
IL-15 Prevents the Development of T-ALL from Aberrant Thymocytes with Impaired DNA Repair Functions and Increased NOTCH1 Activation
 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Monitoring Leukemia Development
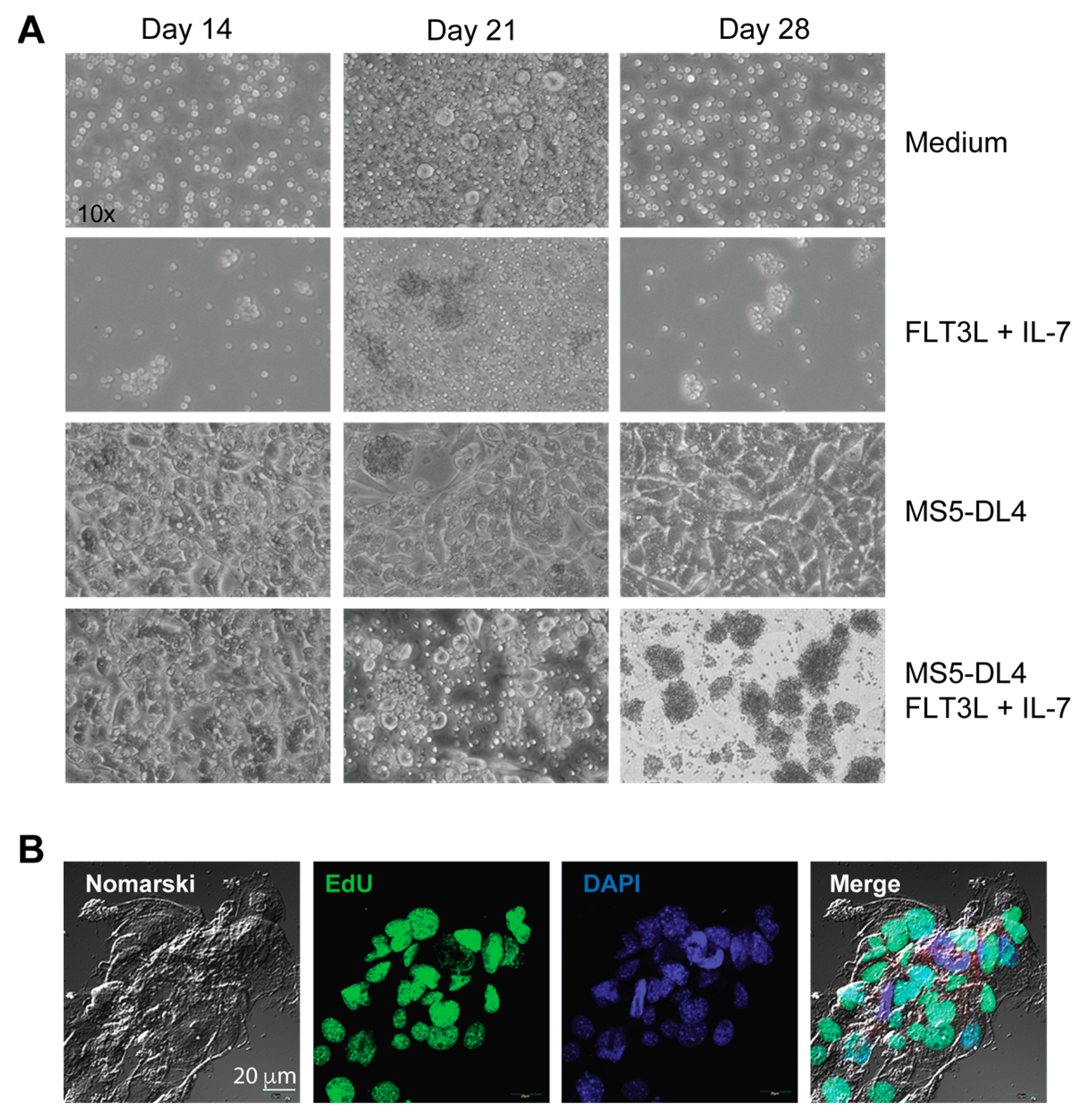
2.3. Cell Lines, Proliferation Assay and Co-Cultures
2.4. Flow Cytometry
2.5. SDS-PAGE and Western Blot
2.6. RNA Extraction and RT–qPCR
2.7. Statistical Analyses
3. Results
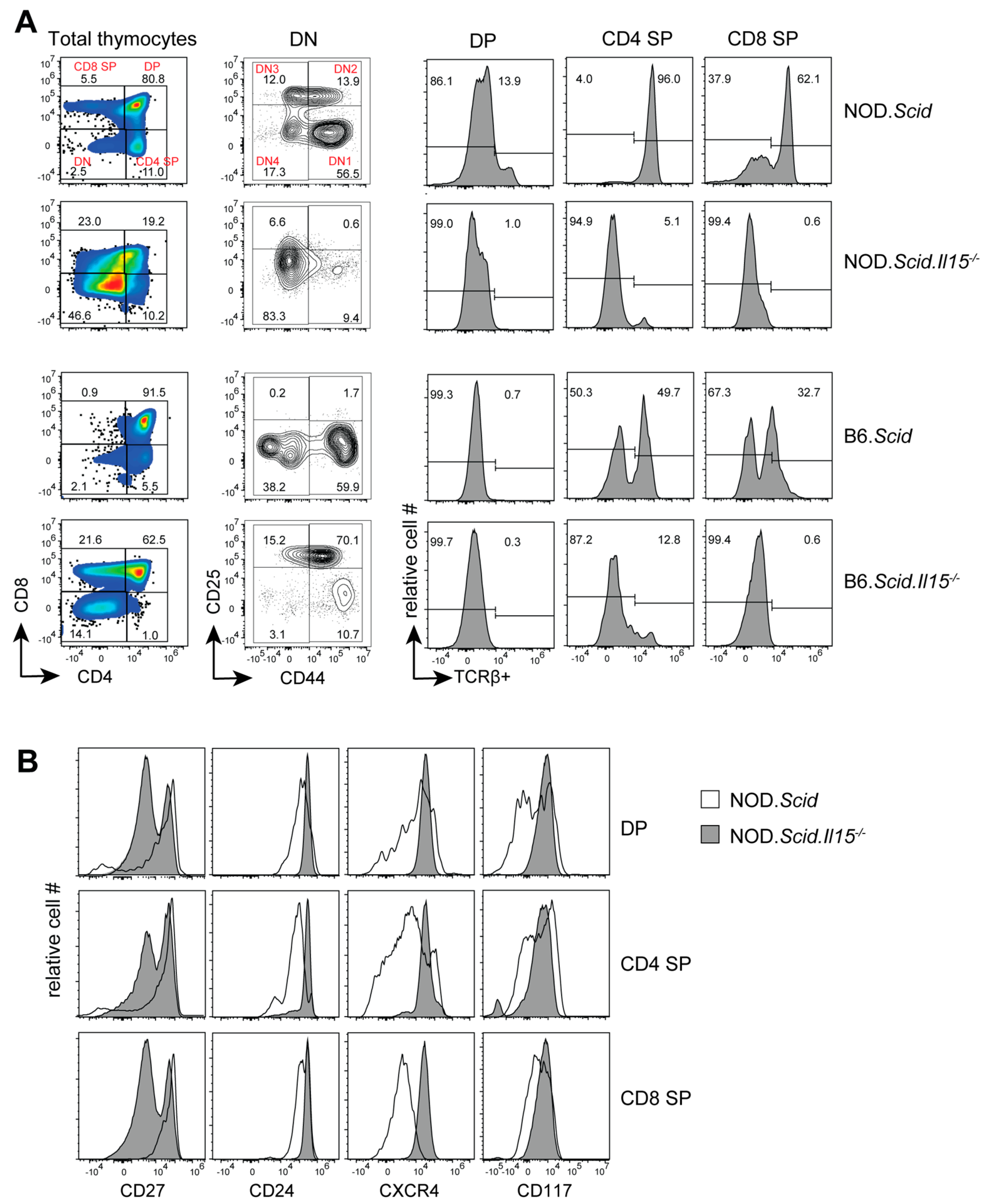
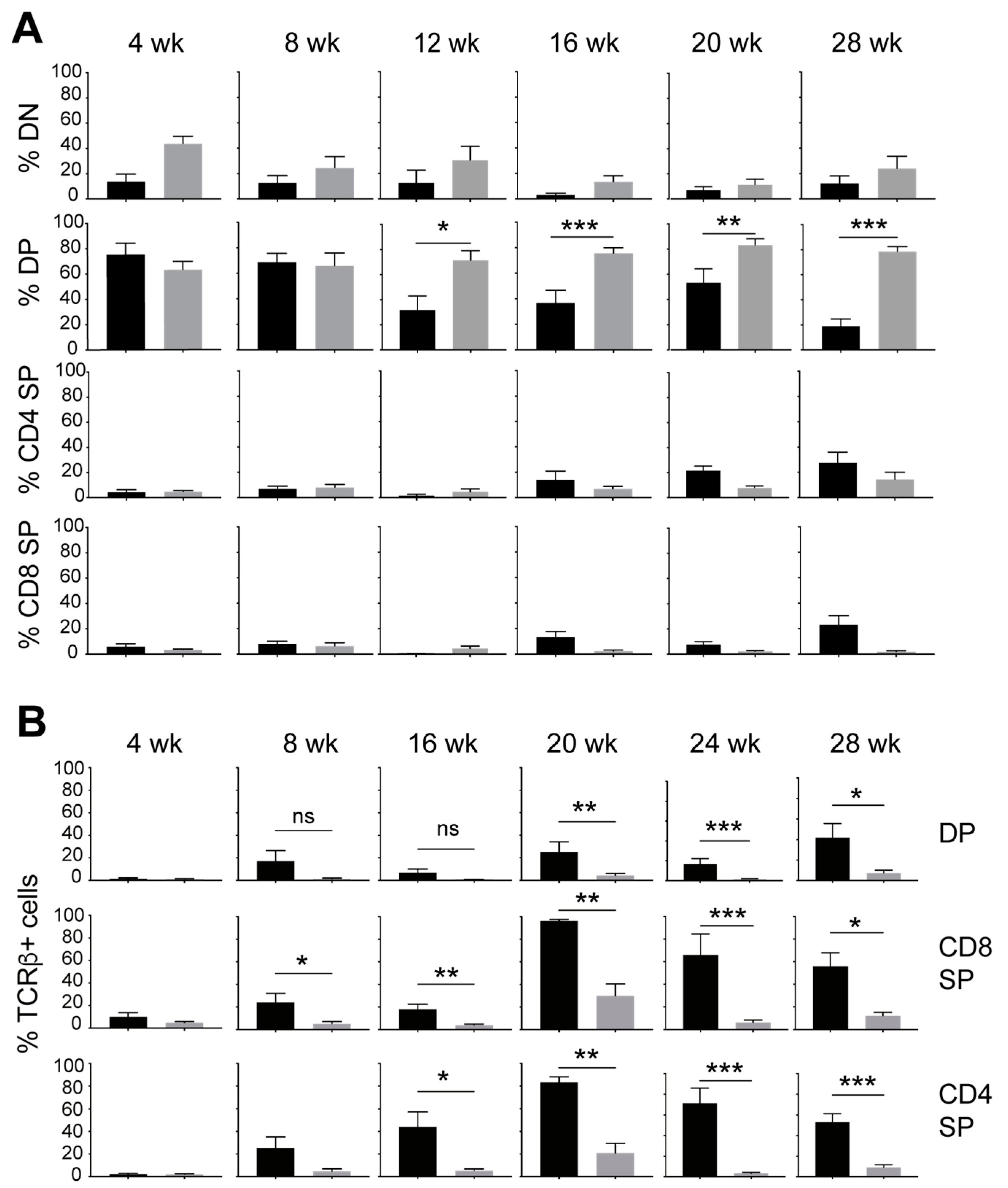
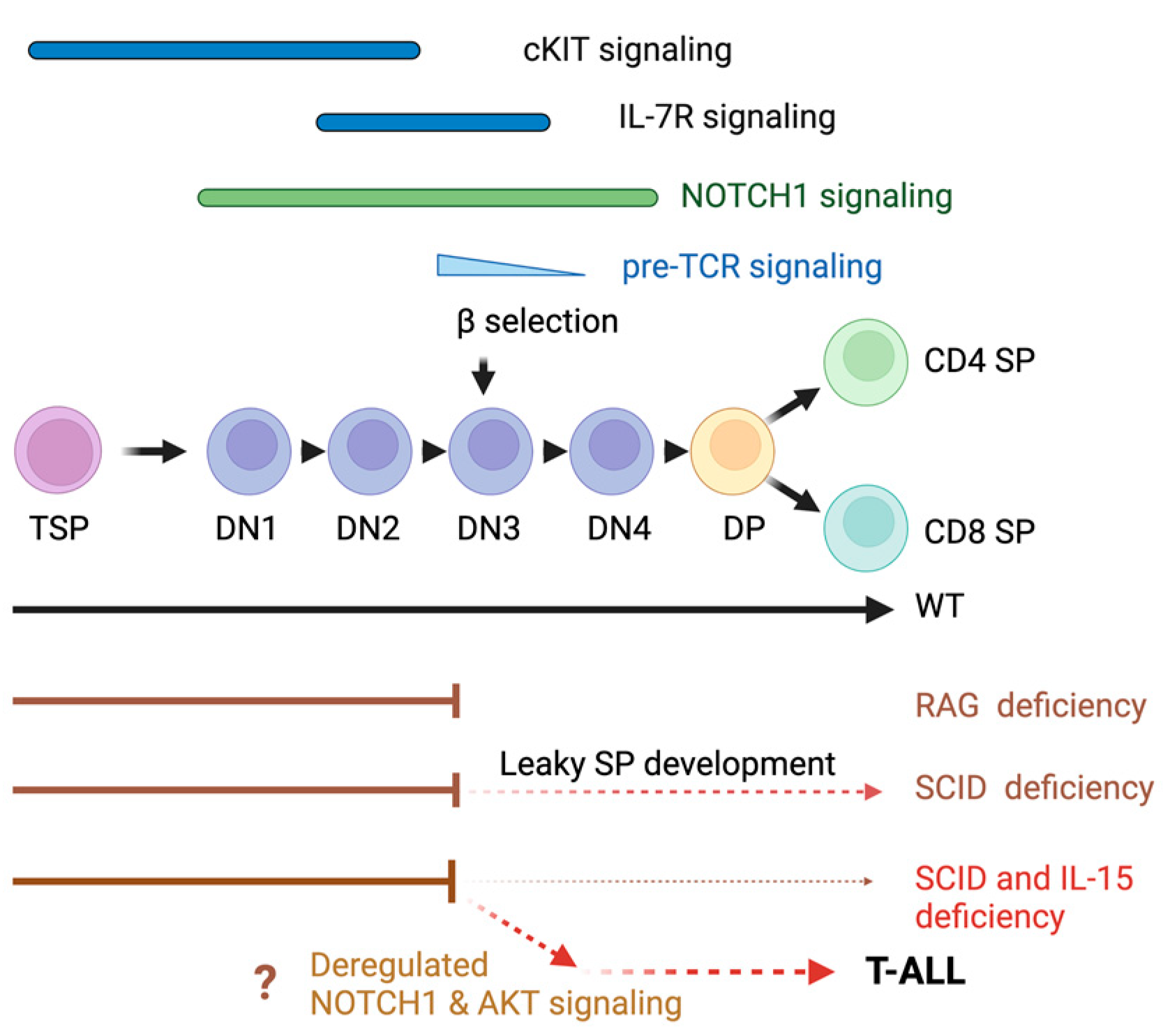
3.1. IL-15 Deficiency in the Scid Genetic Background Facilitates the Development of Aberrant Thymocytes Lacking the TCR
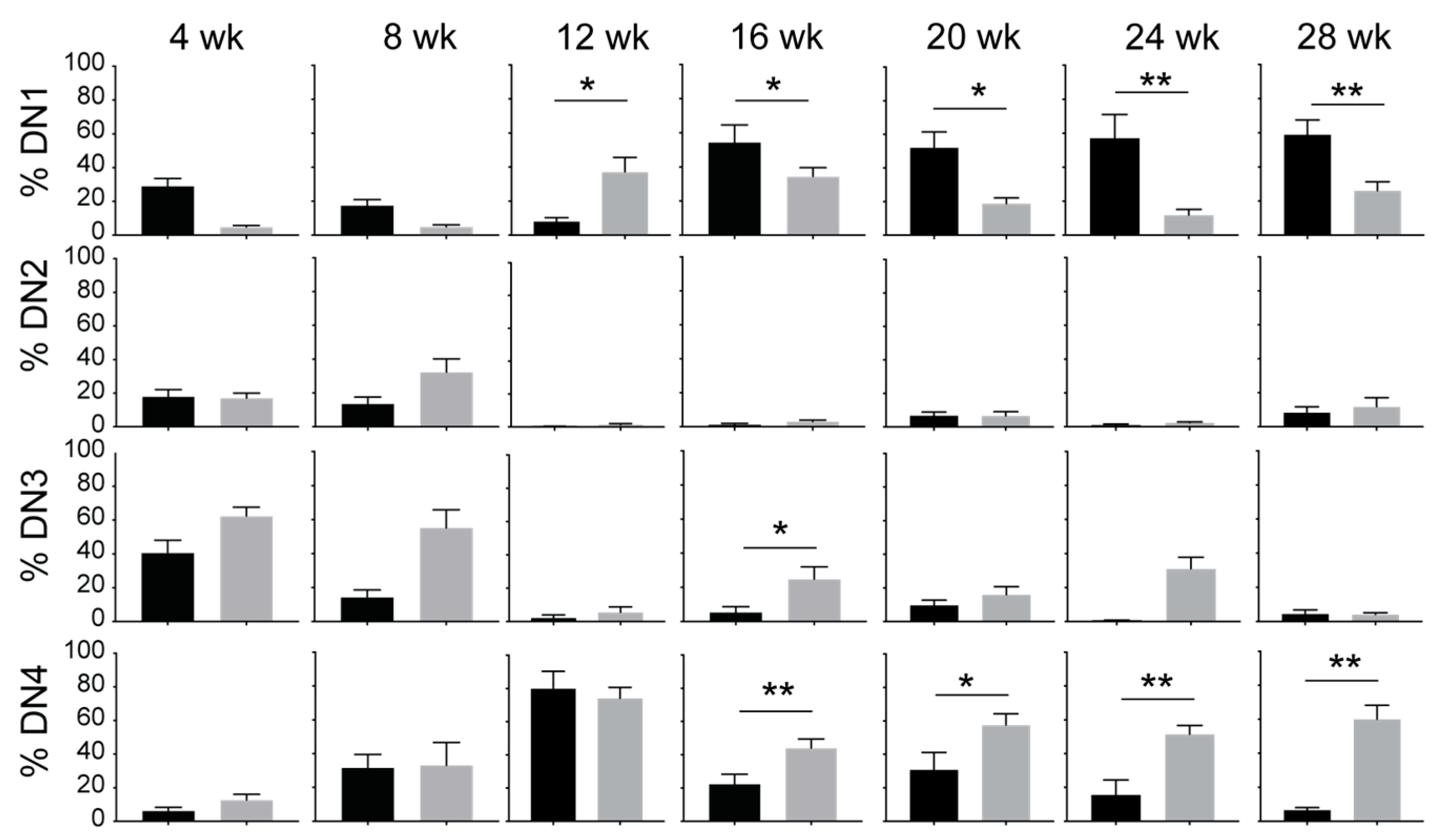
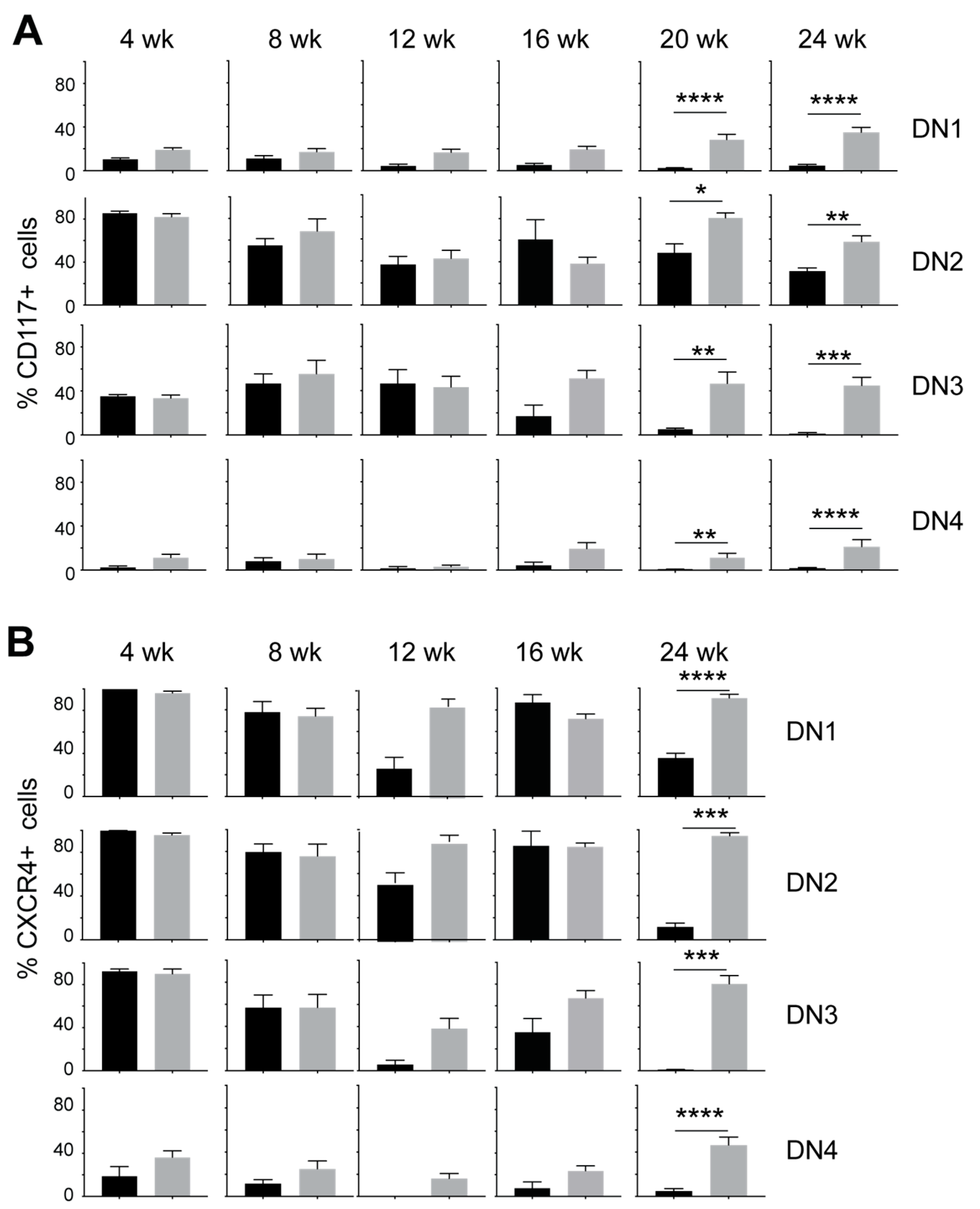
3.2. IL-15 Deficiency in Scid Mice Yields DN Thymocyte Progression towards the DN4 Stage with Elevated Expression of CD117 and CXCR4
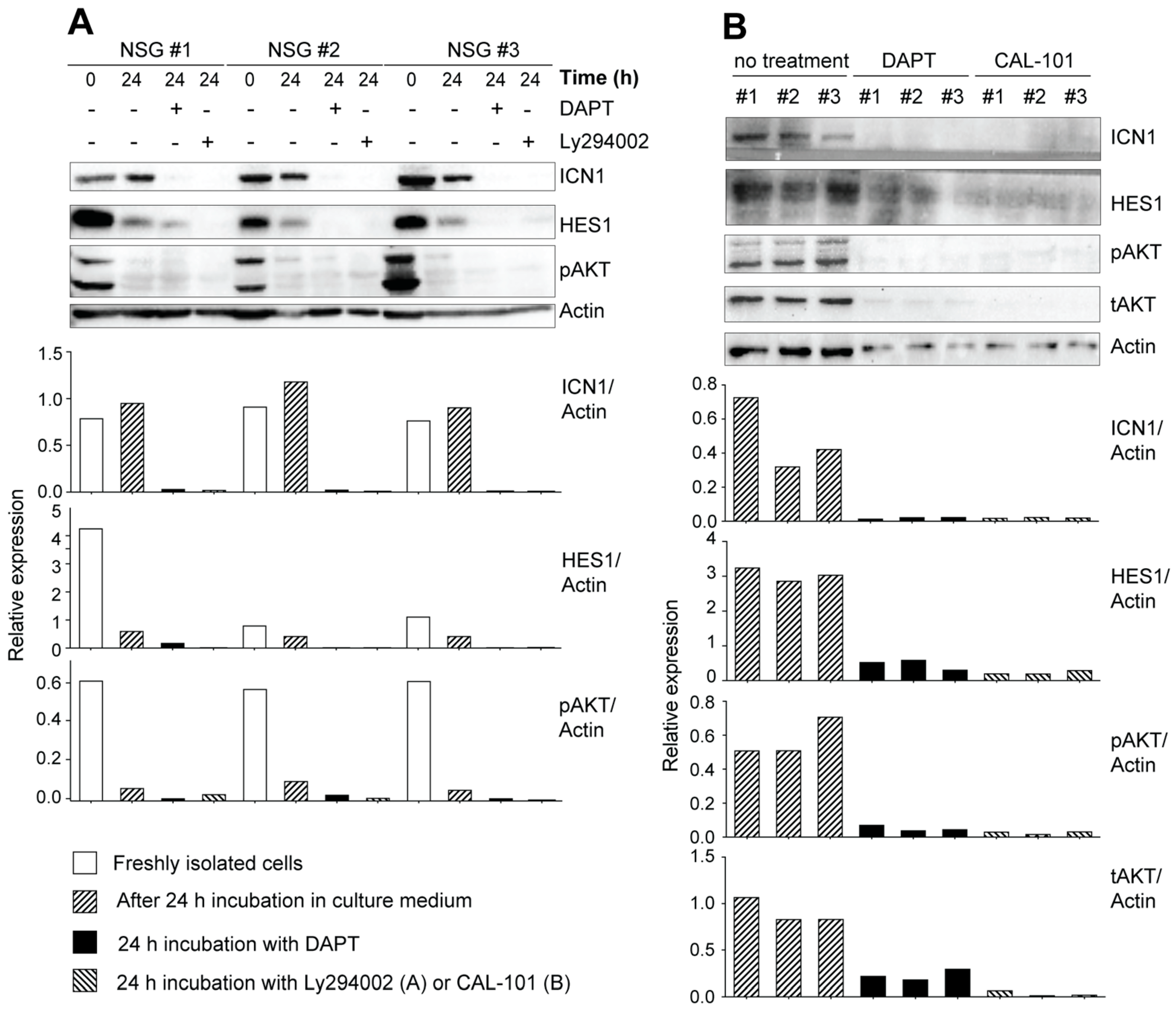
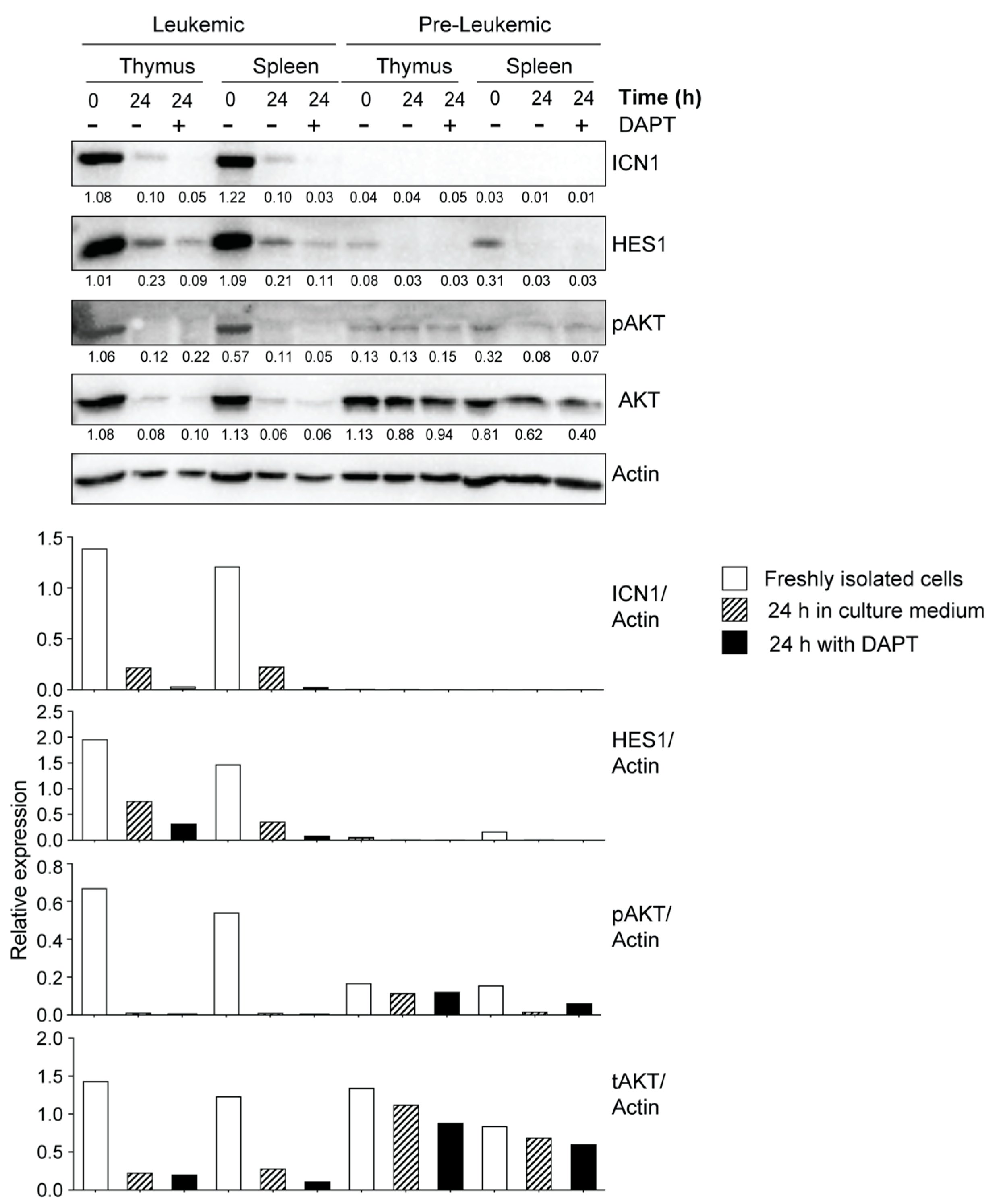
3.3. Leukemic Cells Originating in IL-15-Deficient Scid Mice Display Increased NOTCH1 Activation

3.4. Potential Crosstalk between NOTCH and PI3K/AKT Pathways in Leukemia Development in NOD.Scid.Il15−/− Mice

4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pui, C.H.; Relling, M.V.; Downing, J.R. Acute lymphoblastic leukemia. N. Engl. J. Med. 2004, 350, 1535–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Vlierberghe, P.; Ferrando, A. The molecular basis of T cell acute lymphoblastic leukemia. J. Clin. Investig. 2012, 122, 3398–3406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.M.; Larsen, S.R.; Iland, H.J.; Joshua, D.E.; Gibson, J. Leukaemias into the 21st century: Part 1: The acute leukaemias. Intern. Med. J. 2012, 42, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Tzoneva, G.; Dieck, C.L.; Oshima, K.; Ambesi-Impiombato, A.; Sanchez-Martin, M.; Madubata, C.J.; Khiabanian, H.; Yu, J.; Waanders, E.; Iacobucci, I.; et al. Clonal evolution mechanisms in NT5C2 mutant-relapsed acute lymphoblastic leukaemia. Nature 2018, 553, 511–514. [Google Scholar] [CrossRef]
- Belver, L.; Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef]
- De Bie, J.; Demeyer, S.; Alberti-Servera, L.; Geerdens, E.; Segers, H.; Broux, M.; De Keersmaecker, K.; Michaux, L.; Vandenberghe, P.; Voet, T.; et al. Single-cell sequencing reveals the origin and the order of mutation acquisition in T-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 1358–1369. [Google Scholar] [CrossRef]
- Kourtis, N.; Lazaris, C.; Hockemeyer, K.; Balandran, J.C.; Jimenez, A.R.; Mullenders, J.; Gong, Y.; Trimarchi, T.; Bhatt, K.; Hu, H.; et al. Oncogenic hijacking of the stress response machinery in T cell acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.; Tremblay, C.S.; Herblot, S.; Aplan, P.D.; Hebert, J.; Perreault, C.; Hoang, T. Modeling T-cell acute lymphoblastic leukemia induced by the SCL and LMO1 oncogenes. Genes Dev. 2010, 24, 1093–1105. [Google Scholar] [CrossRef] [Green Version]
- Zuniga-Pflucker, J.C.; Lenardo, M.J. Regulation of thymocyte development from immature progenitors. Curr. Opin. Immunol. 1996, 8, 215–224. [Google Scholar] [CrossRef]
