
Cellular Therapy for Lung Cancer: Focusing on Chimeric Antigen Receptor T (CAR T) Cells and Tumor-Infiltrating Lymphocyte (TIL) Therapy

Abstract
:Simple Summary
Abstract
1. Introduction
Rationale of Use of Cellular Therapy in Lung Cancer
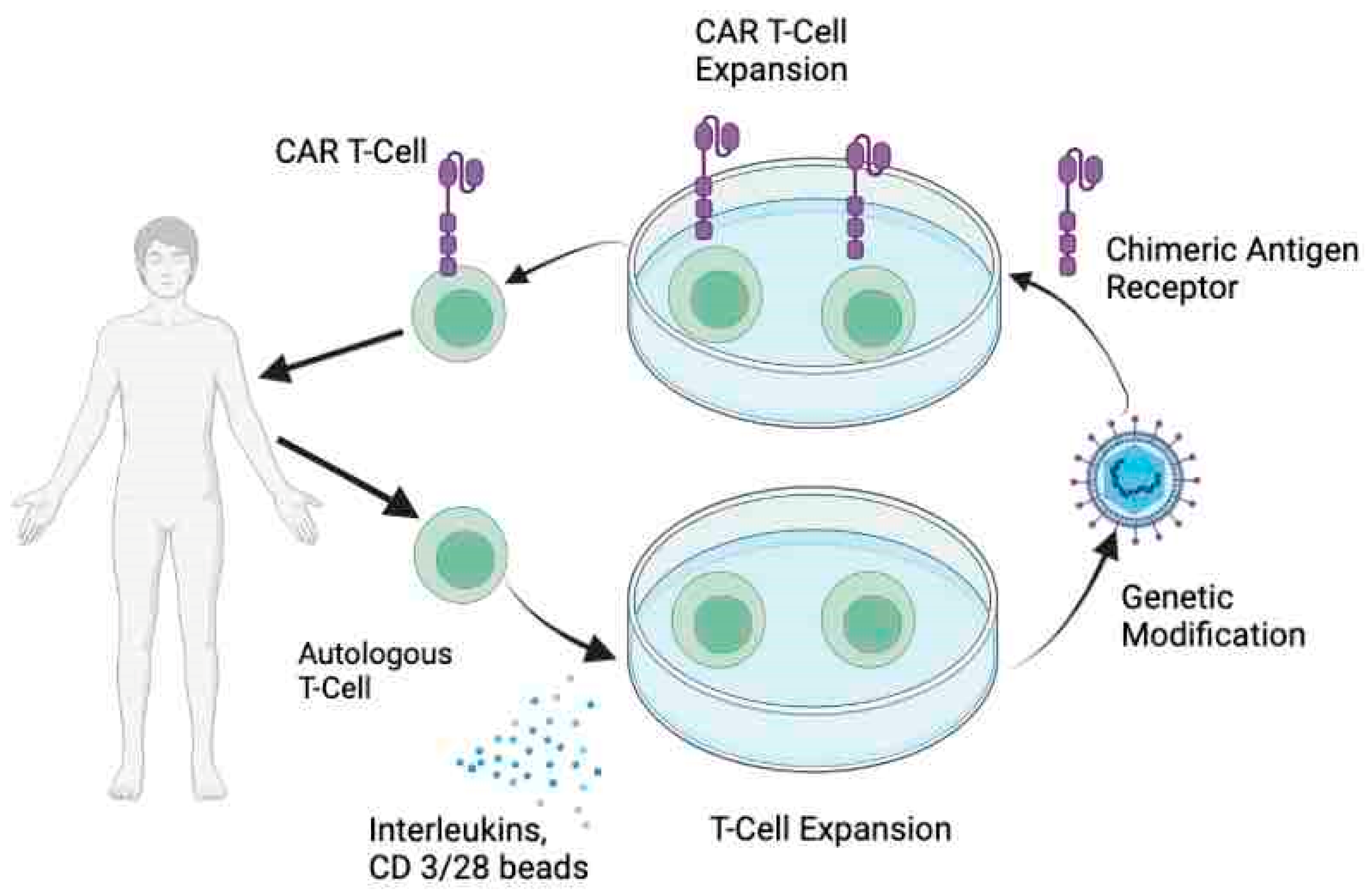
2. CAR T-Cell Therapy
2.1. Toxicities with CAR T Cell Therapy
- Cytokine release syndrome. CRS, a potentially life-threatening toxicity associated with CAR T cell infusion, is caused by widespread activation and proliferation of lymphocytes and myeloid cells which secrete cytokines including IL-6, IFN-γ, IL-8, IL-10, granulocyte-macrophage colony-stimulating factor, and iNOS which in turn creates a state of systemic inflammation [34,35]. It is characterized by fever, hypotension, hypoxia, nausea, fatigue, and cardiac dysfunction. Treatment includes supportive care, corticosteroids, and judicious use of tocilizumab, an Il-6 antagonist [36].
