
Hepatocellular Carcinoma: Molecular Pathogenesis and Therapeutic Advances

 , ,
, ,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Hepatocellular Carcinoma
1.1. Epidemiology
1.2. Molecular Pathogenesis
1.2.1. Cellular Origin
1.2.2. Molecular Drivers
1.2.3. Molecular Classes
1.3. Surveillance
1.4. Diagnosis
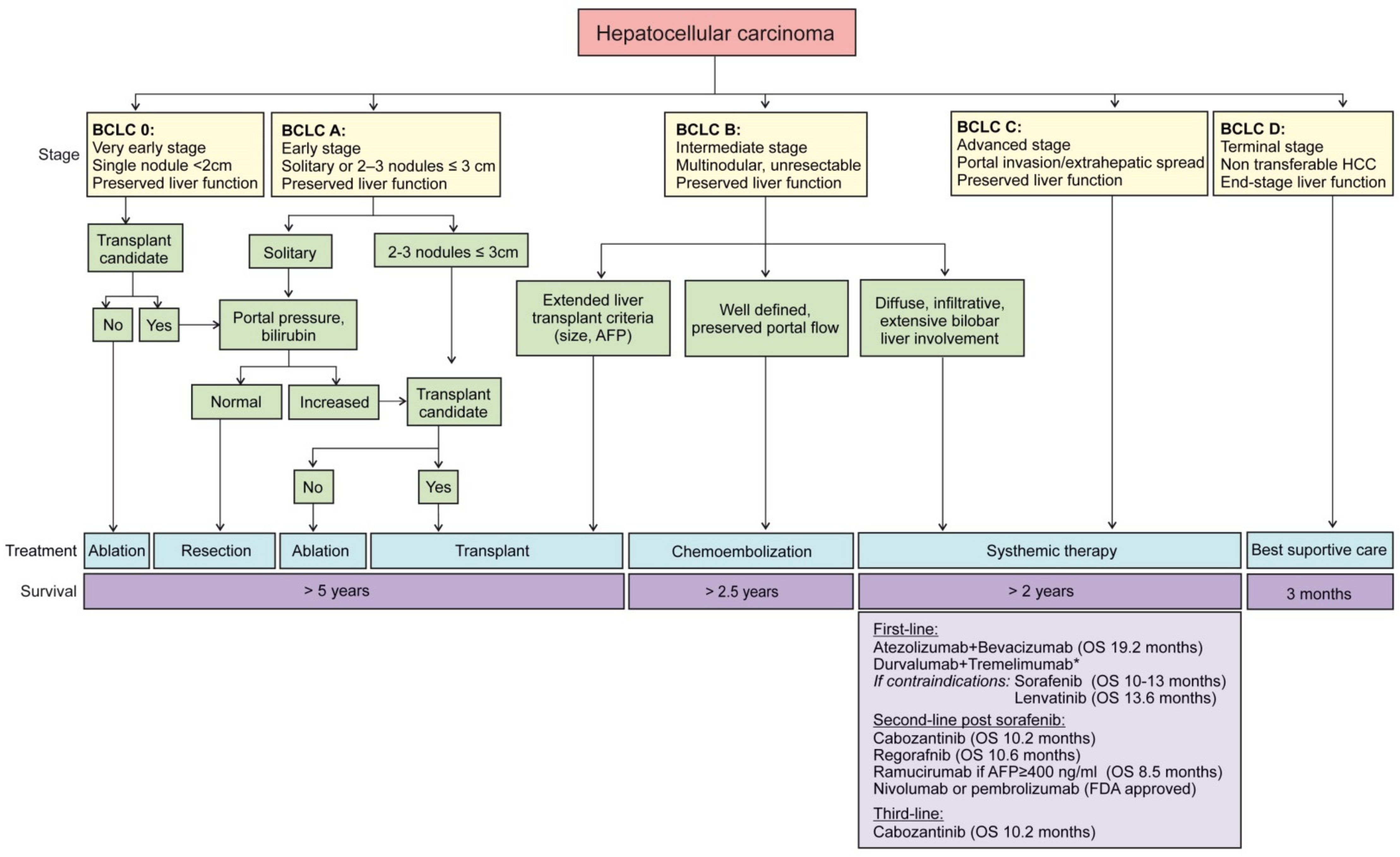
1.5. Staging
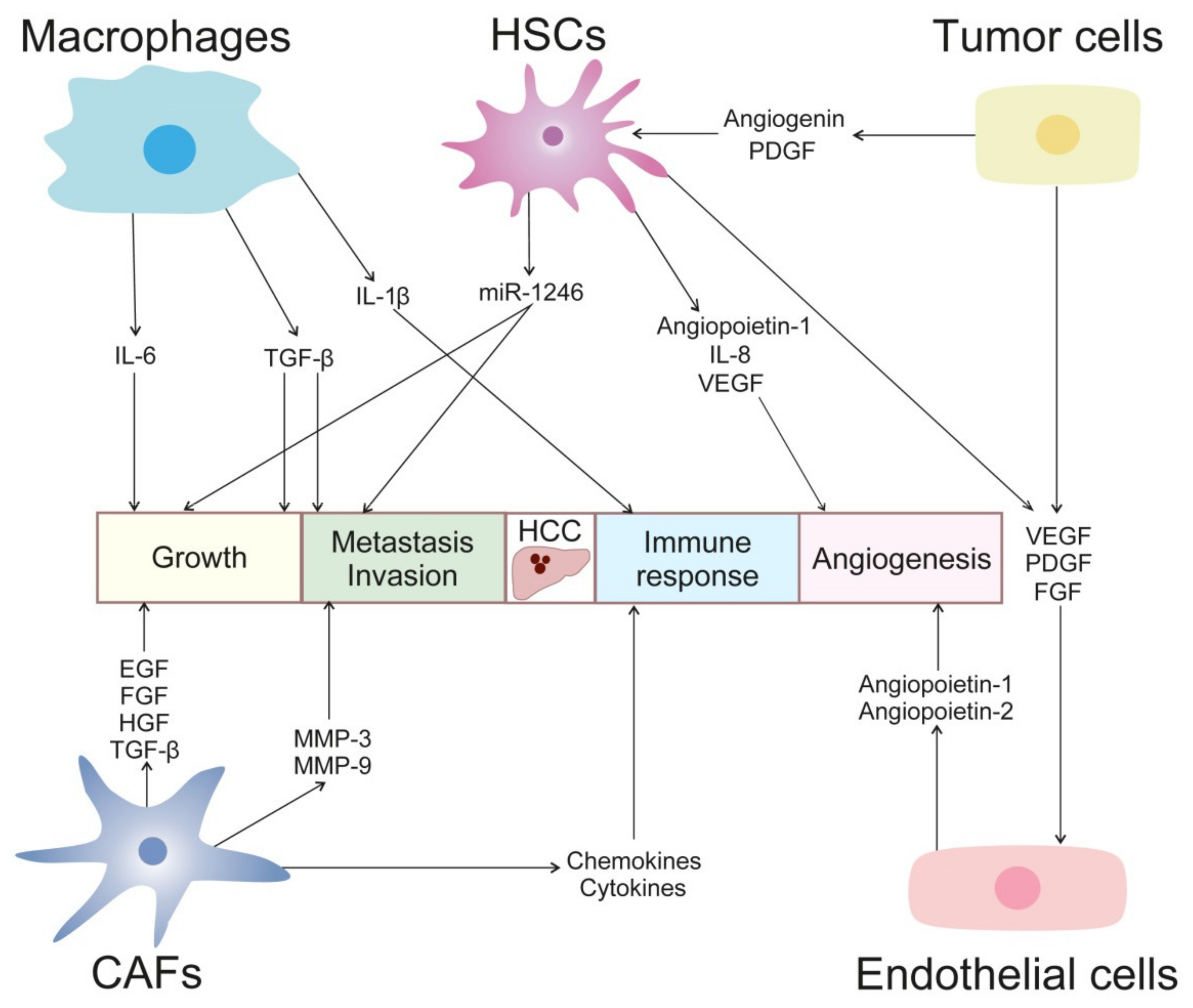
2. Tumor Microenvironment in HCC
2.1. Hepatic Stellate Cells
2.2. Cancer-Associated Fibroblasts
2.3. Tumor-Associated Macrophages
2.4. Endothelial Cells
2.5. Tumor-Associated Cells of the Innate Immune System
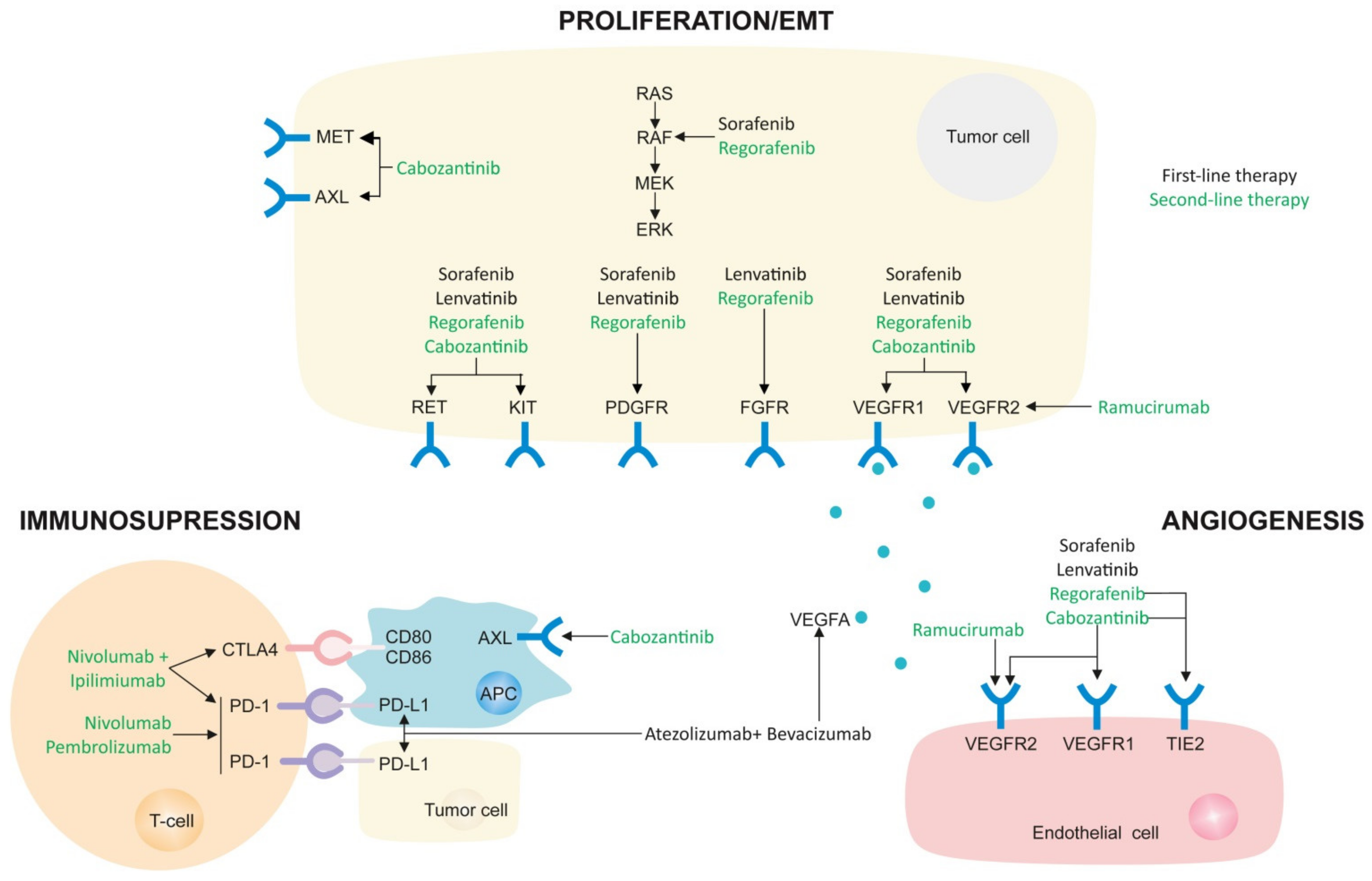
3. Systemic Therapies for HCC
3.1. First-Line Therapies
3.1.1. Atezolizumab-Bevacizumab (Atezo-Bev)
3.1.2. Sorafenib
3.1.3. Lenvatinib
3.2. Second-Line Therapies
3.2.1. Regorafenib
3.2.2. Cabozantinib
3.2.3. Nivolumab
3.2.4. Pembrolizumab
3.2.5. Ramucirumab
3.2.6. Combination Therapies
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2018; pp. 1–6. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.L.; Florio, A.A.; Znaor, A.; Ruggieri, D.; Laversanne, M.; Alvarez, C.S.; Ferlay, J.; Valery, P.C.; Bray, F.; McGlynn, K.A. International trends in hepatocellular carcinoma incidence, 1978–2012. Int. J. Cancer 2020, 147, 317–330. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. 1), 4–13. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; El-Serag, H.B. Epidemiology of hepatocellular carcinoma: Consider the population. J. Clin. Gastroenterol. 2013, 47, S2–S6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, P.P.; Singh, A.G.; Murad, M.H.; Sanchez, W. Statins are associated with a reduced risk of hepatocellular cancer: A systematic review and meta-analysis. Gastroenterology 2013, 144, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Bravi, F.; Bosetti, C.; Tavani, A.; Bagnardi, V.; Gallus, S.; Negri, E.; Franceschi, S.; La Vecchia, C. Coffee drinking and hepatocellular carcinoma risk: A meta-analysis. Hepatology 2007, 46, 430–435. [Google Scholar] [CrossRef]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [Green Version]
- Mu, X.; Español-Suñer, R.; Mederacke, I.; Affò, S.; Manco, R.; Sempoux, C.; Lemaigre, F.P.; Adili, A.; Yuan, D.; Weber, A.; et al. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J. Clin. Investig. 2015, 125, 3891. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nault, J.-C.; Couchy, G.; Balabaud, C.; Morcrette, G.; Caruso, S.; Blanc, J.-F.; Bacq, Y.; Calderaro, J.; Paradis, V.; Ramos, J.; et al. Molecular Classification of Hepatocellular Adenoma Associates With Risk Factors, Bleeding, and Malignant Transformation. Gastroenterology 2017, 152, 880–894.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, K.; Nault, J.-C.; Villanueva, A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. J. Hepatol. 2016, 65, 1031–1042. [Google Scholar] [CrossRef] [Green Version]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Audard, V.; Grimber, G.; Elie, C.; Radenen, B.; Audebourg, A.; Letourneur, F.; Soubrane, O.; Vacher-Lavenu, M.-C.; Perret, C.; Cavard, C.; et al. Cholestasis is a marker for hepatocellular carcinomas displaying beta-catenin mutations. J. Pathol. 2007, 212, 345–352. [Google Scholar] [CrossRef]
- Hsu, I.C.; Metcalf, R.A.; Sun, T.; Welsh, J.A.; Wang, N.J.; Harris, C.C. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 1991, 350, 427–428. [Google Scholar] [CrossRef]
- Bressac, B.; Kew, M.; Wands, J.; Ozturk, M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 1991, 350, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef]
- Sawey, E.T.; Chanrion, M.; Cai, C.; Wu, G.; Zhang, J.; Zender, L.; Zhao, A.; Busuttil, R.W.; Yee, H.; Stein, L.; et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening. Cancer Cell 2011, 19, 347–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwitz, E.; Stein, I.; Andreozzi, M.; Nemeth, J.; Shoham, A.; Pappo, O.; Schweitzer, N.; Tornillo, L.; Kanarek, N.; Quagliata, L.; et al. Human and mouse VEGFA-amplified hepatocellular carcinomas are highly sensitive to sorafenib treatment. Cancer Discov. 2014, 4, 730–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Toffanin, S.; Hoshida, Y.; Lachenmayer, A.; Villanueva, A.; Cabellos, L.; Minguez, B.; Savic, R.; Ward, S.C.; Thung, S.; Chiang, D.Y.; et al. MicroRNA-based classification of hepatocellular carcinoma and oncogenic role of miR-517a. Gastroenterology 2011, 140, 1618–1628.e16. [Google Scholar] [CrossRef] [Green Version]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.-C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [Green Version]
- Hoshida, Y.; Toffanin, S.; Lachenmayer, A.; Villanueva, A.; Minguez, B.; Llovet, J.M. Molecular classification and novel targets in hepatocellular carcinoma: Recent advancements. Semin. Liver Dis. 2010, 30, 35–51. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Villanueva, A.; Lachenmayer, A.; Finn, R.S. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat. Rev. Clin. Oncol. 2015, 12, 408–424. [Google Scholar] [CrossRef]
- Boyault, S.; Rickman, D.S.; de Reyniès, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Hérault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, A.; Hoshida, Y.; Battiston, C.; Tovar, V.; Sia, D.; Alsinet, C.; Cornella, H.; Liberzon, A.; Kobayashi, M.; Kumada, H.; et al. Combining clinical, pathology, and gene expression data to predict recurrence of hepatocellular carcinoma. Gastroenterology 2011, 140, 1501–1512.e2. [Google Scholar] [CrossRef] [Green Version]