- Rothenberg, E.V.; Moore, J.E.; Yui, M.A. Launching the T-cell-lineage developmental programme. Nat. Rev. Immunol. 2008, 8, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K.; Zuniga-Pflucker, J.C. An overview of the intrathymic intricacies of T cell development. J. Immunol. 2014, 192, 4017–4023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Boehmer, H. Unique features of the pre-T-cell receptor alpha-chain: Not just a surrogate. Nat. Rev. Immunol. 2005, 5, 571–577. [Google Scholar] [CrossRef]
- Yui, M.A.; Rothenberg, E.V. Developmental gene networks: A triathlon on the course to T cell identity. Nat. Rev. Immunol. 2014, 14, 529–545. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Meng, Y.; Chen, Y.; Yu, Y.; Wang, H.; Yang, S.; Sun, W. A comprehensive analysis of LMO2 pathogenic regulatory profile during T-lineage development and leukemic transformation. Oncogene 2022, 41, 4079–4090. [Google Scholar] [CrossRef]
- Vadillo, E.; Dorantes-Acosta, E.; Pelayo, R.; Schnoor, M. T cell acute lymphoblastic leukemia (T-ALL ): New insights into the cellular origins and infiltration mechanisms common and unique among hematologic malignancies. Blood Rev. 2018, 32, 36–51. [Google Scholar] [CrossRef]
- Wu, C.; Li, W. Genomics and pharmacogenomics of pediatric acute lymphoblastic leukemia. Crit. Rev. Oncol. Hematol. 2018, 126, 100–111. [Google Scholar] [CrossRef]
- Dai, Y.T.; Zhang, F.; Fang, H.; Li, J.F.; Lu, G.; Jiang, L.; Chen, B.; Mao, D.D.; Liu, Y.F.; Wang, J.; et al. Transcriptome-wide subtyping of pediatric and adult T cell acute lymphoblastic leukemia in an international study of 707 cases. Proc. Natl. Acad. Sci. USA 2022, 119, e2120787119. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P., IV; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. Notch signaling in leukemia. Annu. Rev. Pathol. 2008, 3, 587–613. [Google Scholar] [CrossRef] [PubMed]
- Girardi, T.; Vicente, C.; Cools, J.; De Keersmaecker, K. The genetics and molecular biology of T-ALL. Blood 2017, 129, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Allman, D.; Karnell, F.G.; Punt, J.A.; Bakkour, S.; Xu, L.; Myung, P.; Koretzky, G.A.; Pui, J.C.; Aster, J.C.; Pear, W.S. Separation of Notch1 promoted lineage commitment and expansion/transformation in developing T cells. J. Exp. Med. 2001, 194, 99–106. [Google Scholar] [CrossRef]
- Campese, A.F.; Garbe, A.I.; Zhang, F.; Grassi, F.; Screpanti, I.; von Boehmer, H. Notch1-dependent lymphomagenesis is assisted by but does not essentially require pre-TCR signaling. Blood 2006, 108, 305–310. [Google Scholar] [CrossRef] [Green Version]
- O’Neil, J.; Grim, J.; Strack, P.; Rao, S.; Tibbitts, D.; Winter, C.; Hardwick, J.; Welcker, M.; Meijerink, J.P.; Pieters, R.; et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J. Exp. Med. 2007, 204, 1813–1824. [Google Scholar] [CrossRef] [Green Version]
- Thompson, B.J.; Buonamici, S.; Sulis, M.L.; Palomero, T.; Vilimas, T.; Basso, G.; Ferrando, A.; Aifantis, I. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J. Exp. Med. 2007, 204, 1825–1835. [Google Scholar] [CrossRef] [Green Version]
- Tsunematsu, R.; Nakayama, K.; Oike, Y.; Nishiyama, M.; Ishida, N.; Hatakeyama, S.; Bessho, Y.; Kageyama, R.; Suda, T.; Nakayama, K.I. Mouse Fbw7/Sel-10/Cdc4 is required for notch degradation during vascular development. J. Biol. Chem. 2004, 279, 9417–9423. [Google Scholar] [CrossRef] [Green Version]