- Immune effector cell-associated neurotoxicity syndrome. ICANS is characterized by varied neurological symptoms including but not limited to headache, aphasia, memory loss, delirium, focal weakness, and seizures [37]. Although the pathophysiology is not entirely clear, endothelial activation, and multifocal vascular disruption leading to increased blood–brain barrier (BBB) permeability have been noted in patients with severe neurotoxicity. BBB disruption and consequent elevated concentrations of systemic cytokines in cerebrospinal fluid coupled with CNS-specific production of chemokines are thought to precipitate the neurological side effects [37,38].
2.2. Challenges of CAR T Cell Therapy in Lung Cancer
- Antigen escape. CAR-T cells that infiltrate the tumor are known to rapidly lose their activity as tumor cells evolve after exposure [25]. The loss of target antigens has previously been seen in ALL patients where durability of CAR response is hampered by emergence of CD 19 negative leukemia [39]. Potential pathways for such antigen escape include selection of cells with alternative target expression that lacks the binding site for CARs or lineage switching and phenotypic evolution of cancer cells [40,41]. In addition to hematological malignancies, this phenomenon of initial response and later resistance has also been seen in glioblastoma treated with intracranial CAR T cell therapy targeting IL13Rα2 and it has been hypothesized that decreased tumor burden and immune rejection of CAR T product could be responsible [42]. Thus, identifying a homogenously and steadily expressed target antigen is of utmost importance.
- Tumor heterogeneity. Cancers are dynamic and genomically unstable with spatial and temporal heterogeneity. While spatial heterogeneity refers to unequal distribution of genetically distinct tumor subpopulations across different disease sites or within a single disease site, temporal heterogeneity implies the evolution of tumors over time under different selection pressures [43]. The spatial heterogeneity and its impact on survival have been successfully demonstrated in lung cancer patients [44]. The ability of CARs to recognize a singular target in a constantly changing microenvironment that is also spatially diverse reduces its activity.
- Immunosuppressive TME, physical barrier, and T cell exhaustion. The activity of CAR T-cells is further impeded by immune response suppressive cells including MSCs, cancer-associated fibroblasts, TAMs, and regulatory T cells in TME. The stromal cells along with tumor cells release a host of immunosuppressive cytokines including TGF-β, IL-10, ARG-1, inducible nitric oxide synthase, COX2, PGE2, FAP, and PD-L1 to help the tumor evade CAR T cells [3,45]. Additionally, effective infiltration of T cells into the TME is affected by the density of the stromal extracellular matrix (ECM) with poor migration seen in areas of dense ECM [46]. Even after successful infiltration and engagement, the durability of CAR T response is downregulated by T cell exhaustion mediated by chronic antigen stimulation and upregulation of the NR4A transcription factor family [47].