- Lachenmayer, A.; Alsinet, C.; Savic, R.; Cabellos, L.; Toffanin, S.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Newell, P.; Tsai, H.-W.; et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin. Cancer Res. 2012, 18, 4997–5007. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.-H.; Yang, B.-H.; Tang, Z.-Y. Randomized controlled trial of screening for hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2004, 130, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.G.; Pillai, A.; Tiro, J. Early detection, curative treatment, and survival rates for hepatocellular carcinoma surveillance in patients with cirrhosis: A meta-analysis. PLoS Med. 2014, 11, e1001624. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Mannalithara, A.; Piscitello, A.J.; Kisiel, J.B.; Gores, G.J.; Roberts, L.R.; Kim, W.R. Impact of surveillance for hepatocellular carcinoma on survival in patients with compensated cirrhosis. Hepatology 2018, 68, 78–88. [Google Scholar] [CrossRef]
- Sarasin, F.P.; Giostra, E.; Hadengue, A. Cost-effectiveness of screening for detection of small hepatocellular carcinoma in western patients with Child-Pugh class A cirrhosis. Am. J. Med. 1996, 101, 422–434. [Google Scholar] [CrossRef]
- Yang, H.-I.; Yuen, M.-F.; Chan, H.L.-Y.; Han, K.-H.; Chen, P.-J.; Kim, D.-Y.; Ahn, S.-H.; Chen, C.-J.; Wong, V.W.-S.; Seto, W.-K. Risk estimation for hepatocellular carcinoma in chronic hepatitis B (REACH-B): Development and validation of a predictive score. Lancet. Oncol. 2011, 12, 568–574. [Google Scholar] [CrossRef]
- Singal, A.; Volk, M.L.; Waljee, A.; Salgia, R.; Higgins, P.; Rogers, M.A.M.; Marrero, J.A. Meta-analysis: Surveillance with ultrasound for early-stage hepatocellular carcinoma in patients with cirrhosis. Aliment. Pharmacol. Ther. 2009, 30, 37–47. [Google Scholar] [CrossRef]
- Trevisani, F.; D’Intino, P.E.; Morselli-Labate, A.M.; Mazzella, G.; Accogli, E.; Caraceni, P.; Domenicali, M.; De Notariis, S.; Roda, E.; Bernardi, M. Serum alpha-fetoprotein for diagnosis of hepatocellular carcinoma in patients with chronic liver disease: Influence of HBsAg and anti-HCV status. J. Hepatol. 2001, 34, 570–575. [Google Scholar] [CrossRef]
- Pelizzaro, F.; Cardin, R.; Penzo, B.; Pinto, E.; Vitale, A.; Cillo, U.; Russo, F.P.; Farinati, F. Liquid Biopsy in Hepatocellular Carcinoma: Where Are We Now? Cancers 2021, 13, 2274. [Google Scholar] [CrossRef]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Galle, P.R.; Forner, A.; Llovet, J.M.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.L.; Schirmacher, P.; Vilgrain, V. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, O.; Kobayashi, S.; Sanada, J.; Kouda, W.; Ryu, Y.; Kozaka, K.; Kitao, A.; Nakamura, K.; Gabata, T. Hepatocelluar nodules in liver cirrhosis: Hemodynamic evaluation (angiography-assisted CT) with special reference to multi-step hepatocarcinogenesis. Abdom. Imaging 2011, 36, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremosini, S.; Forner, A.; Boix, L.; Vilana, R.; Bianchi, L.; Reig, M.; Rimola, J.; Rodríguez-Lope, C.; Ayuso, C.; Solé, M.; et al. Prospective validation of an immunohistochemical panel (glypican 3, heat shock protein 70 and glutamine synthetase) in liver biopsies for diagnosis of very early hepatocellular carcinoma. Gut 2012, 61, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Reig, M.; Sherman, M. Evidence-Based Diagnosis, Staging, and Treatment of Patients With Hepatocellular Carcinoma. Gastroenterology 2016, 150, 835–853. [Google Scholar] [CrossRef] [Green Version]
- A new prognostic system for hepatocellular carcinoma: A retrospective study of 435 patients: The Cancer of the Liver Italian Program (CLIP) investigators. Hepatology 1998, 28, 751–755. [CrossRef]
- Yau, T.; Tang, V.Y.F.; Yao, T.-J.; Fan, S.-T.; Lo, C.-M.; Poon, R.T.P. Development of Hong Kong Liver Cancer staging system with treatment stratification for patients with hepatocellular carcinoma. Gastroenterology 2014, 146, 1691–1700.e3. [Google Scholar] [CrossRef]
- Llovet, J.M.; Brú, C.; Bruix, J. Prognosis of hepatocellular carcinoma: The BCLC staging classification. Semin. Liver Dis. 1999, 19, 329–338. [Google Scholar] [CrossRef]
- Child, C.G.; Turcotte, J.G. Surgery and portal hypertension. Major Probl. Clin. Surg. 1964, 1, 1–85. [Google Scholar]
- Ikai, I.; Arii, S.; Kojiro, M.; Ichida, T.; Makuuchi, M.; Matsuyama, Y.; Nakanuma, Y.; Okita, K.; Omata, M.; Takayasu, K.; et al. Reevaluation of prognostic factors for survival after liver resection in patients with hepatocellular carcinoma in a Japanese nationwide survey. Cancer 2004, 101, 796–802. [Google Scholar] [CrossRef]