- King, B.; Trimarchi, T.; Reavie, L.; Xu, L.; Mullenders, J.; Ntziachristos, P.; Aranda-Orgilles, B.; Perez-Garcia, A.; Shi, J.; Vakoc, C.; et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 2013, 153, 1552–1566. [Google Scholar] [CrossRef]
- Sharma, V.M.; Calvo, J.A.; Draheim, K.M.; Cunningham, L.A.; Hermance, N.; Beverly, L.; Krishnamoorthy, V.; Bhasin, M.; Capobianco, A.J.; Kelliher, M.A. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol. Cell. Biol. 2006, 26, 8022–8031. [Google Scholar] [CrossRef] [Green Version]
- Weng, A.P.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.L.; Lau, A.; Wai, C.; Del Bianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, K.; Hattori, M.; Hirai, N.; Shinozuka, Y.; Hirata, H.; Kageyama, R.; Sakai, T.; Minato, N. Hes1 directly controls cell proliferation through the transcriptional repression of p27Kip1. Mol. Cell. Biol. 2005, 25, 4262–4271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerby, B.; Tremblay, C.S.; Tremblay, M.; Rojas-Sutterlin, S.; Herblot, S.; Hebert, J.; Sauvageau, G.; Lemieux, S.; Lecuyer, E.; Veiga, D.F.; et al. SCL, LMO1 and Notch1 reprogram thymocytes into self-renewing cells. PLoS Genet. 2014, 10, e1004768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.W.; Malek, T.R. The structure and function of gamma c-dependent cytokines and receptors: Regulation of T lymphocyte development and homeostasis. Crit. Rev. Immunol. 1998, 18, 503–524. [Google Scholar] [CrossRef]
- Lodolce, J.P.; Burkett, P.R.; Boone, D.L.; Chien, M.; Ma, A. T cell-independent interleukin 15Ralpha signals are required for bystander proliferation. J. Exp. Med. 2001, 194, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Stonier, S.W.; Schluns, K.S. Trans-presentation: A novel mechanism regulating IL–15 delivery and responses. Immunol. Lett. 2010, 127, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Waldmann, T.A. The biology of IL–15: Implications for cancer therapy and the treatment of autoimmune disorders. J. Investig. Dermatol. Symp. Proc. 2013, 16, S28–S30. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.; Li, L.; Liao, N.S.; Schluns, K.S.; Luckhart, S.; Sleasman, J.W.; Zhong, X.P. Thymic Epithelial Cell-Derived IL–15 and IL–15 Receptor alpha Chain Foster Local Environment for Type 1 Innate Like T Cell Development. Front. Immunol. 2021, 12, 623280. [Google Scholar] [CrossRef]
- Bobbala, D.; Kandhi, R.; Chen, X.; Mayhue, M.; Bouchard, E.; Yan, J.; Knecht, H.; Barabe, F.; Ramanathan, S.; Ilangumaran, S. Interleukin-15 deficiency promotes the development of T-cell acute lymphoblastic leukemia in non-obese diabetes mice with severe combined immunodeficiency. Leukemia 2016, 30, 1749–1752. [Google Scholar] [CrossRef]
- Bosma, M.J.; Carroll, A.M. The SCID mouse mutant: Definition, characterization, and potential uses. Annu. Rev. Immunol. 1991, 9, 323–350. [Google Scholar] [CrossRef] [PubMed]
- Danska, J.S.; Holland, D.P.; Mariathasan, S.; Williams, K.M.; Guidos, C.J. Biochemical and genetic defects in the DNA-dependent protein kinase in murine scid lymphocytes. Mol. Cell. Biol. 1996, 16, 5507–5517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulop, G.M.; Phillips, R.A. The scid mutation in mice causes a general defect in DNA repair. Nature 1990, 347, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Dubois, S.; Mueller, J.; Feigenbaum, L.; Waldmann, T.A. Leukemia/lymphoma development in IL–15-deficient TCR-transgenic mice. J. ImmunoTherapy Cancer 2015, 3, P67. [Google Scholar] [CrossRef] [Green Version]
