins of OGT. IJMS 2021, 22, 9620. [Google Scholar] [CrossRef]
- Morris, J.H.; Knudsen, G.M.; Verschueren, E.; Johnson, J.R.; Cimermancic, P.; Greninger, A.L.; Pico, A.R. Affinity Purification–Mass Spectrometry and Network Analysis to Understand Protein-Protein Interactions. Nat. Protoc. 2014, 9, 2539–2554. [Google Scholar] [CrossRef] [Green Version]
- Sears, R.M.; May, D.G.; Roux, K.J. BioID as a Tool for Protein-Proximity Labeling in Living Cells. In Enzyme-Mediated Ligation Methods; Nuijens, T., Schmidt, M., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 2012, pp. 299–313. ISBN 978-1-4939-9545-5. [Google Scholar]
- Jóźwiak, P.; Ciesielski, P.; Zakrzewski, P.K.; Kozal, K.; Oracz, J.; Budryn, G.; Żyżelewicz, D.; Flament, S.; Vercoutter-Edouart, A.-S.; Bray, F.; et al. Mitochondrial O-GlcNAc Transferase Interacts with and Modifies Many Proteins and Its Up-Regulation Affects Mitochondrial Function and Cellular Energy Homeostasis. Cancers 2021, 13, 2956. [Google Scholar] [CrossRef]
- Lazarus, B.D.; Love, D.C.; Hanover, J.A. Recombinant O-GlcNAc Transferase Isoforms: Identification of O-GlcNAcase, Yes Tyrosine Kinase, and Tau as Isoform-Specific Substrates. Glycobiology 2006, 16, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.-H.; Love, D.C.; Hanover, J.A. Elevated O-GlcNAc-Dependent Signaling through Inducible MOGT Expression Selectively Triggers Apoptosis. Amino Acids 2011, 40, 885–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacoman, J.L.; Dagda, R.Y.; Burnham-Marusich, A.R.; Dagda, R.K.; Berninsone, P.M. Mitochondrial O-GlcNAc Transferase (MOGT) Regulates Mitochondrial Structure, Function, and Survival in HeLa Cells. J. Biol. Chem. 2017, 292, 4499–4518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groves, J.A.; Maduka, A.O.; O’Meally, R.N.; Cole, R.N.; Zachara, N.E. Fatty Acid Synthase Inhibits the O-GlcNAcase during Oxidative Stress. J. Biol. Chem. 2017, 292, 6493–6511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A Web Server for Functional Enrichment Analysis and Functional Annotation of Gene Lists (2021 Update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Hu, H.; Miao, Y.-R.; Jia, L.-H.; Yu, Q.-Y.; Zhang, Q.; Guo, A.-Y. AnimalTFDB 3.0: A Comprehensive Resource for Annotation and Prediction of Animal Transcription Factors. Nucleic Acids Res. 2019, 47, D33–D38. [Google Scholar] [CrossRef]
- Eid, S.; Turk, S.; Volkamer, A.; Rippmann, F.; Fulle, S. KinMap: A Web-Based Tool for Interactive Navigation through Human Kinome Data. BMC Bioinform. 2017, 18, 16. [Google Scholar] [CrossRef] [Green Version]
- Damle, N.P.; Köhn, M. The Human DEPhOsphorylation Database DEPOD: 2019 Update. Database 2019, 2019, baz133. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; He, M.; Kong, X.; Jiang, P.; Liu, X.; Diao, L.; Zhang, X.; Li, H.; Ling, X.; et al. UbiBrowser 2.0: A Comprehensive Resource for Proteome-Wide Known and Predicted Ubiquitin Ligase/Deubiquitinase–Substrate Interactions in Eukaryotic Species. Nucleic Acids Res. 2022, 50, D719–D728. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, S.; Jhong, J.-H.; Pang, Y.; Huang, K.-Y.; Li, S.; Lee, T.-Y. UbiNet 2.0: A Verified, Classified, Annotated and Updated Database of E3 Ubiquitin Ligase–Substrate Interactions. Database 2021, 2021, baab010. [Google Scholar] [CrossRef] [PubMed]
- Caudron-Herger, M.; Jansen, R.E.; Wassmer, E.; Diederichs, S. RBP2GO: A Comprehensive Pan-Species Database on RNA-Binding Proteins, Their Interactions and Functions. Nucleic Acids Res. 2021, 49, D425–D436. [Google Scholar] [CrossRef]
- Hanover, J.A.; Krause, M.W.; Love, D.C. Linking Metabolism to Epigenetics through O-GlcNAcylation. Nat. Rev. Mol. Cell Biol. 2012, 13, 312–321. [Google Scholar] [CrossRef]
- Voigt, P.; Tee, W.-W.; Reinberg, D. A Double Take on Bivalent Promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Chen, Y.; Bian, C.; Fujiki, R.; Yu, X. TET2 Promotes Histone O-GlcNAcylation during Gene Transcription. Nature 2013, 493, 561–564. [Google Scholar] [CrossRef] [Green Version]