2.3. Future Perspectives
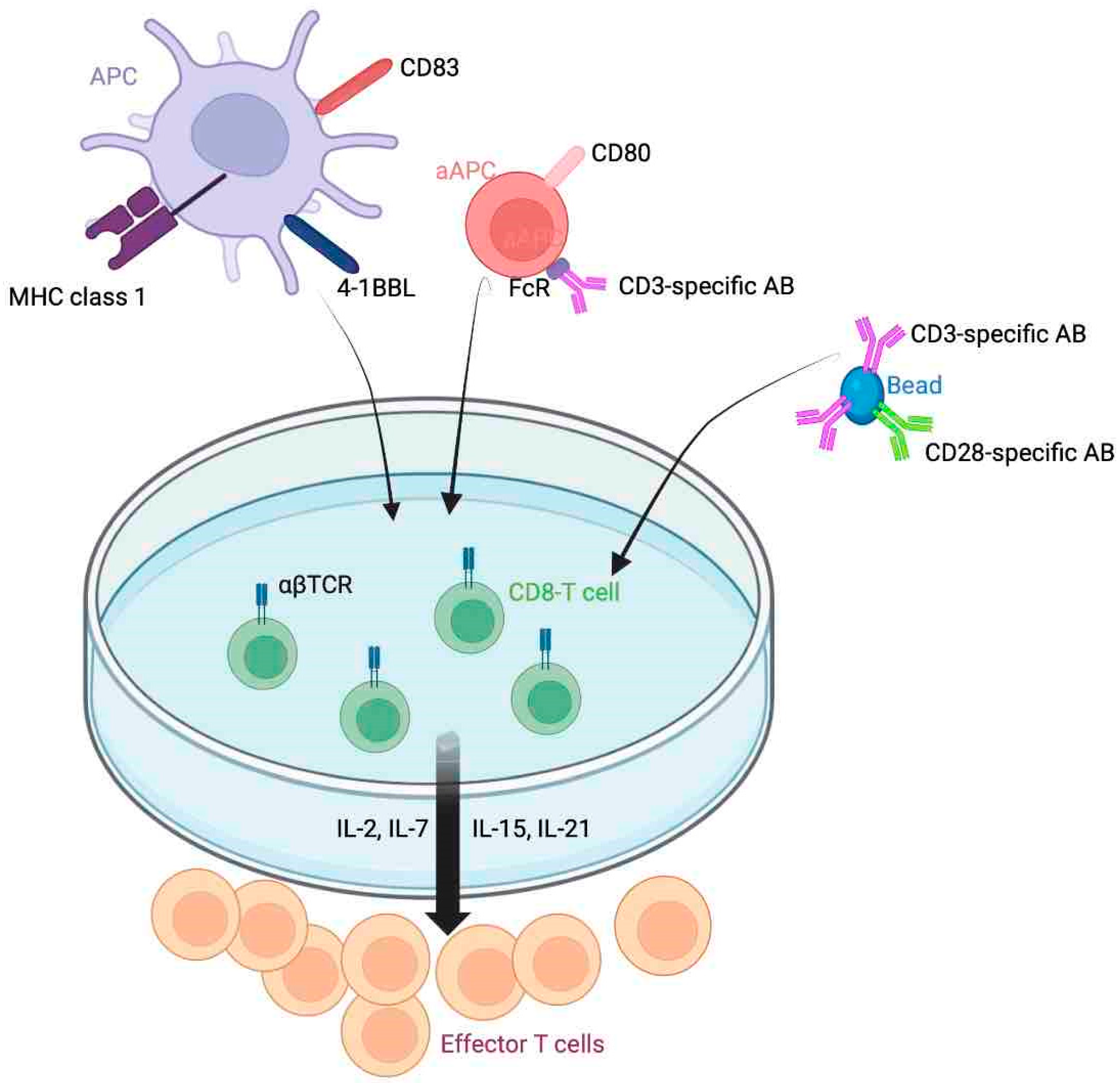
3. Tumor-Infiltrating Lymphocyte Therapy
3.1. Recent Clinical Trials
3.2. Challenges of TIL Therapy in Lung Cancer
3.3. Future Perspectives for TIL Therapy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clin. Cancer Res. 2016, 22, 4585–4593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Jeanson, A.; Tomasini, P.; Souquet-Bressand, M.; Brandone, N.; Boucekine, M.; Grangeon, M.; Chaleat, S.; Khobta, N.; Milia, J.; Mhanna, L.; et al. Efficacy of Immune Checkpoint Inhibitors in KRAS-Mutant Non-Small Cell Lung Cancer (NSCLC). J. Thorac. Oncol. 2019, 14, 1095–1101. [Google Scholar] [CrossRef]
- Hanna, N.; Shepherd, F.A.; Fossella, F.V.; Pereira, J.R.; De Marinis, F.; von Pawel, J.; Gatzemeier, U.; Tsao, T.C.; Pless, M.; Muller, T.; et al. Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. J. Clin. Oncol. 2004, 22, 1589–1597. [Google Scholar] [CrossRef]
- Garon, E.B.; Ciuleanu, T.E.; Arrieta, O.; Prabhash, K.; Syrigos, K.N.; Goksel, T.; Park, K.; Gorbunova, V.; Kowalyszyn, R.D.; Pikiel, J.; et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): A multicentre, double-blind, randomised phase 3 trial. Lancet 2014, 384, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Kraehenbuehl, L.; Weng, C.-H.; Eghbali, S.; Wolchok, J.D.; Merghoub, T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 2022, 19, 37–50. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Champiat, S.; Lai, W.V.; Izumi, H.; Govindan, R.; Boyer, M.; Hummel, H.D.; Borghaei, H.; Johnson, M.L.; Steeghs, N.; et al. Tarlatamab, a First-in-Class DLL3-Targeted Bispecific T-Cell Engager, in Recurrent Small-Cell Lung Cancer: An Open-Label, Phase I Study. J. Clin. Oncol. 2023, 41, 2893–2903. [Google Scholar] [CrossRef] [PubMed]
- Lasvergnas, J.; Naigeon, M.; Chouahnia, K.; Zelek, L.; Chaput, N.; Duchemann, B. Adoptive cell therapies in thoracic malignancies. Cancer Immunol. Immunother. CII 2022, 71, 2077–2098. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Miklos, D.B.; Jacobson, C.A.; Perales, M.A.; Kersten, M.J.; Oluwole, O.O.; Ghobadi, A.; Rapoport, A.P.; McGuirk, J.; Pagel, J.M.; et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 640–654. [Google Scholar] [CrossRef]
- Kamdar, M.; Solomon, S.R.; Arnason, J.; Johnston, P.B.; Glass, B.; Bachanova, V.; Ibrahimi, S.; Mielke, S.; Mutsaers, P.; Hernandez-Ilizaliturri, F.; et al. Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second-line treatment in patients with relapsed or refractory large B-cell lymphoma (TRANSFORM): Results from an interim analysis of an open-label, randomised, phase 3 trial. Lancet 2022, 399, 2294–2308. [Google Scholar] [CrossRef] [PubMed]
