
TNFR1 and TNFR2, Which Link NF-κB Activation, Drive Lung Cancer Progression, Cell Dedifferentiation, and Metastasis


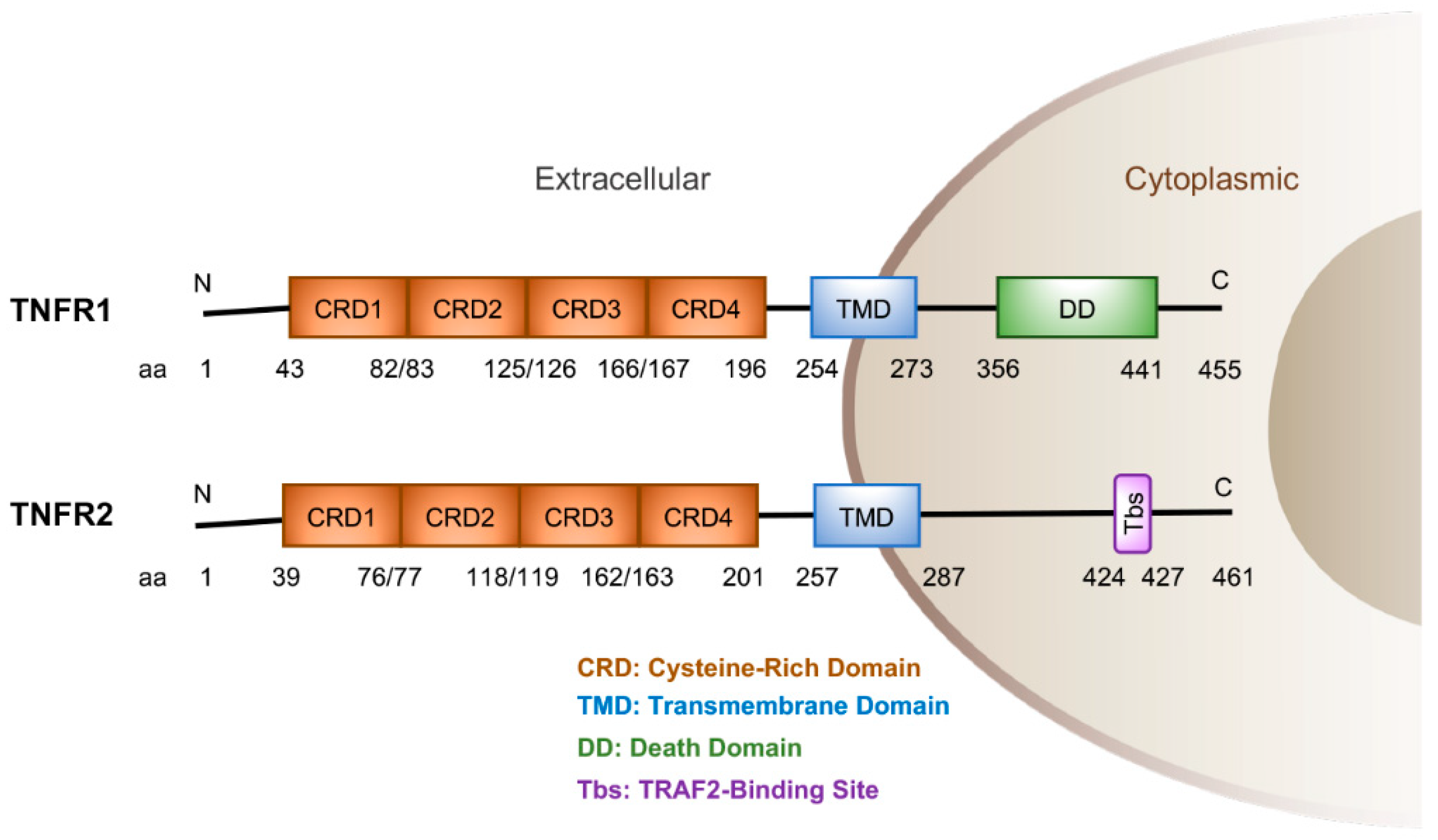{kind=link}
{kind=link}
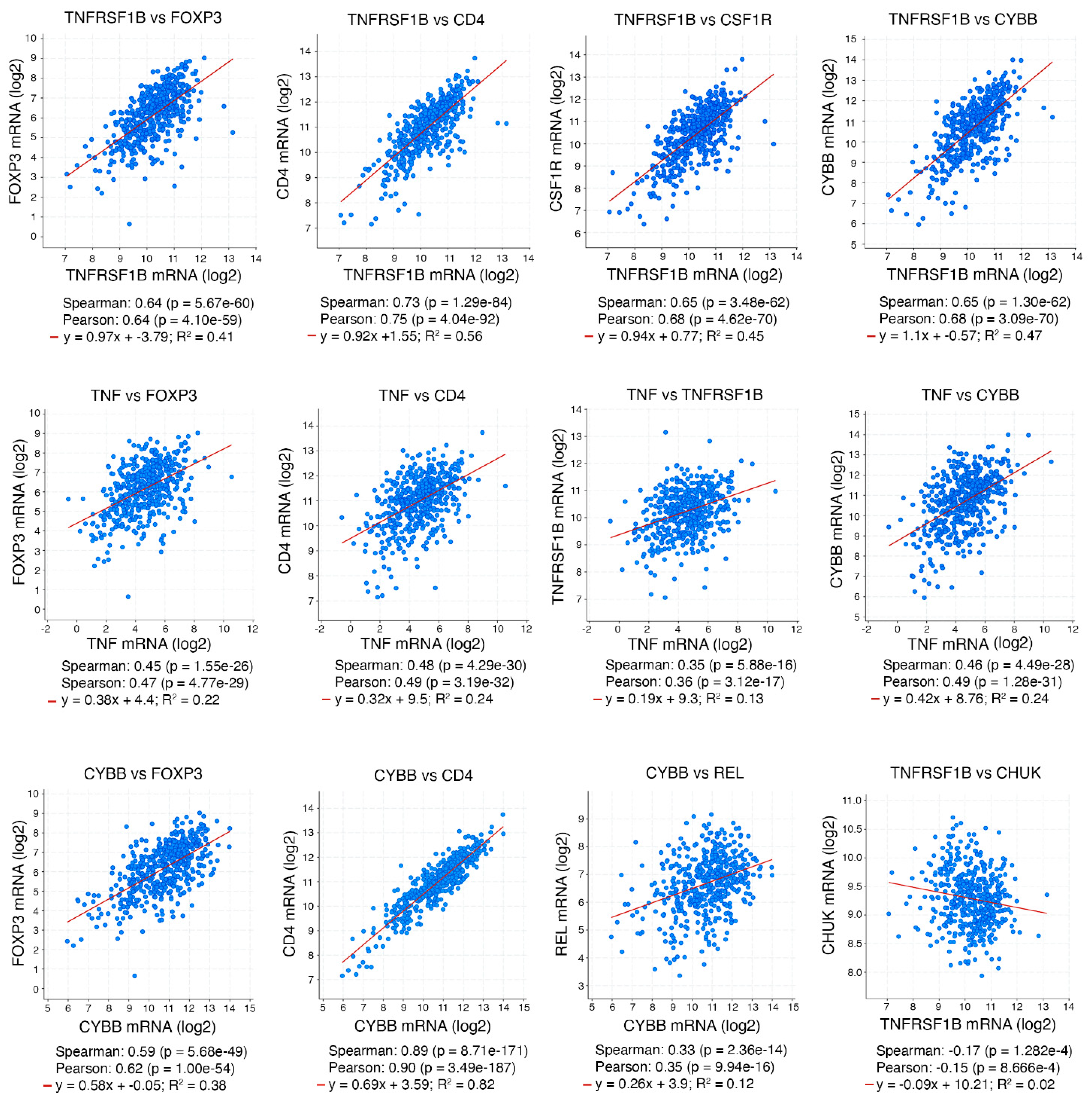{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. TNFR1 Overexpression Promotes Lung SCC Cell Dedifferentiation and Metastasis
2.1. Background
2.2. TNFR1 in Human Lung SCC
2.3. A Lung SCC Mouse Model
2.4. TNFR1 Induction in Lung SCC Development in Human and Mice
2.5. Increased TNFR1 in Lung SCC Cells Drives Cancer Stemness, Dedifferentiation, and Metastasis
2.6. TNFR1 Levels Correlate with Induced NF-κB Activity That is Required for UBE2C/Ube2c Promoter Activity
3. Role of TNFR2 in Lung Adenocarcinoma Progression
3.1. Background
3.2. TNFR2 in CD4 T Cells Is Required for Generating an iTME with Increased Treg Numbers
4. Conclusions and Therapeutic Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xiao, Z.; Jiang, Q.; Willette-Brown, J.; Xi, S.; Zhu, F.; Burkett, S.; Back, T.; Song, N.Y.; Datla, M.; Sun, Z.; et al. The Pivotal Role of IKKalpha in the Development of Spontaneous Lung Squamous Cell Carcinomas. Cancer Cell 2013, 23, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Song, N.Y.; Zhu, F.; Wang, Z.; Willette-Brown, J.; Xi, S.; Sun, Z.; Su, L.; Wu, X.; Ma, B.; Nussinov, R.; et al. IKKalpha inactivation promotes Kras-initiated lung adenocarcinoma development through disrupting major redox regulatory pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E812–E821. [Google Scholar] [CrossRef] [PubMed]
- Hammerman, P.S.; Hayes, D.N.; Wilkerson, M.D.; Schultz, N.; Bose, R.; Chu, A.; Collisson, E.A.; Cope, L.; Creighton, C.J.; Getz, G.; et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar]
- Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Chavdoula, E.; Habiel, D.M.; Roupakia, E.; Markopoulos, G.S.; Vasilaki, E.; Kokkalis, A.; Polyzos, A.P.; Boleti, H.; Thanos, D.; Klinakis, A.; et al. CHUK/IKK-alpha loss in lung epithelial cells enhances NSCLC growth associated with HIF up-regulation. Life Sci. Alliance 2019, 2, e201900460. [Google Scholar] [CrossRef] [PubMed]
- DuPage, M.; Dooley, A.L.; Jacks, T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat. Protoc. 2009, 4, 1064–1072. [Google Scholar] [CrossRef]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef]