- Llovet, J.M.; Peña, C.E.A.; Lathia, C.D.; Shan, M.; Meinhardt, G.; Bruix, J. Plasma biomarkers as predictors of outcome in patients with advanced hepatocellular carcinoma. Clin. Cancer Res. 2012, 18, 2290–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novikova, M.V.; Khromova, N.V.; Kopnin, P.B. Components of the hepatocellular carcinoma microenvironment and their role in tumor progression. Biochemistry 2017, 82, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Cao, Z.-Y.; Liu, S.-Y.; Xu, X.-D. Recent advances regarding tumor microenvironment and immunotherapy in hepatocellular carcinoma. Hepatoma Res. 2020, 6, 24. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Bataller, R.; Brenner, D.A. Human hepatic stellate cells express CCR5 and RANTES to induce proliferation and migration. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G949–G958. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Forbes, S.J.; Parola, M. Liver fibrogenic cells. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 207–217. [Google Scholar] [CrossRef]
- Faouzi, S.; Lepreux, S.; Bedin, C.; Dubuisson, L.; Balabaud, C.; Bioulac-Sage, P.; Desmoulière, A.; Rosenbaum, J. Activation of cultured rat hepatic stellate cells by tumoral hepatocytes. Lab. Investig. 1999, 79, 485–493. [Google Scholar]
- Lv, X.; Fang, C.; Yin, R.; Qiao, B.; Shang, R.; Wang, J.; Song, W.; He, Y.; Chen, Y. Agrin para-secreted by PDGF-activated human hepatic stellate cells promotes hepatocarcinogenesis in vitro and in vivo. Oncotarget 2017, 8, 105340–105355. [Google Scholar] [CrossRef]
- Amann, T.; Bataille, F.; Spruss, T.; Mühlbauer, M.; Gäbele, E.; Schölmerich, J.; Kiefer, P.; Bosserhoff, A.-K.; Hellerbrand, C. Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer Sci. 2009, 100, 646–653. [Google Scholar] [CrossRef]
- Sancho-Bru, P.; Juez, E.; Moreno, M.; Khurdayan, V.; Morales-Ruiz, M.; Colmenero, J.; Arroyo, V.; Brenner, D.A.; Ginès, P.; Bataller, R. Hepatocarcinoma cells stimulate the growth, migration and expression of pro-angiogenic genes in human hepatic stellate cells. Liver Int. 2010, 30, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Coulouarn, C.; Corlu, A.; Glaise, D.; Guénon, I.; Thorgeirsson, S.S.; Clément, B. Hepatocyte-stellate cell cross-talk in the liver engenders a permissive inflammatory microenvironment that drives progression in hepatocellular carcinoma. Cancer Res. 2012, 72, 2533–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carloni, V.; Luong, T.V.; Rombouts, K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: More complicated than ever. Liver Int. 2014, 34, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Bárcena, C.; Stefanovic, M.; Tutusaus, A.; Martinez-Nieto, G.A.; Martinez, L.; García-Ruiz, C.; De Mingo, A.; Caballeria, J.; Fernandez-Checa, J.C.; Marí, M.; et al. Angiogenin secretion from hepatoma cells activates hepatic stellate cells to amplify a self-sustained cycle promoting liver cancer. Sci. Rep. 2015, 5, 7916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, N.; Meng, L.; Lin, J.; Chen, S.; Zhang, P.; Chen, Q.; Lin, Y. Activated hepatic stellate cells promote angiogenesis in hepatocellular carcinoma by secreting angiopoietin-1. J. Cell. Biochem. 2020, 121, 1441–1451. [Google Scholar] [CrossRef]
- Zhu, B.; Lin, N.; Zhang, M.; Zhu, Y.; Cheng, H.; Chen, S.; Ling, Y.; Pan, W.; Xu, R. Activated hepatic stellate cells promote angiogenesis via interleukin-8 in hepatocellular carcinoma. J. Transl. Med. 2015, 13, 365. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro. Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef] [Green Version]
- Iwahasi, S.; Rui, F.; Morine, Y.; Yamada, S.; Saito, Y.U.; Ikemoto, T.; Imura, S.; Shimada, M. Hepatic Stellate Cells Contribute to the Tumor Malignancy of Hepatocellular Carcinoma Through the IL-6 Pathway. Anticancer Res. 2020, 40, 743–749. [Google Scholar] [CrossRef]
- Wen, Q.; Xu, C.; Zhou, J.; Liu, N.-M.; Cui, Y.-H.; Quan, M.-F.; Cao, J.-G.; Ren, K.-Q. 8-bromo-7-methoxychrysin suppress stemness of SMMC-7721 cells induced by co-culture of liver cancer stem-like cells with hepatic stellate cells. BMC Cancer 2019, 19, 224. [Google Scholar] [CrossRef]
- Chiyonobu, N.; Shimada, S.; Akiyama, Y.; Mogushi, K.; Itoh, M.; Akahoshi, K.; Matsumura, S.; Ogawa, K.; Ono, H.; Mitsunori, Y.; et al. Fatty Acid Binding Protein 4 (FABP4) Overexpression in Intratumoral Hepatic Stellate Cells within Hepatocellular Carcinoma with Metabolic Risk Factors. Am. J. Pathol. 2018, 188, 1213–1224. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.-L.; Fu, Y.-P.; Gan, W.; Liu, G.; Zhou, P.-Y.; Zhou, C.; Sun, B.-Y.; Guan, R.-Y.; Zhou, J.; Fan, J.; et al. Hepatic stellate cells promote the progression of hepatocellular carcinoma through microRNA-1246-RORα-Wnt/β-Catenin axis. Cancer Lett. 2020, 476, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Azzariti, A.; Mancarella, S.; Porcelli, L.; Quatrale, A.E.; Caligiuri, A.; Lupo, L.; Dituri, F.; Giannelli, G. Hepatic stellate cells induce hepatocellular carcinoma cell resistance to sorafenib through the laminin-332/α3 integrin axis recovery of focal adhesion kinase ubiquitination. Hepatology 2016, 64, 2103–2117. [Google Scholar] [CrossRef] [PubMed]
- Seitz, T.; Freese, K.; Dietrich, P.; Thasler, W.E.; Bosserhoff, A.; Hellerbrand, C. Fibroblast Growth Factor 9 is expressed by activated hepatic stellate cells and promotes progression of hepatocellular carcinoma. Sci. Rep. 2020, 10, 4546. [Google Scholar] [CrossRef] [PubMed]
- Mogler, C.; König, C.; Wieland, M.; Runge, A.; Besemfelder, E.; Komljenovic, D.; Longerich, T.; Schirmacher, P.; Augustin, H.G. Hepatic stellate cells limit hepatocellular carcinoma progression through the orphan receptor endosialin. EMBO Mol. Med. 2017, 9, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Shimoda, M.; Mellody, K.T.; Orimo, A. Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Semin. Cell Dev. Biol. 2010, 21, 19–25. [Google Scholar] [CrossRef]