- Fowler, N.H.; Dickinson, M.; Dreyling, M.; Martinez-Lopez, J.; Kolstad, A.; Butler, J.; Ghosh, M.; Popplewell, L.; Chavez, J.C.; Bachy, E.; et al. Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: The phase 2 ELARA trial. Nat. Med. 2022, 28, 325–332. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Martin, T.; Usmani, S.Z.; Berdeja, J.G.; Agha, M.; Cohen, A.D.; Hari, P.; Avigan, D.; Deol, A.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, an Anti-B-cell Maturation Antigen Chimeric Antigen Receptor T-Cell Therapy, for Relapsed/Refractory Multiple Myeloma: CARTITUDE-1 2-Year Follow-Up. J. Clin. Oncol. 2023, 41, 1265–1274. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [Green Version]
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Abken, H. TRUCKS, the fourth-generation CAR T cells: Current developments and clinical translation. Adv. Cell Gene Ther. 2020, 3, e84. [Google Scholar] [CrossRef]
- Kandra, P.; Nandigama, R.; Eul, B.; Huber, M.; Kobold, S.; Seeger, W.; Grimminger, F.; Savai, R. Utility and Drawbacks of Chimeric Antigen Receptor T Cell (CAR-T) Therapy in Lung Cancer. Front. Immunol. 2022, 13, 903562. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Woo, E.Y.; Yeh, H.; Chu, C.S.; Schlienger, K.; Carroll, R.G.; Riley, J.L.; Kaiser, L.R.; June, C.H. Cutting edge: Regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation. J. Immunol. 2002, 168, 4272–4276. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Lamers, C.H.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef]
- Koshkin, V.S.; Garcia, J.A.; Reynolds, J.; Elson, P.; Magi-Galluzzi, C.; McKenney, J.K.; Isse, K.; Bishop, E.; Saunders, L.R.; Balyimez, A.; et al. Transcriptomic and Protein Analysis of Small-cell Bladder Cancer (SCBC) Identifies Prognostic Biomarkers and DLL3 as a Relevant Therapeutic Target. Clin. Cancer Res. 2019, 25, 210–221. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Huang, W.; Guo, Y.; Zhou, Y.; Zhi, C.; Chen, J.; Li, J.; He, J.; Lian, H.; Zhou, J.; et al. CAR NK-92 cells targeting DLL3 kill effectively small cell lung cancer cells in vitro and in vivo. J. Leukoc. Biol. 2022, 112, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tacheva-Grigorova, S.K.; Sutton, J.; Melton, Z.; Mak, Y.S.L.; Lay, C.; Smith, B.A.; Sai, T.; Van Blarcom, T.; Sasu, B.J.; et al. Allogeneic CAR T Cells Targeting DLL3 Are Efficacious and Safe in Preclinical Models of Small Cell Lung Cancer. Clin. Cancer Res. 2023, 29, 971–985. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood–Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [Green Version]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Zheng, X.; Weigert, A.; Reu, S.; Guenther, S.; Mansouri, S.; Bassaly, B.; Gattenlöhner, S.; Grimminger, F.; Pullamsetti, S.; Seeger, W.; et al. Spatial Density and Distribution of Tumor-Associated Macrophages Predict Survival in Non-Small Cell Lung Carcinoma. Cancer Res. 2020, 80, 4414–4425. [Google Scholar] [CrossRef]