- Pan, Y.; Han, H.; Hu, H.; Wang, H.; Song, Y.; Hao, Y.; Tong, X.; Patel, A.S.; Misirlioglu, S.; Tang, S.; et al. KMT2D deficiency drives lung squamous cell carcinoma and hypersensitivity to RTK-RAS inhibition. Cancer Cell 2023, 41, 88–105.e108. [Google Scholar] [CrossRef]
- Ferone, G.; Song, J.Y.; Sutherland, K.D.; Bhaskaran, R.; Monkhorst, K.; Lambooij, J.P.; Proost, N.; Gargiulo, G.; Berns, A. SOX2 Is the Determining Oncogenic Switch in Promoting Lung Squamous Cell Carcinoma from Different Cells of Origin. Cancer Cell 2016, 30, 519–532. [Google Scholar] [CrossRef]
- Xiao, Z.; Shi, G.; Xi, S.; Singh, A.K.; Willette-Brown, J.; Li, X.; Zhu, F.; Su, L.; Wu, X.; Schrump, D.S.; et al. A TNFR1-UBCH10 axis drives lung squamous cell carcinoma dedifferentiation and metastasis through a cell-autonomous signaling loop. Cell Death Dis. 2022, 13, 885. [Google Scholar] [CrossRef]
- Song, N.Y.; Li, X.; Ma, B.; Willette-Brown, J.; Zhu, F.; Jiang, C.; Su, L.; Shetty, J.; Zhao, Y.; Shi, G.; et al. IKKalpha-deficient lung adenocarcinomas generate an immunosuppressive microenvironment by overproducing Treg-inducing cytokines. Proc. Natl. Acad. Sci. USA 2022, 119, e2120956119. [Google Scholar] [CrossRef]
- Li, Z.W.; Chu, W.; Hu, Y.; Delhase, M.; Deerinck, T.; Ellisman, M.; Johnson, R.; Karin, M. The IKKb subunit of IkB kinase (IKK) is essential for nuclear factor kB activation and prevention of apoptosis. J. Exp. Med. 1999, 189, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Baud, V.; Oga, T.; Kim, K.I.; Yoshida, K.; Karin, M. IKKa controls formation of the epidermis independently of NF-kB. Nature 2001, 410, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hu, Y. Attribution of NF-kappaB Activity to CHUK/IKKalpha-Involved Carcinogenesis. Cancers 2021, 13, 1411. [Google Scholar] [CrossRef]
- Liu, B.; Xia, X.; Zhu, F.; Park, E.; Carbajal, S.; Kiguchi, K.; DiGiovanni, J.; Fischer, S.M.; Hu, Y. IKKalpha is required to maintain skin homeostasis and prevent skin cancer. Cancer Cell 2008, 14, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Willette-Brown, J.; Song, N.Y.; Lomada, D.; Song, Y.; Xue, L.; Gray, Z.; Zhao, Z.; Davis, S.R.; Sun, Z.; et al. Autoreactive T Cells and Chronic Fungal Infection Drive Esophageal Carcinogenesis. Cell Host Microbe 2017, 21, 478–493.e477. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, M.; Okabe, S.; Marino, M.W.; Sakai, A.; Sueoka, E.; Fujiki, H. Essential role of tumor necrosis factor alpha (TNF-alpha) in tumor promotion as revealed by TNF-alpha-deficient mice. Cancer Res. 1999, 59, 4516–4518. [Google Scholar]
- Moore, R.J.; Owens, D.M.; Stamp, G.; Arnott, C.; Burke, F.; East, N.; Holdsworth, H.; Turner, L.; Rollins, B.; Pasparakis, M.; et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat. Med. 1999, 5, 828–831. [Google Scholar] [CrossRef]
- Hoste, E.; Arwert, E.N.; Lal, R.; South, A.P.; Salas-Alanis, J.C.; Murrell, D.F.; Donati, G.; Watt, F.M. Innate sensing of microbial products promotes wound-induced skin cancer. Nat. Commun. 2015, 6, 5932. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef]
- Gong, K.; Guo, G.; Beckley, N.; Zhang, Y.; Yang, X.; Sharma, M.; Habib, A.A. Tumor necrosis factor in lung cancer: Complex roles in biology and resistance to treatment. Neoplasia 2021, 23, 189–196. [Google Scholar] [CrossRef]
- Wang, X.; Gray, Z.; Willette-Brown, J.; Zhu, F.; Shi, G.; Jiang, Q.; Song, N.Y.; Dong, L.; Hu, Y. Macrophage inducible nitric oxide synthase circulates inflammation and promotes lung carcinogenesis. Cell Death Discov. 2018, 4, 46. [Google Scholar] [CrossRef]
- Gray, Z.; Shi, G.; Wang, X.; Hu, Y. Macrophage inducible nitric oxide synthase promotes the initiation of lung squamous cell carcinoma by maintaining circulated inflammation. Cell Death Dis. 2018, 9, 642. [Google Scholar] [CrossRef]
- Maeda, G.; Chiba, T.; Kawashiri, S.; Satoh, T.; Imai, K. Epigenetic inactivation of IkappaB Kinase-alpha in oral carcinomas and tumor progression. Clin. Cancer Res. 2007, 13, 5041–5047. [Google Scholar] [CrossRef]
- Xia, X.; Park, E.; Liu, B.; Willette-Brown, J.; Gong, W.; Wang, J.; Mitchell, D.; Fischer, S.M.; Hu, Y. Reduction of IKKalpha expression promotes chronic ultraviolet B exposure-induced skin inflammation and carcinogenesis. Am. J. Pathol. 2010, 176, 2500–2508. [Google Scholar] [CrossRef] [PubMed]