- Lau, E.Y.T.; Lo, J.; Cheng, B.Y.L.; Ma, M.K.F.; Lee, J.M.F.; Ng, J.K.Y.; Chai, S.; Lin, C.H.; Tsang, S.Y.; Ma, S.; et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016, 15, 1175–1189. [Google Scholar] [CrossRef] [Green Version]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef]
- Jia, C.-C.; Wang, T.-T.; Liu, W.; Fu, B.-S.; Hua, X.; Wang, G.-Y.; Li, T.-J.; Li, X.; Wu, X.-Y.; Tai, Y.; et al. Cancer-associated fibroblasts from hepatocellular carcinoma promote malignant cell proliferation by HGF secretion. PLoS ONE 2013, 8, e63243. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chen, S.; Wang, W.; Ning, B.-F.; Chen, F.; Shen, W.; Ding, J.; Chen, W.; Xie, W.-F.; Zhang, X. Cancer-associated fibroblasts promote hepatocellular carcinoma metastasis through chemokine-activated hedgehog and TGF-β pathways. Cancer Lett. 2016, 379, 49–59. [Google Scholar] [CrossRef]
- Zhang, Y.; Pan, Q.; Shao, Z. Extracellular vesicle-encapsulated microRNA-1228-3p from cancer-associated fibroblasts promotes the chemoresistance of hepatocellular carcinoma cells via PLAC8. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G638. [Google Scholar] [CrossRef]
- Yugawa, K.; Yoshizumi, T.; Mano, Y.; Itoh, S.; Harada, N.; Ikegami, T.; Kohashi, K.; Oda, Y.; Mori, M. Cancer-associated fibroblasts promote hepatocellular carcinoma progression through downregulation of exosomal miR-150-3p. Eur. J. Surg. Oncol. 2020, 47, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, L.; Piontek, K.; Sakaguchi, M.; Selaru, F.M. Exosome miR-335 as a novel therapeutic strategy in hepatocellular carcinoma. Hepatology 2018, 67, 940–954. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ren, H.; Dai, B.; Li, J.; Shang, L.; Huang, J.; Shi, X. Hepatocellular carcinoma-derived exosomal miRNA-21 contributes to tumor progression by converting hepatocyte stellate cells to cancer-associated fibroblasts. J. Exp. Clin. Cancer Res. 2018, 37, 324. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Shirabe, K.; Mano, Y.; Muto, J.; Matono, R.; Motomura, T.; Toshima, T.; Takeishi, K.; Uchiyama, H.; Yoshizumi, T.; Taketomi, A.; et al. Role of tumor-associated macrophages in the progression of hepatocellular carcinoma. Surg. Today 2012, 42, 1–7. [Google Scholar] [CrossRef]
- Takai, H.; Ashihara, M.; Ishiguro, T.; Terashima, H.; Watanabe, T.; Kato, A.; Suzuki, M. Involvement of glypican-3 in the recruitment of M2-polarized tumor-associated macrophages in hepatocellular carcinoma. Cancer Biol. Ther. 2009, 8, 2329–2338. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.-M.; Jing, Y.-Y.; Yu, G.-F.; Kou, X.-R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.-D.; Yang, Y.; Lu, Z.-H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef]
- Gupta, D.K.; Singh, N.; Sahu, D.K. TGF-β Mediated Crosstalk Between Malignant Hepatocyte and Tumor Microenvironment in Hepatocellular Carcinoma. Cancer Growth Metastasis 2014, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roderfeld, M.; Rath, T.; Lammert, F.; Dierkes, C.; Graf, J.; Roeb, E. Innovative immunohistochemistry identifies MMP-9 expressing macrophages at the invasive front of murine HCC. World J. Hepatol. 2010, 2, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Nishie, A.; Aishima, S.; Kubo, Y.; Asayama, Y.; Ishigami, K.; Kakihara, D.; Ushijima, Y.; Takayama, Y.; Shirabe, K.; et al. Role of tumor-associated macrophages in the angiogenesis of well-differentiated hepatocellular carcinoma: Pathological-radiological correlation. Oncol. Rep. 2014, 31, 2499–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Li, Q.; Qin, L.; Zhao, S.; Wang, J.; Chen, X. Transition of tumor-associated macrophages from MHC class II(hi) to MHC class II(low) mediates tumor progression in mice. BMC Immunol. 2011, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, K.; Koletsa, T.; Mitroulis, I.; Germanidis, G. Tumor-Associated Macrophages in Hepatocellular Carcinoma Pathogenesis, Prognosis and Therapy. Cancers 2022, 14, 226. [Google Scholar] [CrossRef]
- Yang, Y.; Ye, Y.-C.; Chen, Y.; Zhao, J.-L.; Gao, C.-C.; Han, H.; Liu, W.-C.; Qin, H.-Y. Crosstalk between hepatic tumor cells and macrophages via Wnt/β-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. 2018, 9, 793. [Google Scholar] [CrossRef]
- Sprinzl, M.F.; Reisinger, F.; Puschnik, A.; Ringelhan, M.; Ackermann, K.; Hartmann, D.; Schiemann, M.; Weinmann, A.; Galle, P.R.; Schuchmann, M.; et al. Sorafenib perpetuates cellular anticancer effector functions by modulating the crosstalk between macrophages and natural killer cells. Hepatology 2013, 57, 2358–2368. [Google Scholar] [CrossRef]
- Chen, J.; Li, G.; Meng, H.; Fan, Y.; Song, Y.; Wang, S.; Zhu, F.; Guo, C.; Zhang, L.; Shi, Y. Upregulation of B7-H1 expression is associated with macrophage infiltration in hepatocellular carcinomas. Cancer Immunol. Immunother. 2012, 61, 101–108. [Google Scholar] [CrossRef]