- Mittal, V.; El Rayes, T.; Narula, N.; McGraw, T.E.; Altorki, N.K.; Barcellos-Hoff, M.H. The Microenvironment of Lung Cancer and Therapeutic Implications. Adv. Exp. Med. Biol. 2016, 890, 75–110. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; López-Moyado, I.F.; Seo, H.; Lio, C.-W.J.; Hempleman, L.J.; Sekiya, T.; Yoshimura, A.; Scott-Browne, J.P.; Rao, A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019, 567, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438.e1411. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Nandakumar, K.S.; Cheng, K. Optimization of CAR-T Cell-Based Therapies Using Small-Molecule-Based Safety Switches. J. Med. Chem. 2021, 64, 9577–9591. [Google Scholar] [CrossRef]
- Cao, Y.J.; Wang, X.; Wang, Z.; Zhao, L.; Li, S.; Zhang, Z.; Wei, X.; Yun, H.; Choi, S.-h.; Liu, Z.; et al. Switchable CAR-T Cells Outperformed Traditional Antibody-Redirected Therapeutics Targeting Breast Cancers. ACS Synth. Biol. 2021, 10, 1176–1183. [Google Scholar] [CrossRef]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, M.H.; Sadri, M.; Najafi, A.; Rahimi, A.; Baghernejadan, Z.; Khorramdelazad, H.; Falak, R. Tumor-infiltrating lymphocytes for treatment of solid tumors: It takes two to tango? Front. Immunol. 2022, 13, 1018962. [Google Scholar] [CrossRef] [PubMed]
- Creelan, B.C.; Wang, C.; Teer, J.K.; Toloza, E.M.; Yao, J.; Kim, S.; Landin, A.M.; Mullinax, J.E.; Saller, J.J.; Saltos, A.N.; et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: A phase 1 trial. Nat. Med. 2021, 27, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Aoki, Y.; Takakuwa, K.; Kodama, S.; Tanaka, K.; Takahashi, M.; Tokunaga, A.; Takahashi, T. Use of adoptive transfer of tumor-infiltrating lymphocytes alone or in combination with cisplatin-containing chemotherapy in patients with epithelial ovarian cancer. Cancer Res. 1991, 51, 1934–1939. [Google Scholar]
- Kradin, R.L.; Boyle, L.A.; Preffer, F.I.; Callahan, R.J.; Barlai-Kovach, M.; Strauss, H.W.; Dubinett, S.; Kurnick, J.T. Tumor-derived interleukin-2-dependent lymphocytes in adoptive immunotherapy of lung cancer. Cancer Immunol. Immunother. 1987, 24, 76–85. [Google Scholar] [CrossRef]
- Stevanović, S.; Draper, L.M.; Langhan, M.M.; Campbell, T.E.; Kwong, M.L.; Wunderlich, J.R.; Dudley, M.E.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J. Clin. Oncol. 2015, 33, 1543–1550. [Google Scholar] [CrossRef] [Green Version]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef]
- Galvano, A.; Gristina, V.; Malapelle, U.; Pisapia, P.; Pepe, F.; Barraco, N.; Castiglia, M.; Perez, A.; Rolfo, C.; Troncone, G.; et al. The prognostic impact of tumor mutational burden (TMB) in the first-line management of advanced non-oncogene addicted non-small-cell lung cancer (NSCLC): A systematic review and meta-analysis of randomized controlled trials. ESMO Open 2021, 6, 100124. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.B.