- Bass, A.J.; Watanabe, H.; Mermel, C.H.; Yu, S.; Perner, S.; Verhaak, R.G.; Kim, S.Y.; Wardwell, L.; Tamayo, P.; Gat-Viks, I.; et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat. Genet. 2009, 41, 1238–1242. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Unternaehrer, J.J. Epithelial-mesenchymal Transition and Cancer Stem Cells: At the Crossroads of Differentiation and Dedifferentiation. Dev. Dyn. 2019, 248, 10–20. [Google Scholar] [CrossRef]
- Lobito, A.A.; Kimberley, F.C.; Muppidi, J.R.; Komarow, H.; Jackson, A.J.; Hull, K.M.; Kastner, D.L.; Screaton, G.R.; Siegel, R.M. Abnormal disulfide-linked oligomerization results in ER retention and altered signaling by TNFR1 mutants in TNFR1-associated periodic fever syndrome (TRAPS). Blood 2006, 108, 1320–1327. [Google Scholar] [CrossRef]
- Karathanasis, C.; Medler, J.; Fricke, F.; Smith, S.; Malkusch, S.; Widera, D.; Fulda, S.; Wajant, H.; van Wijk, S.J.L.; Dikic, I.; et al. Single-molecule imaging reveals the oligomeric state of functional TNFalpha-induced plasma membrane TNFR1 clusters in cells. Sci. Signal 2020, 13, eaax5647. [Google Scholar] [CrossRef]
- van Ree, J.H.; Jeganathan, K.B.; Malureanu, L.; van Deursen, J.M. Overexpression of the E2 ubiquitin-conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. J. Cell Biol. 2010, 188, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Zhang, H.; Cowell, J. Ubiquitin-conjugating enzyme UBE2C: Molecular biology, role in tumorigenesis, and potential as a biomarker. Tumour Biol. 2012, 33, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yin, L.; Yang, L.; Zheng, Y.; Liu, S.; Yang, J.; Cui, H.; Wang, H. Silencing ubiquitin-conjugating enzyme 2C inhibits proliferation and epithelial-mesenchymal transition in pancreatic ductal adenocarcinoma. FEBS J. 2019, 286, 4889–4909. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, R.; Chi, S.; Zhang, W.; Xiao, C.; Zhou, X.; Zhao, Y.; Wang, H. UBE2C Is Upregulated by Estrogen and Promotes Epithelial-Mesenchymal Transition via p53 in Endometrial Cancer. Mol. Cancer Res. 2020, 18, 204–215. [Google Scholar] [CrossRef]
- Liu, W.; Lu, X.; Shi, P.; Yang, G.; Zhou, Z.; Li, W.; Mao, X.; Jiang, D.; Chen, C. TNF-alpha increases breast cancer stem-like cells through up-regulating TAZ expression via the non-canonical NF-kappaB pathway. Sci. Rep. 2020, 10, 1804. [Google Scholar] [CrossRef] [PubMed]