- Bobbala, D.; Chen, X.L.; Leblanc, C.; Mayhue, M.; Stankova, J.; Tanaka, T.; Chen, Y.G.; Ilangumaran, S.; Ramanathan, S. Interleukin-15 plays an essential role in the pathogenesis of autoimmune diabetes in the NOD mouse. Diabetologia 2012, 55, 3010–3020. [Google Scholar] [CrossRef] [Green Version]
- Bobbala, D.; Mayhue, M.; Menendez, A.; Ilangumaran, S.; Ramanathan, S. Trans-presentation of interleukin-15 by interleukin-15 receptor alpha is dispensable for the pathogenesis of autoimmune type 1 diabetes. Clin. Exp. Immunol. 2017, 14, 590–596. [Google Scholar] [CrossRef] [Green Version]
- Nandi, M.; Moyo, M.M.; Orkhis, S.; Mobulakani, J.M.F.; Limoges, M.A.; Rexhepi, F.; Mayhue, M.; Cayarga, A.A.; Marrero, G.C.; Ilangumaran, S.; et al. IL–15Ralpha-Independent IL–15 Signaling in Non-NK Cell-Derived IFNgamma Driven Control of Listeria monocytogenes. Front. Immunol. 2021, 12, 793918. [Google Scholar] [CrossRef]
- Ghosh, A.; Ihsan, A.U.; Nandi, M.; Cloutier, M.; Khan, M.G.M.; Ramanathan, S.; Ilangumaran, S. Application of EdU-Based DNA Synthesis Assay to Measure Hepatocyte Proliferation In Situ During Liver Regeneration. Methods Mol. Biol. 2022, 2544, 195–206. [Google Scholar] [CrossRef]
- Ribot, J.C.; deBarros, A.; Pang, D.J.; Neves, J.F.; Peperzak, V.; Roberts, S.J.; Girardi, M.; Borst, J.; Hayday, A.C.; Pennington, D.J.; et al. CD27 is a thymic determinant of the balance between interferon-gamma- and interleukin 17-producing gammadelta T cell subsets. Nat. Immunol. 2009, 10, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Rodewald, H.R.; Waskow, C.; Haller, C. Essential requirement for c-kit and common gamma chain in thymocyte development cannot be overruled by enforced expression of Bcl-2. J. Exp. Med. 2001, 193, 1431–1437. [Google Scholar] [CrossRef]
- Trampont, P.C.; Tosello-Trampont, A.C.; Shen, Y.; Duley, A.K.; Sutherland, A.E.; Bender, T.P.; Littman, D.R.; Ravichandran, K.S. CXCR4 acts as a costimulator during thymic beta-selection. Nat. Immunol. 2010, 11, 162–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabher, C.; von Boehmer, H.; Look, A.T. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2006, 6, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Lobbardi, R.; Pinder, J.; Martinez-Pastor, B.; Theodorou, M.; Blackburn, J.S.; Abraham, B.J.; Namiki, Y.; Mansour, M.; Abdelfattah, N.S.; Molodtsov, A.; et al. TOX Regulates Growth, DNA Repair, and Genomic Instability in T-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2017, 7, 1336–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadwick, N.; Zeef, L.; Portillo, V.; Boros, J.; Hoyle, S.; van Doesburg, J.C.; Buckle, A.M. Notch protection against apoptosis in T-ALL cells mediated by GIMAP5. Blood Cells Mol. Dis. 2010, 45, 201–209. [Google Scholar] [CrossRef]
- Chadwick, N.; Zeef, L.; Portillo, V.; Fennessy, C.; Warrander, F.; Hoyle, S.; Buckle, A.M. Identification of novel Notch target genes in T cell leukaemia. Mol. Cancer 2009, 8, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liau, W.S.; Tan, S.H.; Ngoc, P.C.T.; Wang, C.Q.; Tergaonkar, V.; Feng, H.; Gong, Z.; Osato, M.; Look, A.T.; Sanda, T. Aberrant activation of the GIMAP enhancer by oncogenic transcription factors in T-cell acute lymphoblastic leukemia. Leukemia 2017, 31, 1798–1807. [Google Scholar] [CrossRef] [PubMed]