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852.e844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kradin, R.L.; Kurnick, J.T.; Lazarus, D.S.; Preffer, F.I.; Dubinett, S.M.; Pinto, C.E.; Gifford, J.; Davidson, E.; Grove, B.; Callahan, R.J.; et al. Tumour-infiltrating lymphocytes and interleukin-2 in treatment of advanced cancer. Lancet 1989, 1, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Ratto, G.B.; Zino, P.; Mirabelli, S.; Minuti, P.; Aquilina, R.; Fantino, G.; Spessa, E.; Ponte, M.; Bruzzi, P.; Melioli, G. A randomized trial of adoptive immunotherapy with tumor-infiltrating lymphocytes and interleukin-2 versus standard therapy in the postoperative treatment of resected nonsmall cell lung carcinoma. Cancer 1996, 78, 244–251. [Google Scholar] [CrossRef]
- Geng, Y.; Shao, Y.; He, W.; Hu, W.; Xu, Y.; Chen, J.; Wu, C.; Jiang, J. Prognostic Role of Tumor-Infiltrating Lymphocytes in Lung Cancer: A Meta-Analysis. Cell Physiol. Biochem. 2015, 37, 1560–1571. [Google Scholar] [CrossRef]
- Hiraoka, K.; Miyamoto, M.; Cho, Y.; Suzuoki, M.; Oshikiri, T.; Nakakubo, Y.; Itoh, T.; Ohbuchi, T.; Kondo, S.; Katoh, H. Concurrent infiltration by CD8+ T cells and CD4+ T cells is a favourable prognostic factor in non-small-cell lung carcinoma. Br. J. Cancer 2006, 94, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Erfani, N.; Mehrabadi, S.M.; Ghayumi, M.A.; Haghshenas, M.R.; Mojtahedi, Z.; Ghaderi, A.; Amani, D. Increase of regulatory T cells in metastatic stage and CTLA-4 over expression in lymphocytes of patients with non-small cell lung cancer (NSCLC). Lung Cancer 2012, 77, 306–311. [Google Scholar] [CrossRef]
- Schneider, T.; Kimpfler, S.; Warth, A.; Schnabel, P.A.; Dienemann, H.; Schadendorf, D.; Hoffmann, H.; Umansky, V. Foxp3(+) regulatory T cells and natural killer cells distinctly infiltrate primary tumors and draining lymph nodes in pulmonary adenocarcinoma. J. Thorac. Oncol. 2011, 6, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Ben-Avi, R.; Farhi, R.; Ben-Nun, A.; Gorodner, M.; Greenberg, E.; Markel, G.; Schachter, J.; Itzhaki, O.; Besser, M.J. Establishment of adoptive cell therapy with tumor infiltrating lymphocytes for non-small cell lung cancer patients. Cancer Immunol. Immunother. 2018, 67, 1221–1230. [Google Scholar] [CrossRef]
- Schoenfeld, A.; Lee, S.; Paz-Ares, L.; Doger, B.; Gettinger, S.; Haefliger, S.; Orcurto, A.; Sukari, A.; Papa, S.; Moreno, J.F.R.; et al. 458 First phase 2 results of autologous tumor-infiltrating lymphocyte (TIL; LN-145) monotherapy in patients with advanced, immune checkpoint inhibitor-treated, non-small cell lung cancer (NSCLC). J. Immunother. Cancer 2021, 9, A1–A1054. [Google Scholar] [CrossRef]
- Zhao, Y.; Deng, J.; Rao, S.; Guo, S.; Shen, J.; Du, F.; Wu, X.; Chen, Y.; Li, M.; Chen, M.; et al. Tumor Infiltrating Lymphocyte (TIL) Therapy for Solid Tumor Treatment: Progressions and Challenges. Cancers 2022, 14, 4160. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, C.A.; Bennett, E.P.; Kverneland, A.H.; Svane, I.M.; Donia, M.; Met, Ö. Highly efficient PD-1-targeted CRISPR-Cas9 for tumor-infiltrating lymphocyte-based adoptive T cell therapy. Mol. Ther. Oncolytics 2022, 24, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.; Wise-Draper, T.; Sarnaik, A.A.; Graf Finckenstein, F.; Hari, P.; Jagasia, M.; Desai, A.; Suzuki, A.; Wu, X.; Betof Warner, A. 883TiP A phase I/II open-label study (IOV-GM1-201) of TALEN-mediated PD-1 inactivated autologous tumor-infiltrating lymphocytes (TIL; IOV-4001) in patients with advanced melanoma and NSCLC. Ann. Oncol. 2022, 33, S952. [Google Scholar] [CrossRef]