- Deplus, R.; Delatte, B.; Schwinn, M.K.; Defrance, M.; Méndez, J.; Murphy, N.; Dawson, M.A.; Volkmar, M.; Putmans, P.; Calonne, E.; et al. TET2 and TET3 Regulate GlcNAcylation and H3K4 Methylation through OGT and SET1/COMPASS. EMBO J. 2013, 32, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Katsura, S.; Shimada, H.; Tsuchiya, H.; Hada, M.; Okumura, T.; Sugawara, A.; Yokoyama, A. TET3-OGT Interaction Increases the Stability and the Presence of OGT in Chromatin. Genes Cells 2014, 19, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, F.; Kudlow, J.E. Recruitment of O-GlcNAc Transferase to Promoters by Corepressor MSin3A. Cell 2002, 110, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Vella, P.; Scelfo, A.; Jammula, S.; Chiacchiera, F.; Williams, K.; Cuomo, A.; Roberto, A.; Christensen, J.; Bonaldi, T.; Helin, K.; et al. Tet Proteins Connect the O-Linked N-Acetylglucosamine Transferase Ogt to Chromatin in Embryonic Stem Cells. Mol. Cell 2013, 49, 645–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannenberg, J.-H.; David, G.; Zhong, S.; van der Torre, J.; Wong, W.H.; DePinho, R.A. MSin3A Corepressor Regulates Diverse Transcriptional Networks Governing Normal and Neoplastic Growth and Survival. Genes Dev. 2005, 19, 1581–1595. [Google Scholar] [CrossRef]
- Yang, Y.; Yin, X.; Yang, H.; Xu, Y. Histone Demethylase LSD2 Acts as an E3 Ubiquitin Ligase and Inhibits Cancer Cell Growth through Promoting Proteasomal Degradation of OGT. Mol. Cell 2015, 58, 47–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, H.G.; Kim, H.B.; Yoon, J.Y.; Kweon, T.H.; Park, Y.S.; Kang, J.; Jung, J.; Son, S.; Yi, E.C.; Lee, T.H.; et al. Mutual Regulation between OGT and XIAP to Control Colon Cancer Cell Growth and Invasion. Cell Death Dis. 2020, 11, 815. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Robichaud, N.; Sonenberg, N.; Ruggero, D.; Schneider, R.J. Translational Control in Cancer. Cold Spring Harb. Perspect. Biol. 2019, 11, a032896. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Su, K.; Roos, M.D.; Chang, Q.; Paterson, A.J.; Kudlow, J.E. O-Linkage of N-Acetylglucosamine to Sp1 Activation Domain Inhibits Its Transcriptional Capability. Proc. Natl. Acad. Sci. USA 2001, 98, 6611–6616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Housley, M.P.; Rodgers, J.T.; Udeshi, N.D.; Kelly, T.J.; Shabanowitz, J.; Hunt, D.F.; Puigserver, P.; Hart, G.W. O-GlcNAc Regulates FoxO Activation in Response to Glucose. J. Biol. Chem. 2008, 283, 16283–16292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.H.; Kim, J.E.; Nam, H.W.; Ju, J.W.; Kim, H.S.; Kim, Y.S.; Cho, J.W. Modification of P53 with O-Linked N-Acetylglucosamine Regulates P53 Activity and Stability. Nat. Cell Biol. 2006, 8, 1074–1083. [Google Scholar] [CrossRef]
- Ramakrishnan, P.; Clark, P.M.; Mason, D.E.; Peters, E.C.; Hsieh-Wilson, L.C.; Baltimore, D. Activation of the Transcriptional Function of the NF-ΚB Protein c-Rel by O-GlcNAc Glycosylation. Sci. Signal. 2013, 6, ra75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Li, Y.; Zhu, K.S.; Wang, H.; Zhu, W.-G. Advances in Cellular Characterization of the Sirtuin Isoform, SIRT7. Front. Endocrinol. 2018, 9, 652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Li, Y.; Chen, Q.; Zheng, L.; Lou, J.; Lin, C.; Gong, J.; Zhu, Y.; Wu, Y. O-GlcNAcylation and Stablization of SIRT7 Promote Pancreatic Cancer Progression by Blocking the SIRT7-REGγ Interaction. Cell Death Differ. 2022, 29, 1970–1981. [Google Scholar] [CrossRef]
- Duan, F.; Wu, H.; Jia, D.; Wu, W.; Ren, S.; Wang, L.; Song, S.; Guo, X.; Liu, F.; Ruan, Y.; et al. O-GlcNAcylation of RACK1 Promotes Hepatocellular Carcinogenesis. J. Hepatol. 2018, 68, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zhu, Y.; Zhang, W.; Liao, Q.; Chen, Y.; Zhao, X.; Guo, Q.; Shen, P.; Zhen, B.; Qian, X.; et al. Regulation of the Hippo-YAP Pathway by Glucose Sensor O-GlcNAcylation. Mol. Cell 2017, 68, 591–604.e5. [Google Scholar] [CrossRef]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 2019, 5, 297–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X. Tumor Suppressor LATS1 Is a Negative Regulator of Oncogene YAP. J. Biol. Chem. 2008, 283, 5496–5509. [Google Scholar] [CrossRef] [Green Version]
