

Polycomb Alterations in Acute Myeloid Leukaemia: From Structure to Function

Abstract
:Simple Summary
Abstract
1. Polycomb Repressive Complexes
2. Structural and Functional Regions of the PRC2 Complex
2.1. Catalytic and Regulatory Regions of PRC2
2.2. Interaction Regions of PRC2
3. PRC2 Alterations in Acute Myeloid Leukaemia
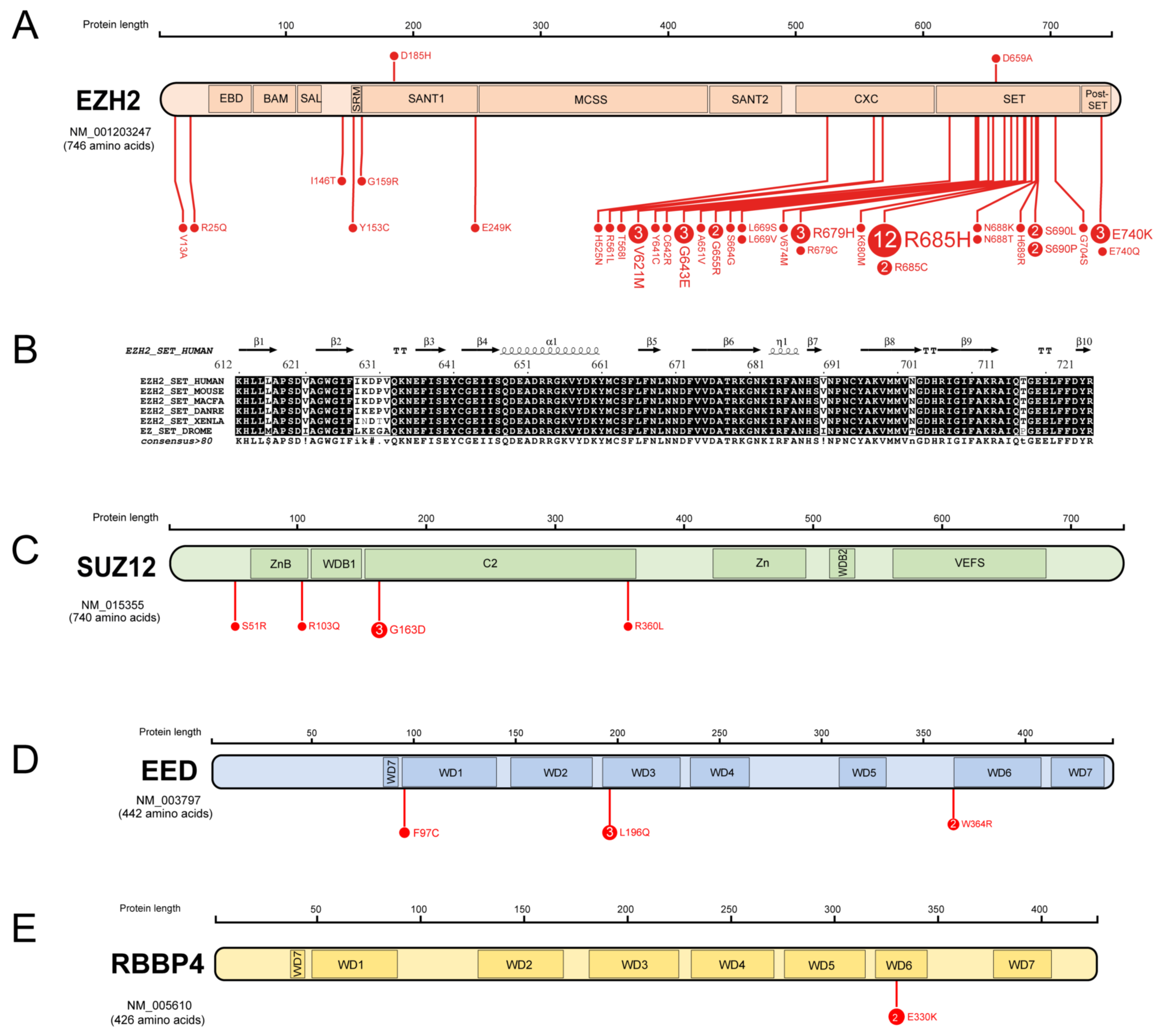
3.1. Missense Mutations of PRC2 Components in AML
3.2. PRC2 Genetic Interactions in Childhood and Adult AML
4. Therapeutic Implications of PRC2 Alterations in AML
4.1. Prognostic Associations of PRC2 Alterations in AML
4.2. Epigenetic Therapeutic Avenues towards Improved Treatments for AML
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Paro, R.; Hogness, D.S. The Polycomb protein shares a homologous domain with a heterochromatin-associated protein of Drosophila. Proc. Natl. Acad. Sci. USA 1991, 88, 263–267. [Google Scholar] [CrossRef] [Green Version]
- DeCamillis, M.; Cheng, N.S.; Pierre, D.; Brock, H.W. The polyhomeotic gene of Drosophila encodes a chromatin protein that shares polytene chromosome-binding sites with Polycomb. Genes Dev. 1992, 6, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Struhl, G. A gene product required for correct initiation of segmental determination in Drosophila. Nature 1981, 293, 36–41. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF Homologs, CBX Proteins, and RYBP Define Functionally Distinct PRC1 Family Complexes. Mol. Cell 2012, 45, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Owen, B.M.; Davidovich, C. DNA binding by polycomb-group proteins: Searching for the link to CpG islands. Nucleic Acids Res. 2022, 50, 4813–4839. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Kingston, R.E. Occupying Chromatin: Polycomb Mechanisms for Getting to Genomic Targets, Stopping Transcriptional Traffic, and Staying Put. Mol. Cell 2013, 49, 808–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizán, P.; Beringer, M.; Ballaré, C.; Di Croce, L. Role of PRC2-associated factors in stem cells and disease. FEBS J. 2014, 282, 1723–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Jiao, L.; Liu, X.; Yang, X.; Liu, X. A Dimeric Structural Scaffold for PRC2-PCL Targeting to CpG Island Chromatin. Mol. Cell 2020, 77, 1265–1278.e1267. [Google Scholar] [CrossRef] [PubMed]
- Pirrotta, V. Polycomb Mechanisms and Epigenetic Control of Gene Activity. In Handbook of Epigenetics, 2nd ed.; 2017; pp. 93–110. [Google Scholar] [CrossRef]
- Son, J.; Shen, S.S.; Margueron, R.; Reinberg, D. Nucleosome-binding activities within JARID2 and EZH1 regulate the function of PRC2 on chromatin. Genes Dev. 2013, 27, 2663–2677. [Google Scholar] [CrossRef] [Green Version]
- Kouznetsova, V.L.; Tchekanov, A.; Li, X.; Yan, X.; Tsigelny, I.F. Polycomb repressive 2 complex—Molecular mechanisms of function. Protein Sci. 2019, 28, 1387–1399. [Google Scholar] [CrossRef]