- Kunsch, C.; Ruben, S.M.; Rosen, C.A. Selection of optimal kappa B/Rel DNA-binding motifs: Interaction of both subunits of NF-kappa B with DNA is required for transcriptional activation. Mol. Cell Biol. 1992, 12, 4412–4421. [Google Scholar] [CrossRef] [PubMed]
- van Hogerlinden, M.; Rozell, B.L.; Ahrlund-Richter, L.; Toftgard, R. Squamous cell carcinomas and increased apoptosis in skin with inhibited Rel/nuclear factor-kB signaling. Cancer Res. 1999, 59, 3299–3303. [Google Scholar]
- Lind, M.H.; Rozell, B.; Wallin, R.P.; van Hogerlinden, M.; Ljunggren, H.G.; Toftgard, R.; Sur, I. Tumor necrosis factor receptor 1-mediated signaling is required for skin cancer development induced by NF-kappaB inhibition. Proc. Natl. Acad. Sci. USA 2004, 101, 4972–4977. [Google Scholar] [CrossRef]
- Park, E.; Zhu, F.; Liu, B.; Xia, X.; Shen, J.; Bustos, T.; Fischer, S.M.; Hu, Y. Reduction in IkappaB kinase alpha expression promotes the development of skin papillomas and carcinomas. Cancer Res. 2007, 67, 9158–9168. [Google Scholar] [CrossRef]
- Liu, B.; Park, E.; Zhu, F.; Bustos, T.; Liu, J.; Shen, J.; Fischer, S.M.; Hu, Y. A critical role for I{kappa}B kinase {alpha} in the development of human and mouse squamous cell carcinomas. Proc. Natl. Acad. Sci. USA 2006, 103, 17202–17207. [Google Scholar] [CrossRef]
- Meylan, E.; Dooley, A.L.; Feldser, D.M.; Shen, L.; Turk, E.; Ouyang, C.; Jacks, T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 2009, 462, 104–107. [Google Scholar] [CrossRef]
- Xue, W.; Meylan, E.; Oliver, T.G.; Feldser, D.M.; Winslow, M.M.; Bronson, R.; Jacks, T. Response and resistance to NF-kappaB inhibitors in mouse models of lung adenocarcinoma. Cancer Discov. 2011, 1, 236–247. [Google Scholar] [CrossRef]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Muppidi, J.R.; Tschopp, J.; Siegel, R.M. Life and death decisions: Secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity 2004, 21, 461–465. [Google Scholar] [CrossRef]
- Chen, X.; Wu, X.; Zhou, Q.; Howard, O.M.; Netea, M.G.; Oppenheim, J.J. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype in the inflammatory environment. J. Immunol. 2013, 190, 1076–1084. [Google Scholar] [CrossRef]