- Heemskerk, B.; Liu, K.; Dudley, M.E.; Johnson, L.A.; Kaiser, A.; Downey, S.; Zheng, Z.; Shelton, T.E.; Matsuda, K.; Robbins, P.F.; et al. Adoptive cell therapy for patients with melanoma, using tumor-infiltrating lymphocytes genetically engineered to secrete interleukin-2. Hum. Gene Ther. 2008, 19, 496–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Ye, Y.; Rabinovich, B.A.; Liu, C.; Lou, Y.; Zhang, M.; Whittington, M.; Yang, Y.; Overwijk, W.W.; Lizée, G.; et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin. Cancer Res. 2010, 16, 5458–5468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yu, Z.; Muranski, P.; Palmer, D.C.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Inhibition of TGF-β signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther. 2013, 20, 575–580. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Trial Identifier | Title | Phase | Types of Cancers |
|---|---|---|---|
| NCT05341492 | EGFR/B7H3 CAR T on lung cancer lung cancer and triple negative breast cancer | I | EGFR-B7H3 Positive cancers |
| NCT03198052 | GPC3/mesothelin/claudin18.2/GUCY2C/B7-H3/PSCA/PSMA/MUC1/TGFβ/HER2/Lewis-Y/AXL/EGFR-CAR T cells against cancers | I | Lung cancer |
| NCT05060796 | Study of CXCR5 modified EGFR targeted CAR T cells for advanced NSCLC | I | NSCLC |
| NCT05239143 | P-MUC1C-ALLO1 allogeneic CAR T cells in the treatment of subjects with advanced or metastatic solid tumors | I | NSCLC and multiple cancers |
| NCT05483491 | KK-LC-1 TCR-T cell therapy for gastric, breast, cervical, and lung cancer | I | Lung cancer and multiple cancers |
| NCT05274451 | A study to investigate LYL797 in adults with solid tumors | I | Breast cancer Lung cancer |
| NCT05120271 | BOXR1030 T cells in subjects with advanced GPC3-positive solid tumors | I | Squamous NSCLC |
| NCT03740256 | Binary oncolytic adenovirus in combination with HER2-specific autologous CAR T VST, advanced HER2 positive solid tumors (VISTA) | I | HER 2 positive lung and multiple cancers |
| NCT05736731 | A study to evaluate the safety and efficacy of A2B530, a logic-gated CAR T, in subjects with solid tumors that express CEA and have lost HLA-A*02 expression (EVEREST-1) | I/II | NSCLC and multiple cancers |
| NCT04348643 | Safety and efficacy of CEA-targeted CAR T therapy for relapsed/refractory CEA+ cancer | I/II | NSCLC and multiple cancers |
| NCT03198546 | GPC3-CAR T cells for immunotherapy of cancer with GPC3 expression | I | Squamous cell lung cancer, HCC |
| NCT02414269 | Malignant pleural disease treated with autologous T cells genetically engineered to target the cancer-cell surface antigen mesothelin | I/II | NSCLC and multiple cancers |