- Andricovich, J.; Kai, Y.; Peng, W.; Foudi, A.; Tzatsos, A. Histone demethylase KDM2B regulates lineage commitment in normal and malignant hematopoiesis. J. Clin. Investig. 2016, 126, 905–920. [Google Scholar] [CrossRef]
- Cao, Q.; Gearhart, M.D.; Gery, S.; Shojaee, S.; Yang, H.; Sun, H.; Lin, D.-C.; Bai, J.-W.; Mead, M.; Zhao, Z.; et al. BCOR regulates myeloid cell proliferation and differentiation. Leukemia 2016, 30, 1155–1165. [Google Scholar] [CrossRef] [Green Version]
- van den Boom, V.; Maat, H.; Geugien, M.; Rodriguez Lopez, A.; Sotoca, A.M.; Jaques, J.; Brouwers-Vos, A.Z.; Fusetti, F.; Groen, R.W.; Yuan, H.; et al. Non-canonical PRC1.1 Targets Active Genes Independent of H3K27me3 and Is Essential for Leukemogenesis. Cell Rep. 2015, 14, 332–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamminga, L.M.; Bystrykh, L.V.; de Boer, A.; Houwer, S.; Douma, J.; Weersing, E.; Dontje, B.; de Haan, G. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood 2006, 107, 2170–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochizuki-Kashio, M.; Mishima, Y.; Miyagi, S.; Negishi, M.; Saraya, A.; Konuma, T.; Shinga, J.; Koseki, H.; Iwama, A. Dependency on the polycomb gene Ezh2 distinguishes fetal from adult hematopoietic stem cells. Blood 2011, 118, 6553–6561. [Google Scholar] [CrossRef]
- Lee, S.C.W.; Miller, S.; Hyland, C.; Kauppi, M.; Lebois, M.; Di Rago, L.; Metcalf, D.; Kinkel, S.A.; Josefsson, E.C.; Blewitt, M.E.; et al. Polycomb repressive complex 2 component Suz12 is required for hematopoietic stem cell function and lymphopoiesis. Blood 2015, 126, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Herviou, L.; Cavalli, G.; Cartron, G.; Klein, B.; Moreaux, J. EZH2 in normal hematopoiesis and hematological malignancies. Oncotarget 2015, 7, 2284–2296. [Google Scholar] [CrossRef] [Green Version]
- Su, I.-H.; Basavaraj, A.; Krutchinsky, A.N.; Hobert, O.; Ullrich, A.; Chait, B.T.; Tarakhovsky, A. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat. Immunol. 2002, 4, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Ueda, T.; Yamasaki, N.; Nakata, Y.; Sera, Y.; Nagamachi, A.; Miyama, T.; Kobayashi, H.; Takubo, K.; Kanai, A.; et al. Maintenance of the functional integrity of mouse hematopoiesis by EED and promotion of leukemogenesis by EED haploinsufficiency. Sci. Rep. 2016, 6, 29454. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Zhang, F.; Wang, S.; Fu, Y.; Chen, J.; Liang, X.; Le, H.; Pu, W.T.; Zhang, B. Depletion of polycomb repressive complex 2 core component EED impairs fetal hematopoiesis. Cell Death Dis. 2017, 8, e2744. [Google Scholar] [CrossRef] [Green Version]
- Jones, L.; McCarthy, P.; Bond, J. Epigenetics of paediatric acute myeloid leukaemia. Br. J. Haematol. 2020, 188, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Lefeivre, T.; Jones, L.; Trinquand, A.; Pinton, A.; Macintyre, E.; Laurenti, E.; Bond, J. Immature acute leukaemias: Lessons from the haematopoietic roadmap. FEBS J. 2021, 289, 4355–4370. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.; Labis, E.; Marceau-Renaut, A.; Duployez, N.; Labopin, M.; Hypolite, G.; Michel, G.; Ducassou, S.; Boutroux, H.; Nelken, B.; et al. Polycomb repressive complex 2 haploinsufficiency identifies a high-risk subgroup of pediatric acute myeloid leukemia. Leukemia 2018, 32, 1878–1882. [Google Scholar] [CrossRef] [PubMed]