- Long, M.; Park, S.G.; Strickland, I.; Hayden, M.S.; Ghosh, S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 2009, 31, 921–931. [Google Scholar] [CrossRef]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.S.; Ghosh, S. NF-kappaB c-Rel Is Crucial for the Regulatory T Cell Immune Checkpoint in Cancer. Cell 2017, 170, 1096–1108.e1013. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Grinberg-Bleyer, Y.; Liao, W.; Maloney, D.; Wang, P.; Wu, Z.; Wang, J.; Bhatt, D.M.; Heise, N.; Schmid, R.M.; et al. An NF-kappaB Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity 2017, 47, 450–465.e455. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.S.; Akama-Garren, E.H.; Lu, Y.; Lee, D.Y.; Chang, G.P.; Li, A.; DuPage, M.; Tammela, T.; Kerper, N.R.; Farago, A.F.; et al. Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses. Immunity 2015, 43, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Langowski, J.L.; Zhang, X.; Wu, L.; Mattson, J.D.; Chen, T.; Smith, K.; Basham, B.; McClanahan, T.; Kastelein, R.A.; Oft, M. IL-23 promotes tumour incidence and growth. Nature 2006, 442, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Benoot, T.; Piccioni, E.; De Ridder, K.; Goyvaerts, C. TNFalpha and Immune Checkpoint Inhibition: Friend or Foe for Lung Cancer? Int. J. Mol. Sci. 2021, 22, 8691. [Google Scholar] [CrossRef]
- Steeland, S.; Libert, C.; Vandenbroucke, R.E. A New Venue of TNF Targeting. Int. J. Mol. Sci. 2018, 19, 1442. [Google Scholar] [CrossRef]
- Creaven, P.J.; Plager, J.E.; Dupere, S.; Huben, R.P.; Takita, H.; Mittelman, A.; Proefrock, A. Phase I clinical trial of recombinant human tumor necrosis factor. Cancer Chemother. Pharmacol. 1987, 20, 137–144. [Google Scholar] [CrossRef]
- Feinberg, B.; Kurzrock, R.; Talpaz, M.; Blick, M.; Saks, S.; Gutterman, J.U. A phase I trial of intravenously-administered recombinant tumor necrosis factor-alpha in cancer patients. J. Clin. Oncol. 1988, 6, 1328–1334. [Google Scholar] [CrossRef]
- Roberts, N.J.; Zhou, S.; Diaz, L.A., Jr.; Holdhoff, M. Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget 2011, 2, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Schipper, J.H.; Frixen, U.H.; Behrens, J.; Unger, A.; Jahnke, K.; Birchmeier, W. E-cadherin expression in squamous cell carcinomas of head and neck: Inverse correlation with tumor dedifferentiation and lymph node metastasis. Cancer Res. 1991, 51, 6328–6337. [Google Scholar] [PubMed]