| Trial Identifier | Title | Phase | Types of Cancer |
|---|---|---|---|
| NCT04614103 | Autologous LN-145 in patients with metastatic non-small-cell lung cancer | II | NSCLC |
| NCT05681780 | Clinical trial of CD40L-augmented TIL for patients with EGFR, ALK, ROS1, or HER2-Driven NSCLC | I/II | NSCLC |
| NCT02133196 | T cell receptor immunotherapy for patients with metastatic non-small cell lung cancer | II | NSCLC |
| NCT05366478 | A clinical study of LM103 injection in the treatment of advanced solid tumors | I | NSCLC Cervical cancer Melanoma |
| NCT03645928 | Study of autologous tumors infiltrating lymphocytes in patients with solid tumors | II | Lung Head and neck Melanoma |
| NCT05361174 | A study to investigate the efficacy and safety of an infusion of IOV-4001 in adult participants with unresectable or metastatic melanoma or stage III or IV non-small-cell lung cancer | I/II | Melanoma NSCLC |
| NCT05573035 | A study to investigate LYL845 in adults with solid tumors | I | Melanoma Lung cancer Colorectal cancer |
| NCT03778814 | TCR-T cell immunotherapy of lung cancer and other solid tumors | I | NSCLC Solid tumors |
| NCT03215810 | Nivolumab and tumor infiltrating lymphocytes (TIL) in advanced non-small cell lung cancer | I | NSCLC |
| NCT05878028 | L-TIL plus tislelizumab for PD1 antibody resistant aNSCLC | II | NSCLC |
| NCT05397093 | ITIL-306 in advanced solid tumors | I | NSCLC Epithelial ovarian and Renal cell carcinoma |
| NCT05576077 | A study of TBio-4101 (TIL) and pembrolizumab in patients with advanced solid tumors (STARLING) | I | NSCLC and multiple other cancers |
| NCT05680922 | DLL3-directed chimeric antigen receptor T cells in subjects with extensive stage small cell lung cancer | I | SCLC and large cell neuroendocrine carcinoma of lung |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katiyar, V.; Chesney, J.; Kloecker, G. Cellular Therapy for Lung Cancer: Focusing on Chimeric Antigen Receptor T (CAR T) Cells and Tumor-Infiltrating Lymphocyte (TIL) Therapy. Cancers 2023, 15, 3733. https://doi.org/10.3390/cancers15143733
Katiyar V, Chesney J, Kloecker G. Cellular Therapy for Lung Cancer: Focusing on Chimeric Antigen Receptor T (CAR T) Cells and Tumor-Infiltrating Lymphocyte (TIL) Therapy. Cancers. 2023; 15(14):3733. https://doi.org/10.3390/cancers15143733
Chicago/Turabian StyleKatiyar, Vatsala, Jason Chesney, and Goetz Kloecker. 2023. "Cellular Therapy for Lung Cancer: Focusing on Chimeric Antigen Receptor T (CAR T) Cells and Tumor-Infiltrating Lymphocyte (TIL) Therapy" Cancers 15, no. 14: 3733. https://doi.org/10.3390/cancers15143733
APA StyleKatiyar, V., Chesney, J., & Kloecker, G. (2023). Cellular Therapy for Lung Cancer: Focusing on Chimeric Antigen Receptor T (CAR T) Cells and Tumor-Infiltrating Lymphocyte (TIL) Therapy. Cancers, 15(14), 3733. https://doi.org/10.3390/cancers15143733