- Göllner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.-U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Ariës, I.M.; Bodaar, K.; Karim, S.A.; Ni Chonghaile, T.; Hinze, L.; Burns, M.A.; Pfirrmann, M.; Degar, J.; Landrigan, J.T.; Balbach, S.; et al. PRC2 loss induces chemoresistance by repressing apoptosis in T cell acute lymphoblastic leukemia. J. Exp. Med. 2018, 215, 3094–3114. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Xing, X.; Hu, M.; Zhang, Y.; Liu, P.; Chai, J. Structural Basis of EZH2 Recognition by EED. Structure 2007, 15, 1306–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciferri, C.; Lander, G.C.; Maiolica, A.; Herzog, F.; Aebersold, R.; Nogales, E. Molecular architecture of human polycomb repressive complex 2. Elife 2012, 1, e00005. [Google Scholar] [CrossRef]
- Jani, K.S.; Jain, S.U.; Ge, E.J.; Diehl, K.L.; Lundgren, S.M.; Müller, M.M.; Lewis, P.W.; Muir, T.W. Histone H3 tail binds a unique sensing pocket in EZH2 to activate the PRC2 methyltransferase. Proc. Natl. Acad. Sci. USA 2019, 116, 8295–8300. [Google Scholar] [CrossRef] [Green Version]
- Finogenova, K.; Bonnet, J.; Poepsel, S.; Schäfer, I.B.; Finkl, K.; Schmid, K.; Litz, C.; Strauss, M.; Benda, C.; Müller, J. Structural basis for PRC2 decoding of active histone methylation marks H3K36me2/3. Elife 2020, 9, e61964. [Google Scholar] [CrossRef]
- Kasinath, V.; Beck, C.; Sauer, P.; Poepsel, S.; Kosmatka, J.; Faini, M.; Toso, D.; Aebersold, R.; Nogales, E. JARID2 and AEBP2 regulate PRC2 in the presence of H2AK119ub1 and other histone modifications. Science 2021, 371, eabc3393. [Google Scholar] [CrossRef]
- Brien, G.; Gambero, G.; O’Connell, D.; Jerman, E.; A Turner, S.; Egan, C.M.; Dunne, E.J.; Jurgens, M.C.; Wynne, K.; Piao, L.; et al. Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat. Struct. Mol. Biol. 2012, 19, 1273–1281. [Google Scholar] [CrossRef]
- Bolouri, H.; Farrar, J.E.; Triche, T., Jr.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nat. Cell Biol. 2018, 562, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Marceau-Renaut, A.; Duployez, N.; Ducourneau, B.; Labopin, M.; Petit, A.; Rousseau, A.; Geffroy, S.; Bucci, M.; Cuccuini, W.; Fenneteau, O.; et al. Molecular Profiling Defines Distinct Prognostic Subgroups in Childhood AML: A Report From the French ELAM02 Study Group. Hemasphere 2018, 2, e31. [Google Scholar] [CrossRef]
- Alexander, T.B.; Gu, Z.; Iacobucci, I.; Dickerson, K.; Choi, J.K.; Xu, B.; Payne-Turner, D.; Yoshihara, H.; Loh, M.L.; Horan, J.; et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 2018, 562, 373–379. [Google Scholar] [CrossRef]