- Zong, Z.; Zou, J.; Mao, R.; Ma, C.; Li, N.; Wang, J.; Wang, X.; Zhou, H.; Zhang, L.; Shi, Y. M1 Macrophages Induce PD-L1 Expression in Hepatocellular Carcinoma Cells Through IL-1β Signaling. Front. Immunol. 2019, 10, 1643. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Kryczek, I.; Chen, L.; Zou, W.; Welling, T.H. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res. 2009, 69, 8067–8075. [Google Scholar] [CrossRef] [Green Version]
- Fujii, H.; Kawada, N. Fibrogenesis in alcoholic liver disease. World J. Gastroenterol. 2014, 20, 8048–8054. [Google Scholar] [CrossRef] [PubMed]
- Dudley, A.C. Tumor endothelial cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006536. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.E.; Senger, D.R. Endothelial extracellular matrix: Biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pralhad, T.; Madhusudan, S.; Rajendrakumar, K. Concept, mechanisms and therapeutics of angiogenesis in cancer and other diseases. J. Pharm. Pharmacol. 2003, 55, 1045–1053. [Google Scholar] [CrossRef]
- Baluk, P.; Morikawa, S.; Haskell, A.; Mancuso, M.; McDonald, D.M. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2003, 163, 1801–1815. [Google Scholar] [CrossRef] [Green Version]
- Von Marschall, Z.; Cramer, T.; Höcker, M.; Finkenzeller, G.; Wiedenmann, B.; Rosewicz, S. Dual mechanism of vascular endothelial growth factor upregulation by hypoxia in human hepatocellular carcinoma. Gut 2001, 48, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-R.; Moon, H.-E.; Kim, K.-W. Hypoxia-induced angiogenesis in human hepatocellular carcinoma. J. Mol. Med. 2002, 80, 703–714. [Google Scholar] [CrossRef]
- Xiong, X.X.; Qiu, X.Y.; Hu, D.X.; Chen, X.Q. Advances in Hypoxia-Mediated Mechanisms in Hepatocellular Carcinoma. Mol. Pharmacol. 2017, 92, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Morse, M.A.; Sun, W.; Kim, R.; He, A.R.; Abada, P.B.; Mynderse, M.; Finn, R.S. The role of angiogenesis in hepatocellular carcinoma. Clin. Cancer Res. 2019, 25, 912–920. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, R.; Yano, H.; Iemura, A.; Ogasawara, S.; Haramaki, M.; Kojiro, M. Expression of vascular endothelial growth factor in human hepatocellular carcinoma. Hepatology 1998, 28, 68–77. [Google Scholar] [CrossRef]
- Amini, A.; Masoumi Moghaddam, S.; Morris, D.L.; Pourgholami, M.H. The critical role of vascular endothelial growth factor in tumor angiogenesis. Curr. Cancer Drug Targets 2012, 12, 23–43. [Google Scholar] [CrossRef] [PubMed]
- Poon, R.T.P.; Ho, J.W.Y.; Tong, C.S.W.; Lau, C.; Ng, I.O.L.; Fan, S.-T. Prognostic significance of serum vascular endothelial growth factor and endostatin in patients with hepatocellular carcinoma. Br. J. Surg. 2004, 91, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Tang, Z.Y.; Qin, L.X.; Zhou, J.; Sun, H.C. Serum vascular endothelial growth factor is a predictor of invasion and metastasis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 1999, 18, 511–517. [Google Scholar] [PubMed]
- Heldin, C.-H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 2013, 11, 97. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Pan, Q.; Zhang, X.; Kong, L.-Q.; Fan, J.; Dai, Z.; Wang, L.; Yang, X.-R.; Hu, J.; Wan, J.-L.; et al. MiR-146a enhances angiogenic activity of endothelial cells in hepatocellular carcinoma by promoting PDGFRA expression. Carcinogenesis 2013, 34, 2071–2079. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Cao, R.; Hedlund, E.-M. R Regulation of tumor angiogenesis and metastasis by FGF and PDGF signaling pathways. J. Mol. Med. 2008, 86, 785–789. [Google Scholar] [CrossRef]
- Imura, S.; Miyake, H.; Izumi, K.; Tashiro, S.; Uehara, H. Correlation of vascular endothelial cell proliferation with microvessel density and expression of vascular endothelial growth factor and basic fibroblast growth factor in hepatocellular carcinoma. J. Med. Investig. 2004, 51, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Bupathi, M.; Kaseb, A.; Janku, F. Angiopoietin 2 as a therapeutic target in hepatocellular carcinoma treatment: Current perspectives. Onco. Targets. Ther. 2014, 7, 1927–1932. [Google Scholar] [CrossRef] [Green Version]
- Torimura, T.; Ueno, T.; Kin, M.; Harada, R.; Taniguchi, E.; Nakamura, T.; Sakata, R.; Hashimoto, O.; Sakamoto, M.; Kumashiro, R.; et al. Overexpression of angiopoietin-1 and angiopoietin-2 in hepatocellular carcinoma. J. Hepatol. 2004, 40, 799–807. [Google Scholar] [CrossRef]
- He, Y.-F.; Wang, C.-Q.; Yu, Y.; Qian, J.; Song, K.; Sun, Q.-M.; Zhou, J. Tie2-Expressing Monocytes Are Associated with Identification and Prognoses of Hepatitis B Virus Related Hepatocellular Carcinoma after Resection. PLoS ONE 2015, 10, e0143657. [Google Scholar] [CrossRef] [Green Version]
- Scholz, A.; Rehm, V.A.; Rieke, S.; Derkow, K.; Schulz, P.; Neumann, K.; Koch, I.; Pascu, M.; Wiedenmann, B.; Berg, T.; et al. Angiopoietin-2 serum levels are elevated in patients with liver cirrhosis and hepatocellular carcinoma. Am. J. Gastroenterol. 2007, 102, 2471–2481. [Google Scholar] [CrossRef] [PubMed]
- Kuboki, S.; Shimizu, H.; Mitsuhashi, N.; Kusashio, K.; Kimura, F.; Yoshidome, H.; Ohtsuka, M.; Kato, A.; Yoshitomi, H.; Miyazaki, M. Angiopoietin-2 levels in the hepatic vein as a useful predictor of tumor invasiveness and prognosis in human hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2008, 23, e157–e164. [Google Scholar] [CrossRef] [PubMed]