- Curtis, D.J.; McCormack, M.P. The molecular basis of Lmo2-induced T-cell acute lymphoblastic leukemia. Clin. Cancer Res. 2010, 16, 5618–5623. [Google Scholar] [CrossRef] [Green Version]
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.H.; Downing, J.R.; Gilliland, D.G.; et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002, 1, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Ramirez, I.; Bhatia, S.; Rodriguez-Hernandez, G.; Gonzalez-Herrero, I.; Walter, C.; Gonzalez de Tena-Davila, S.; Parvin, S.; Haas, O.; Woessmann, W.; Stanulla, M.; et al. Lmo2 expression defines tumor cell identity during T-cell leukemogenesis. EMBO J. 2018, 37, e98783. [Google Scholar] [CrossRef]
- McCormack, M.P.; Shields, B.J.; Jackson, J.T.; Nasa, C.; Shi, W.; Slater, N.J.; Tremblay, C.S.; Rabbitts, T.H.; Curtis, D.J. Requirement for Lyl1 in a model of Lmo2-driven early T-cell precursor ALL. Blood 2013, 122, 2093–2103. [Google Scholar] [CrossRef]
- Tatarek, J.; Cullion, K.; Ashworth, T.; Gerstein, R.; Aster, J.C.; Kelliher, M.A. Notch1 inhibition targets the leukemia-initiating cells in a Tal1/Lmo2 mouse model of T-ALL. Blood 2011, 118, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, P.; van Grotel, M.; Beverloo, H.B.; Lee, C.; Helgason, T.; Buijs-Gladdines, J.; Passier, M.; van Wering, E.R.; Veerman, A.J.; Kamps, W.A.; et al. The cryptic chromosomal deletion del(11)(p12p13) as a new activation mechanism of LMO2 in pediatric T-cell acute lymphoblastic leukemia. Blood 2006, 108, 3520–3529. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.; Lambert, J.A.; Martin, R. SCL/TAL1 in Hematopoiesis and Cellular Reprogramming. Curr. Top. Dev. Biol. 2016, 118, 163–204. [Google Scholar] [CrossRef]
- Sanda, T.; Leong, W.Z. TAL1 as a master oncogenic transcription factor in T-cell acute lymphoblastic leukemia. Exp. Hematol. 2017, 53, 7–15. [Google Scholar] [CrossRef]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerby, B.; Veiga, D.F.; Krosl, J.; Nourreddine, S.; Ouellette, J.; Haman, A.; Lavoie, G.; Fares, I.; Tremblay, M.; Litalien, V.; et al. High-throughput screening in niche-based assay identifies compounds to target preleukemic stem cells. J. Clin. Investig. 2016, 126, 4569–4584. [Google Scholar] [CrossRef]
- Fehniger, T.A.; Caligiuri, M.A. Interleukin 15: Biology and relevance to human disease. Blood 2001, 97, 14–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpdogan, O.; Eng, J.M.; Muriglan, S.J.; Willis, L.M.; Hubbard, V.M.; Tjoe, K.H.; Terwey, T.H.; Kochman, A.; van den Brink, M.R. Interleukin-15 enhances immune reconstitution after allogeneic bone marrow transplantation. Blood 2005, 105, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Waldmann, T.A. Interleukin-15 in the treatment of cancer. Expert Rev. Clin. Immunol. 2014, 10, 1689–1701. [Google Scholar] [CrossRef]
- Barrett, A.J.; Battiwalla, M. Relapse after allogeneic stem cell transplantation. Expert Rev. Hematol. 2010, 3, 429–441. [Google Scholar] [CrossRef] [Green Version]