- Richter, F.; Williams, S.K.; John, K.; Huber, C.; Vaslin, C.; Zanker, H.; Fairless, R.; Pichi, K.; Marhenke, S.; Vogel, A.; et al. The TNFR1 Antagonist Atrosimab Is Therapeutic in Mouse Models of Acute and Chronic Inflammation. Front. Immunol. 2021, 12, 705485. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Colangelo, J. Small-molecule inhibitors of the interaction between TNF and TNFR. Future Med. Chem. 2013, 5, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Wu, H.; Zhang, X.; Huang, J.; He, X.; Chen, L.; Guo, W.; Guo, X.; Hao, B.; Li, Y. Pre-clinical study of a TNFR1-targeted (18)F probe for PET imaging of breast cancer. Amino Acids 2018, 50, 409–419. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; Waddelow, T.; Kantarjian, H.M.; Albitar, M.; Giles, F.J. Pilot study of recombinant human soluble tumor necrosis factor (TNF) receptor (p75) fusion protein (TNFR:Fc; Enbrel) in patients with refractory multiple myeloma: Increase in plasma TNF alpha levels during treatment. Leuk. Res. 2003, 27, 375–380. [Google Scholar] [CrossRef]
- Sprott, H.; Glatzel, M.; Michel, B.A. Treatment of myositis with etanercept (Enbrel), a recombinant human soluble fusion protein of TNF-alpha type II receptor and IgG1. Rheumatology 2004, 43, 524–526. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, G.; Hu, Y. TNFR1 and TNFR2, Which Link NF-κB Activation, Drive Lung Cancer Progression, Cell Dedifferentiation, and Metastasis. Cancers 2023, 15, 4299. https://doi.org/10.3390/cancers15174299
Shi G, Hu Y. TNFR1 and TNFR2, Which Link NF-κB Activation, Drive Lung Cancer Progression, Cell Dedifferentiation, and Metastasis. Cancers. 2023; 15(17):4299. https://doi.org/10.3390/cancers15174299
Chicago/Turabian StyleShi, Gongping, and Yinling Hu. 2023. "TNFR1 and TNFR2, Which Link NF-κB Activation, Drive Lung Cancer Progression, Cell Dedifferentiation, and Metastasis" Cancers 15, no. 17: 4299. https://doi.org/10.3390/cancers15174299
APA StyleShi, G., & Hu, Y. (2023). TNFR1 and TNFR2, Which Link NF-κB Activation, Drive Lung Cancer Progression, Cell Dedifferentiation, and Metastasis. Cancers, 15(17), 4299. https://doi.org/10.3390/cancers15174299