- Yoshiji, H.; Kuriyama, S.; Noguchi, R.; Yoshii, J.; Ikenaka, Y.; Yanase, K.; Namisaki, T.; Kitade, M.; Uemura, M.; Masaki, T.; et al. Angiopoietin 2 displays a vascular endothelial growth factor dependent synergistic effect in hepatocellular carcinoma development in mice. Gut 2005, 54, 1768–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, E.; Critelli, R.; Lei, B.; Marzocchi, G.; Cammà, C.; Giannelli, G.; Pontisso, P.; Cabibbo, G.; Enea, M.; Colopi, S.; et al. Neoangiogenesis-related genes are hallmarks of fast-growing hepatocellular carcinomas and worst survival. Results from a prospective study. Gut 2016, 65, 861–869. [Google Scholar] [CrossRef]
- Gao, L.; Ge, C.; Fang, T.; Zhao, F.; Chen, T.; Yao, M.; Li, J.; Li, H. ANGPTL2 promotes tumor metastasis in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2015, 30, 396–404. [Google Scholar] [CrossRef]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- Roderburg, C.; Wree, A.; Demir, M.; Schmelzle, M.; Tacke, F. The role of the innate immune system in the development and treatment of hepatocellular carcinoma. Hepatic Oncol. 2020, 7, HEP17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, S.; Kuo, N.; Kryczek, I.; Zou, W.; Welling, T.H. Myeloid cells in hepatocellular carcinoma. Hepatology 2015, 62, 1304–1312. [Google Scholar] [CrossRef]
- Barnes, T.A.; Amir, E. HYPE or HOPE: The prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 2017, 117, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalathil, S.G.; Thanavala, Y. Natural Killer Cells and T Cells in Hepatocellular Carcinoma and Viral Hepatitis: Current Status and Perspectives for Future Immunotherapeutic Approaches. Cells 2021, 10, 1332. [Google Scholar] [CrossRef]
- Bozward, A.G.; Warricker, F.; Oo, Y.H.; Khakoo, S.I. Natural Killer Cells and Regulatory T Cells Cross Talk in Hepatocellular Carcinoma: Exploring Therapeutic Options for the Next Decade. Front. Immunol. 2021, 12, 1493. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Chan, S.L.; Galle, P.R.; Rimassa, L.; Sangro, B. Systemic treatment of hepatocellular carcinoma: An EASL position paper. J. Hepatol. 2021, 75, 960–974. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 2016, 7, 12624. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.-H.; Harding, J.J.; Merle, P.; et al. CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs. sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2019, 30, v874–v875. [Google Scholar] [CrossRef]
- Lee, M.S.; Ryoo, B.-Y.; Hsu, C.-H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.P.; Spahn, J.; Liu, B.; Abdullah, H.; et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): An open-label, multicentre, phase 1b study. Lancet Oncol. 2020, 21, 808–820. [Google Scholar] [CrossRef]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.-X.; Wang, T.; Deng, Y.-Z.; Yang, P.; Li, J.-J.; Guan, D.-X.; Yao, F.; Zhu, Y.-Q.; Qin, Y.; Wang, H.; et al. Sorafenib suppresses postsurgical recurrence and metastasis of hepatocellular carcinoma in an orthotopic mouse model. Hepatology 2011, 53, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Geng, Z.; Jha, R.K.; Li, B.; Chen, C.; Li, W.; Zheng, J.; Wang, L.; Huanchen, S. Sorafenib inhibition of hepatic stellate cell proliferation in tumor microenvironment of hepatocellular carcinoma: A study of the sorafenib mechanisms. Cell Biochem. Biophys. 2014, 69, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.V.; Maguire, O.; Kim, M.; Curtin, L.I.; Sexton, S.; Fisher, D.T.; Schihl, S.A.; Fetterly, G.; Menne, S.; Minderman, H. Dose-Dependent Sorafenib-Induced Immunosuppression Is Associated with Aberrant NFAT Activation and Expression of PD-1 in T Cells. Cancers 2019, 11, 681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Reig, M.; Torres, F.; Rodriguez-Lope, C.; Forner, A.; LLarch, N.; Rimola, J.; Darnell, A.; Ríos, J.; Ayuso, C.; Bruix, J. Early dermatologic adverse events predict better outcome in HCC patients treated with sorafenib. J. Hepatol. 2014, 61, 318–324. [Google Scholar] [CrossRef]
- Rahmani, M.; Davis, E.M.; Bauer, C.; Dent, P.; Grant, S. Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. J. Biol. Chem. 2005, 280, 35217–35227. [Google Scholar] [CrossRef] [Green Version]
- Stefanovic, M.; Tutusaus, A.; Martinez-Nieto, G.A.; Bárcena, C.; De Gregorio, E.; Moutinho, C.; Barbero-Camps, E.; Villanueva, A.; Colell, A.; Marí, M.; et al. Targeting glucosylceramide synthase upregulation reverts sorafenib resistance in experimental hepatocellular carcinoma. Oncotarget 2016, 7, 8253–8267. [Google Scholar] [CrossRef] [Green Version]
- Tutusaus, A.; Stefanovic, M.; Boix, L.; Cucarull, B.; Zamora, A.; Blasco, L.; de Frutos, P.G.; Reig, M.; Fernandez-Checa, J.C.; Marí, M.; et al. Antiapoptotic BCL-2 proteins determine sorafenib/regorafenib resistance and BH3-mimetic efficacy in hepatocellular carcinoma. Oncotarget 2018, 9, 16701–16717. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Takehara, T.; Hikita, H.; Kodama, T.; Tsunematsu, H.; Miyagi, T.; Hosui, A.; Ishida, H.; Tatsumi, T.; Kanto, T.; et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int. J. Cancer 2012, 131, 548–557. [Google Scholar] [CrossRef]