- Fehniger, T.A.; Suzuki, K.; Ponnappan, A.; VanDeusen, J.B.; Cooper, M.A.; Florea, S.M.; Freud, A.G.; Robinson, M.L.; Durbin, J.; Caligiuri, M.A. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J. Exp. Med. 2001, 193, 219–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehniger, T.A.; Suzuki, K.; VanDeusen, J.B.; Cooper, M.A.; Freud, A.G.; Caligiuri, M.A. Fatal leukemia in interleukin-15 transgenic mice. Blood Cells Mol. Dis. 2001, 27, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Yokohama, A.; Mishra, A.; Mitsui, T.; Becknell, B.; Johns, J.; Curphey, D.; Blaser, B.W.; Vandeusen, J.B.; Mao, H.; Yu, J.; et al. A novel mouse model for the aggressive variant of NK cell and T cell large granular lymphocyte leukemia. Leuk. Res. 2010, 34, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Ettersperger, J.; Montcuquet, N.; Malamut, G.; Guegan, N.; Lopez-Lastra, S.; Gayraud, S.; Reimann, C.; Vidal, E.; Cagnard, N.; Villarese, P.; et al. Interleukin-15-Dependent T-Cell-like Innate Intraepithelial Lymphocytes Develop in the Intestine and Transform into Lymphomas in Celiac Disease. Immunity 2016, 45, 610–625. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Liu, C.; Xue, M.; Liu, R.; Jiang, L.; Yu, X.; Bao, G.; Deng, F.; Yu, M.; Ao, J.; et al. The role of interleukin-15 polymorphisms in adult acute lymphoblastic leukemia. PLoS ONE 2010, 5, e13626. [Google Scholar] [CrossRef] [Green Version]
- Aly, R.M.; Taalab, M.M.; Ghazy, H.F. Influence of interleukin-15 polymorphism on the survival of adult patients with acute lymphoblastic leukemia in Egypt. Leuk. Lymphoma 2015, 56, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.W.; Roberts, K.G.; Gu, Z.; Shi, L.; Pounds, S.; Pei, D.; Cheng, C.; Dai, Y.; Devidas, M.; Qu, C.; et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat. Genet. 2022, 54, 1376–1389. [Google Scholar] [CrossRef]
- Custer, R.P.; Bosma, G.C.; Bosma, M.J. Severe combined immunodeficiency (SCID) in the mouse. Pathology, reconstitution, neoplasms. Am. J. Pathol. 1985, 120, 464–477. [Google Scholar] [PubMed]
- Zipris, D.; Crow, A.R.; Delovitch, T.L. Altered thymic and peripheral T-lymphocyte repertoire preceding onset of diabetes in NOD mice. Diabetes 1991, 40, 429–435. [Google Scholar] [CrossRef]
- Prochazka, M.; Gaskins, H.R.; Shultz, L.D.; Leiter, E.H. The nonobese diabetic scid mouse: Model for spontaneous thymomagenesis associated with immunodeficiency. Proc. Natl. Acad. Sci. USA 1992, 89, 3290–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, H.C., 3rd; Anver, M.R.; Fredrickson, T.N.; Haines, D.C.; Harris, A.W.; Harris, N.L.; Jaffe, E.S.; Kogan, S.C.; MacLennan, I.C.; Pattengale, P.K.; et al. Bethesda proposals for classification of lymphoid neoplasms in mice. Blood 2002, 100, 246–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.H.; Bertulfo, F.C.; Sanda, T. Leukemia-Initiating Cells in T-Cell Acute Lymphoblastic Leukemia. Front. Oncol. 2017, 7, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, C.L.; Poursine-Laurent, J.; Yang, L.; Yokoyama, W.M. Development of thymic NK cells from double negative 1 thymocyte precursors. Blood 2011, 118, 3570–3578. [Google Scholar] [CrossRef] [Green Version]
- Lauzon, R.J.; Siminovitch, K.A.; Fulop, G.M.; Phillips, R.A.; Roder, J.C. An expanded population of natural killer cells in mice with severe combined immunodeficiency (SCID) lack rearrangement and expression of T cell receptor genes. J. Exp. Med. 1986, 164, 1797–1802. [Google Scholar] [CrossRef]