- Blum, R.H. Adriamycin: A New Anticancer Drug with Significant Clinical Activity. Ann. Intern. Med. 1974, 80, 249. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Gui, B.; Kumar, R. Role of MTA1 in Cancer Progression and Metastasis. Cancer Metastasis Rev. 2014, 33, 879–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Wu, Q.; Zhang, K.; Liu, Y.; Zhang, N.; Chen, Q.; Wang, L.; Li, W.; Zhang, J.; Liu, Y. O-GlcNAc Modification Regulates MTA1 Transcriptional Activity during Breast Cancer Cell Genotoxic Adaptation. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129930. [Google Scholar] [CrossRef]
- King, D.T.; Serrano-Negrón, J.E.; Zhu, Y.; Moore, C.L.; Shoulders, M.D.; Foster, L.J.; Vocadlo, D.J. Thermal Proteome Profiling Reveals the O-GlcNAc-Dependent Meltome. J. Am. Chem. Soc. 2022, 144, 3833–3842. [Google Scholar] [CrossRef]
- Ruan, H.-B.; Han, X.; Li, M.-D.; Singh, J.P.; Qian, K.; Azarhoush, S.; Zhao, L.; Bennett, A.M.; Samuel, V.T.; Wu, J.; et al. O-GlcNAc Transferase/Host Cell Factor C1 Complex Regulates Gluconeogenesis by Modulating PGC-1α Stability. Cell Metab. 2012, 16, 226–237. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Qin, J.; Zhao, J.; Li, J.; Li, D.; Popp, M.; Popp, F.; Alakus, H.; Kong, B.; Dong, Q.; et al. Inflammatory IFIT3 Renders Chemotherapy Resistance by Regulating Post-Translational Modification of VDAC2 in Pancreatic Cancer. Theranostics 2020, 10, 7178–7192. [Google Scholar] [CrossRef] [PubMed]
- Chin, H.S.; Li, M.X.; Tan, I.K.L.; Ninnis, R.L.; Reljic, B.; Scicluna, K.; Dagley, L.F.; Sandow, J.J.; Kelly, G.L.; Samson, A.L.; et al. VDAC2 Enables BAX to Mediate Apoptosis and Limit Tumor Development. Nat. Commun. 2018, 9, 4976. [Google Scholar] [CrossRef] [Green Version]
- Hua, Q.; Zhang, B.; Xu, G.; Wang, L.; Wang, H.; Lin, Z.; Yu, D.; Ren, J.; Zhang, D.; Zhao, L.; et al. CEMIP, a Novel Adaptor Protein of OGT, Promotes Colorectal Cancer Metastasis through Glutamine Metabolic Reprogramming via Reciprocal Regulation of β-Catenin. Oncogene 2021, 40, 6443–6455. [Google Scholar] [CrossRef] [PubMed]
- Pon, J.R.; Marra, M.A. Driver and Passenger Mutations in Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The Cancer Genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Sahni, N.; Yi, S.; Taipale, M.; Fuxman Bass, J.I.; Coulombe-Huntington, J.; Yang, F.; Peng, J.; Weile, J.; Karras, G.I.; Wang, Y.; et al. Widespread Macromolecular Interaction Perturbations in Human Genetic Disorders. Cell 2015, 161, 647–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Zhao, J.; Wang, Y.; Lu, W.; Liu, Z.; Zhou, Y.; Martin, W.R.; Wang, R.; Huang, J.; Hao, T.; et al. Comprehensive Characterization of Protein–Protein Interactions Perturbed by Disease Mutations. Nat. Genet. 2021, 53, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, C.-W.; Xie, J.; Jiang, J. The Emerging Roles of Protein Interactions with O-GlcNAc Cycling Enzymes in Cancer. Cancers 2022, 14, 5135. https://doi.org/10.3390/cancers14205135
Hu C-W, Xie J, Jiang J. The Emerging Roles of Protein Interactions with O-GlcNAc Cycling Enzymes in Cancer. Cancers. 2022; 14(20):5135. https://doi.org/10.3390/cancers14205135
Chicago/Turabian StyleHu, Chia-Wei, Jinshan Xie, and Jiaoyang Jiang. 2022. "The Emerging Roles of Protein Interactions with O-GlcNAc Cycling Enzymes in Cancer" Cancers 14, no. 20: 5135. https://doi.org/10.3390/cancers14205135
APA StyleHu, C. -W., Xie, J., & Jiang, J. (2022). The Emerging Roles of Protein Interactions with O-GlcNAc Cycling Enzymes in Cancer. Cancers, 14(20), 5135. https://doi.org/10.3390/cancers14205135