- Tai, W.-T.; Shiau, C.-W.; Chen, H.-L.; Liu, C.-Y.; Lin, C.-S.; Cheng, A.-L.; Chen, P.-J.; Chen, K.-F. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. 2013, 4, e485. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Domínguez, N.; Ordóñez, R.; Fernández, A.; Méndez-Blanco, C.; Baulies, A.; Garcia-Ruiz, C.; Fernández-Checa, J.C.; Mauriz, J.L.; González-Gallego, J. Melatonin-induced increase in sensitivity of human hepatocellular carcinoma cells to sorafenib is associated with reactive oxygen species production and mitophagy. J. Pineal Res. 2016, 61, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Cucarull, B.; Tutusaus, A.; Hernáez-Alsina, T.; García de Frutos, P.; Reig, M.; Colell, A.; Marí, M.; Morales, A. Antioxidants Threaten Multikinase Inhibitor Efficacy against Liver Cancer by Blocking Mitochondrial Reactive Oxygen Species. Antioxidants 2021, 10, 1336. [Google Scholar] [CrossRef] [PubMed]
- Van Malenstein, H.; Dekervel, J.; Verslype, C.; Van Cutsem, E.; Windmolders, P.; Nevens, F.; van Pelt, J. Long-term exposure to sorafenib of liver cancer cells induces resistance with epithelial-to-mesenchymal transition, increased invasion and risk of rebound growth. Cancer Lett. 2013, 329, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.K.-M.; Ng, L.; Lam, C.S.-C.; Wong, S.K.-M.; Wan, T.M.-H.; Cheng, N.S.-M.; Yau, T.C.-C.; Poon, R.T.-P.; Pang, R.W.-C. The Enhanced metastatic potential of hepatocellular carcinoma (HCC) cells with sorafenib resistance. PLoS ONE 2013, 8, e78675. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Zheng, T.; Song, R.; Wang, J.; Yin, D.; Wang, L.; Liu, H.; Tian, L.; Fang, X.; Meng, X.; et al. Hypoxia-mediated sorafenib resistance can be overcome by EF24 through Von Hippel-Lindau tumor suppressor-dependent HIF-1α inhibition in hepatocellular carcinoma. Hepatology 2013, 57, 1847–1857. [Google Scholar] [CrossRef]
- Dong, N.; Shi, X.; Wang, S.; Gao, Y.; Kuang, Z.; Xie, Q.; Li, Y.; Deng, H.; Wu, Y.; Li, M.; et al. M2 macrophages mediate sorafenib resistance by secreting HGF in a feed-forward manner in hepatocellular carcinoma. Br. J. Cancer 2019, 121, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Matsui, J.; Funahashi, Y.; Uenaka, T.; Watanabe, T.; Tsuruoka, A.; Asada, M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin. Cancer Res. 2008, 14, 5459–5465. [Google Scholar] [CrossRef] [Green Version]
- Adachi, Y.; Matsuki, M.; Watanabe, H.; Takase, K.; Kodama, K.; Matsui, J.; Funahashi, Y.; Nomoto, K. Antitumor and Antiangiogenic Activities of Lenvatinib in Mouse Xenograft Models of Vascular Endothelial Growth Factor-Induced Hypervascular Human Hepatocellular Carcinoma. Cancer Investig. 2019, 37, 185–198. [Google Scholar] [CrossRef]
- Hoshi, T.; Watanabe Miyano, S.; Watanabe, H.; Sonobe, R.M.K.; Seki, Y.; Ohta, E.; Nomoto, K.; Matsui, J.; Funahashi, Y. Lenvatinib induces death of human hepatocellular carcinoma cells harboring an activated FGF signaling pathway through inhibition of FGFR-MAPK cascades. Biochem. Biophys. Res. Commun. 2019, 513, 1–7. [Google Scholar] [CrossRef]
- Kimura, T.; Kato, Y.; Ozawa, Y.; Kodama, K.; Ito, J.; Ichikawa, K.; Yamada, K.; Hori, Y.; Tabata, K.; Takase, K.; et al. Immunomodulatory activity of lenvatinib contributes to antitumor activity in the Hepa1-6 hepatocellular carcinoma model. Cancer Sci. 2018, 109, 3993–4002. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Kodama, K.; Kawaoka, T.; Namba, M.; Uchikawa, S.; Ohya, K.; Morio, K.; Nakahara, T.; Murakami, E.; Yamauchi, M.; Hiramatsu, A.; et al. Correlation between Early Tumor Marker Response and Imaging Response in Patients with Advanced Hepatocellular Carcinoma Treated with Lenvatinib. Oncology 2019, 97, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Chuma, M.; Uojima, H.; Numata, K.; Hidaka, H.; Toyoda, H.; Hiraoka, A.; Tada, T.; Hirose, S.; Atsukawa, M.; Itokawa, N.; et al. Early Changes in Circulating FGF19 and Ang-2 Levels as Possible Predictive Biomarkers of Clinical Response to Lenvatinib Therapy in Hepatocellular Carcinoma. Cancers 2020, 12, 293. [Google Scholar] [CrossRef] [Green Version]
- Saeki, I.; Yamasaki, T.; Yamashita, S.; Hanazono, T.; Urata, Y.; Furutani, T.; Yokoyama, Y.; Oishi, T.; Maeda, M.; Kimura, T.; et al. Early Predictors of Objective Response in Patients with Hepatocellular Carcinoma Undergoing Lenvatinib Treatment. Cancers 2020, 12, 779. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Moriguchi, M.; Seko, Y.; Shima, T.; Mitsumoto, Y.; Takashima, H.; Kimura, H.; Fujii, H.; Ishikawa, H.; Takaharu, Y.; et al. Early Tumor Shrinkage as a Predictive Factor for Outcomes in Hepatocellular Carcinoma Patients Treated with Lenvatinib: A Multicenter Analysis. Cancers 2020, 12, 754. [Google Scholar] [CrossRef] [Green Version]
- Fu, R.; Jiang, S.; Li, J.; Chen, H.; Zhang, X. Activation of the HGF/c-MET axis promotes lenvatinib resistance in hepatocellular carcinoma cells with high c-MET expression. Med. Oncol. 2020, 37, 24. [Google Scholar] [CrossRef]
- Strumberg, D.; Schultheis, B. Regorafenib for cancer. Expert Opin. Investig. Drugs 2012, 21, 879–889. [Google Scholar] [CrossRef]
- Carr, B.I.; D’Alessandro, R.; Refolo, M.G.; Iacovazzi, P.A.; Lippolis, C.; Messa, C.; Cavallini, A.; Correale, M.; Di Carlo, A. Effects of low concentrations of regorafenib and sorafenib on human HCC cell AFP, migration, invasion, and growth in vitro. J. Cell. Physiol. 2013, 228, 1344–1350. [Google Scholar] [CrossRef] [Green Version]