- Tremblay, C.S.; Hoang, T.; Hoang, T. Early T cell differentiation lessons from T-cell acute lymphoblastic leukemia. Prog. Mol. Biol. Transl. Sci. 2010, 92, 121–156. [Google Scholar] [CrossRef]
- Zhong, Y.; Jiang, L.; Hiai, H.; Toyokuni, S.; Yamada, Y. Overexpression of a transcription factor LYL1 induces T- and B-cell lymphoma in mice. Oncogene 2007, 26, 6937–6947. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.L.; Wadman, I.; Baer, R. Formation of in vivo complexes between the TAL1 and E2A polypeptides of leukemic T cells. Proc. Natl. Acad. Sci. USA 1994, 91, 3181–3185. [Google Scholar] [CrossRef] [Green Version]
- Curtis, D.J.; Robb, L.; Strasser, A.; Begley, C.G. The CD2-scl transgene alters the phenotype and frequency of T-lymphomas in N-ras transgenic or p53 deficient mice. Oncogene 1997, 15, 2975–2983. [Google Scholar] [CrossRef] [Green Version]
- Condorelli, G.L.; Facchiano, F.; Valtieri, M.; Proietti, E.; Vitelli, L.; Lulli, V.; Huebner, K.; Peschle, C.; Croce, C.M. T-cell-directed TAL-1 expression induces T-cell malignancies in transgenic mice. Cancer Res. 1996, 56, 5113–5119. [Google Scholar]
- Dadi, S.; Le Noir, S.; Payet-Bornet, D.; Lhermitte, L.; Zacarias-Cabeza, J.; Bergeron, J.; Villarese, P.; Vachez, E.; Dik, W.A.; Millien, C.; et al. TLX homeodomain oncogenes mediate T cell maturation arrest in T-ALL via interaction with ETS1 and suppression of TCRalpha gene expression. Cancer Cell 2012, 21, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Veiga, D.F.T.; Tremblay, M.; Gerby, B.; Herblot, S.; Haman, A.; Gendron, P.; Lemieux, S.; Zuniga-Pflucker, J.C.; Hebert, J.; Cohen, J.P.; et al. Monoallelic Heb/Tcf12 Deletion Reduces the Requirement for NOTCH1 Hyperactivation in T-Cell Acute Lymphoblastic Leukemia. Front. Immunol. 2022, 13, 867443. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Lopez, C.; Varas, A.; Sacedon, R.; Jimenez, E.; Munoz, J.J.; Zapata, A.G.; Vicente, A. Stromal cell-derived factor 1/CXCR4 signaling is critical for early human T-cell development. Blood 2002, 99, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Passaro, D.; Irigoyen, M.; Catherinet, C.; Gachet, S.; Da Costa De Jesus, C.; Lasgi, C.; Quang, C.T.; Ghysdael, J. CXCR4 Is Required for Leukemia-Initiating Cell Activity in T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2015, 27, 769–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrandino, F.; Bernardini, G.; Tsaouli, G.; Grazioli, P.; Campese, A.F.; Noce, C.; Ciuffetta, A.; Vacca, A.; Besharat, Z.M.; Bellavia, D.; et al. Intrathymic Notch3 and CXCR4 combinatorial interplay facilitates T-cell leukemia propagation. Oncogene 2018, 37, 6285–6298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, L.A.; Tikhonova, A.N.; Hu, H.; Trimarchi, T.; King, B.; Gong, Y.; Sanchez-Martin, M.; Tsirigos, A.; Littman, D.R.; Ferrando, A.A.; et al. CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell 2015, 27, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Jost, T.R.; Borga, C.; Radaelli, E.; Romagnani, A.; Perruzza, L.; Omodho, L.; Cazzaniga, G.; Biondi, A.; Indraccolo, S.; Thelen, M.; et al. Role of CXCR4-mediated bone marrow colonization in CNS infiltration by T cell acute lymphoblastic leukemia. J. Leukoc. Biol. 2016, 99, 1077–1087. [Google Scholar] [CrossRef] [Green Version]