- Carr, B.I.; Cavallini, A.; Lippolis, C.; D’Alessandro, R.; Messa, C.; Refolo, M.G.; Tafaro, A. Fluoro-Sorafenib (Regorafenib) effects on hepatoma cells: Growth inhibition, quiescence, and recovery. J. Cell. Physiol. 2013, 228, 292–297. [Google Scholar] [CrossRef] [Green Version]
- Han, R.; Li, S. Regorafenib delays the proliferation of hepatocellular carcinoma by inducing autophagy. Pharmazie 2018, 73, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Cucarull, B.; Tutusaus, A.; Subías, M.; Stefanovic, M.; Hernáez-Alsina, T.; Boix, L.; Reig, M.; de Frutos, P.G.; Marí, M.; Colell, A.; et al. Regorafenib alteration of the BCL-xL/MCL-1 ratio provides a therapeutic opportunity for BH3-mimetics in hepatocellular carcinoma models. Cancers 2020, 12, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.-J.; Pan, P.-J.; Hsu, F.-T. Regorafenib induces extrinsic and intrinsic apoptosis through inhibition of ERK/NF-κB activation in hepatocellular carcinoma cells. Oncol. Rep. 2017, 37, 1036–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-C.; Wu, R.-H.; Wang, W.-S. Regorafenib diminishes the expression and secretion of angiogenesis and metastasis associated proteins and inhibits cell invasion via NF-κB inactivation in SK-Hep1 cells. Oncol. Lett. 2017, 14, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Yang, J.; Zhang, Y.; Cai, H.; Chen, X.; Sun, D. Regorafenib reverses HGF-induced sorafenib resistance by inhibiting epithelial-mesenchymal transition in hepatocellular carcinoma. FEBS Open Bio. 2019, 9, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Gerolami, R.; Caparello, C.; et al. Outcomes of sequential treatment with sorafenib followed by regorafenib for HCC: Additional analyses from the phase III RESORCE trial. J. Hepatol. 2018, 69, 353–358. [Google Scholar] [CrossRef]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Q.; Chen, W.; Ren, M.; Wang, J.; Zhang, H.; Deng, D.Y.B.; Zhang, L.; Shang, C.; Chen, Y. Cabozantinib suppresses tumor growth and metastasis in hepatocellular carcinoma by a dual blockade of VEGFR2 and MET. Clin. Cancer Res. 2014, 20, 2959–2970. [Google Scholar] [CrossRef] [Green Version]
- Kelley, R.K.; Verslype, C.; Cohn, A.L.; Yang, T.-S.; Su, W.-C.; Burris, H.; Braiteh, F.; Vogelzang, N.; Spira, A.; Foster, P.; et al. Cabozantinib in hepatocellular carcinoma: Results of a phase 2 placebo-controlled randomized discontinuation study. Ann. Oncol. 2017, 28, 528–534. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.H.; Park, J.W.; et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Sangro, B.; Melero, I.; Wadhawan, S.; Finn, R.S.; Abou-Alfa, G.K.; Cheng, A.-L.; Yau, T.; Furuse, J.; Park, J.-W.; Boyd, Z.; et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol. 2020, 73, 1460–1469. [Google Scholar] [CrossRef]
- Kim, C.G.; Kim, C.; Yoon, S.E.; Kim, K.H.; Choi, S.J.; Kang, B.; Kim, H.R.; Park, S.-H.; Shin, E.-C.; Kim, Y.-Y.; et al. Hyperprogressive disease during PD-1 blockade in patients with advanced hepatocellular carcinoma. J. Hepatol. 2020, 74, 350–359. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet. Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Kudo, M.; Hatano, E.; Ohkawa, S.; Fujii, H.; Masumoto, A.; Furuse, J.; Wada, Y.; Ishii, H.; Obi, S.; Kaneko, S.; et al. Ramucirumab as second-line treatment in patients with advanced hepatocellular carcinoma: Japanese subgroup analysis of the REACH trial. J. Gastroenterol. 2017, 52, 494–503. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Kudo, M.; Okusaka, T.; Motomura, K.; Ohno, I.; Morimoto, M.; Seo, S.; Wada, Y.; Sato, S.; Yamashita, T.; Furukawa, M.; et al. Ramucirumab after prior sorafenib in patients with advanced hepatocellular carcinoma and elevated alpha-fetoprotein: Japanese subgroup analysis of the REACH-2 trial. J. Gastroenterol. 2020, 55, 627–639. [Google Scholar] [CrossRef] [Green Version]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fábrega, J.; Burrel, M.; Garcia-Criado, A.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC strategy for prognosis prediction and treatment recommendation Barcelona Clinic Liver Cancer (BCLC) staging system. The 2022 update. J. Hepatol. 2022. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cucarull, B.; Tutusaus, A.; Rider, P.; Hernáez-Alsina, T.; Cuño, C.; García de Frutos, P.; Colell, A.; Marí, M.; Morales, A. Hepatocellular Carcinoma: Molecular Pathogenesis and Therapeutic Advances. Cancers 2022, 14, 621. https://doi.org/10.3390/cancers14030621
Cucarull B, Tutusaus A, Rider P, Hernáez-Alsina T, Cuño C, García de Frutos P, Colell A, Marí M, Morales A. Hepatocellular Carcinoma: Molecular Pathogenesis and Therapeutic Advances. Cancers. 2022; 14(3):621. https://doi.org/10.3390/cancers14030621
Chicago/Turabian StyleCucarull, Blanca, Anna Tutusaus, Patricia Rider, Tania Hernáez-Alsina, Carlos Cuño, Pablo García de Frutos, Anna Colell, Montserrat Marí, and Albert Morales. 2022. "Hepatocellular Carcinoma: Molecular Pathogenesis and Therapeutic Advances" Cancers 14, no. 3: 621. https://doi.org/10.3390/cancers14030621
APA StyleCucarull, B., Tutusaus, A., Rider, P., Hernáez-Alsina, T., Cuño, C., García de Frutos, P., Colell, A., Marí, M., & Morales, A. (2022). Hepatocellular Carcinoma: Molecular Pathogenesis and Therapeutic Advances. Cancers, 14(3), 621. https://doi.org/10.3390/cancers14030621