- Tsaouli, G.; Ferretti, E.; Bellavia, D.; Vacca, A.; Felli, M.P. Notch/CXCR4 Partnership in Acute Lymphoblastic Leukemia Progression. J. Immunol. Res. 2019, 2019, 5601396. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Walker, K.L.; Rinella, S.P.; Hess, N.J.; Turicek, D.P.; Kabakov, S.A.; Zhu, F.; Bouchlaka, M.N.; Olson, S.L.; Cho, M.M.; Quamine, A.E.; et al. CXCR4 allows T cell acute lymphoblastic leukemia to escape from JAK1/2 and BCL2 inhibition through CNS infiltration. Leuk. Lymphoma 2021, 62, 1167–1177. [Google Scholar] [CrossRef]
- Ashworth, T.D.; Pear, W.S.; Chiang, M.Y.; Blacklow, S.C.; Mastio, J.; Xu, L.; Kelliher, M.; Kastner, P.; Chan, S.; Aster, J.C. Deletion-based mechanisms of Notch1 activation in T-ALL: Key roles for RAG recombinase and a conserved internal translational start site in Notch1. Blood 2010, 116, 5455–5464. [Google Scholar] [CrossRef] [PubMed]
- Jeannet, R.; Mastio, J.; Macias-Garcia, A.; Oravecz, A.; Ashworth, T.; Geimer Le Lay, A.S.; Jost, B.; Le Gras, S.; Ghysdael, J.; Gridley, T.; et al. Oncogenic activation of the Notch1 gene by deletion of its promoter in Ikaros-deficient T-ALL. Blood 2010, 116, 5443–5454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, P.P.; Jiang, H.; Dick, J.E. Leukemia-initiating cells in human T-lymphoblastic leukemia exhibit glucocorticoid resistance. Blood 2010, 116, 5268–5279. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Leukemia Incidence 1 | |
|---|---|---|
| Males | Females | |
| NOD.Scid | 0/33 | 0/12 |
| NOD.Scid.Il15−/− | 12/12 | 17/17 |
| NOD.Rag1 −/− | 0/28 | 0/32 |
| NOD.Rag1−/−Il15−/− | 0/39 | 0/21 |
| C57BL/6.Scid | 0/9 | 0/6 |
| C57BL/6.Scid.Il15−/− | 5/8 | 3/4 |
| C57BL/6.Rag1−/− | 0/29 | 0/46 |
| C57BL/6.Rag1−/−Il15−/− | 0/62 | 0/38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nandi, M.; Ghosh, A.; Akbari, S.A.; Bobbala, D.; Boucher, M.-J.; Menendez, A.; Hoang, T.; Ilangumaran, S.; Ramanathan, S. IL-15 Prevents the Development of T-ALL from Aberrant Thymocytes with Impaired DNA Repair Functions and Increased NOTCH1 Activation. Cancers 2023, 15, 671. https://doi.org/10.3390/cancers15030671
Nandi M, Ghosh A, Akbari SA, Bobbala D, Boucher M-J, Menendez A, Hoang T, Ilangumaran S, Ramanathan S. IL-15 Prevents the Development of T-ALL from Aberrant Thymocytes with Impaired DNA Repair Functions and Increased NOTCH1 Activation. Cancers. 2023; 15(3):671. https://doi.org/10.3390/cancers15030671
Chicago/Turabian StyleNandi, Madhuparna, Amit Ghosh, Sara Ali Akbari, Diwakar Bobbala, Marie-Josée Boucher, Alfredo Menendez, Trang Hoang, Subburaj Ilangumaran, and Sheela Ramanathan. 2023. "IL-15 Prevents the Development of T-ALL from Aberrant Thymocytes with Impaired DNA Repair Functions and Increased NOTCH1 Activation" Cancers 15, no. 3: 671. https://doi.org/10.3390/cancers15030671
APA StyleNandi, M., Ghosh, A., Akbari, S. A., Bobbala, D., Boucher, M. -J., Menendez, A., Hoang, T., Ilangumaran, S., & Ramanathan, S. (2023). IL-15 Prevents the Development of T-ALL from Aberrant Thymocytes with Impaired DNA Repair Functions and Increased NOTCH1 Activation. Cancers, 15(3), 671. https://doi.org/10.3390/cancers15030671