- Bond, J.; Graux, C.; Lhermitte, L.; Lara, D.; Cluzeau, T.; Leguay, T.; Cieslak, A.; Trinquand, A.; Pastoret, C.; Belhocine, M.; et al. Early Response–Based Therapy Stratification Improves Survival in Adult Early Thymic Precursor Acute Lymphoblastic Leukemia: A Group for Research on Adult Acute Lymphoblastic Leukemia Study. J. Clin. Oncol. 2017, 35, 2683–2691. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Edmonson, M.N.; Wilkinson, M.R.; Patel, A.; Wu, G.; Liu, Y.; Li, Y.; Zhang, Z.; Rusch, M.C.; Parker, M.; et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat. Genet. 2016, 48, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Wigle, T.J.; Knutson, S.K.; Jin, L.; Kuntz, K.W.; Pollock, R.M.; Richon, V.M.; Copeland, R.A.; Scott, M.P. The Y641C mutation of EZH2 alters substrate specificity for histone H3 lysine 27 methylation states. FEBS Lett. 2011, 585, 3011–3014. [Google Scholar] [CrossRef] [PubMed]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Duployez, N.; Marceau-Renaut, A.; Boissel, N.; Petit, A.; Bucci, M.; Geffroy, S.; Lapillonne, H.; Renneville, A.; Ragu, C.; Figeac, M.; et al. Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood 2016, 127, 2451–2459. [Google Scholar] [CrossRef] [Green Version]
- Cordonnier, G.; Mandoli, A.; Cagnard, N.; Hypolite, G.; Lhermitte, L.; Verhoeyen, E.; Asnafi, V.; Dillon, N.; Macintyre, E.; Martens, J.H.; et al. CBFβ-SMMHC Affects Genome-wide Polycomb Repressive Complex 1 Activity in Acute Myeloid Leukemia. Cell Rep. 2020, 30, 299–307.e293. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Mazor, T.; Huang, H.; Huang, H.-T.; Kathrein, K.L.; Woo, A.J.; Chouinard, C.R.; Labadorf, A.; Akie, T.E.; Moran, T.B.; et al. Direct Recruitment of Polycomb Repressive Complex 1 to Chromatin by Core Binding Transcription Factors. Mol. Cell 2012, 45, 330–343. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lichtenberg, T.M.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Wen, Y.; Jin, R.; Chen, H. Epigenetic modifications and targeted therapy in pediatric acute myeloid leukemia. Front. Pediatr. 2022, 10, 975819. [Google Scholar] [CrossRef] [PubMed]
- Gallipoli, P.; Giotopoulos, G.; Huntly, B. Epigenetic regulators as promising therapeutic targets in acute myeloid leukemia. Ther. Adv. Hematol. 2015, 6, 103–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrieu, G.P.; Kohn, M.; Simonin, M.; Smith, C.L.; Cieslak, A.; Dourthe, M.; Charbonnier, G.; Graux, C.; Rigal-Huguet, F.; Lhéritier, V.; et al. PRC2 loss of function confers a targetable vulnerability to BET proteins in T-ALL. Blood 2021, 138, 1855–1869. [Google Scholar] [CrossRef]
- Perner, F.; Gadrey, J.Y.; Xiong, Y.; Hatton, C.; Eschle, B.K.; Weiss, A.; Stauffer, F.; Gaul, C.; Tiedt, R.; Perry, J.A.; et al. Novel inhibitors of the histone methyltransferase DOT1L show potent antileukemic activity in patient-derived xenografts. Blood 2020, 136, 1983–1988. [Google Scholar] [CrossRef]
- Dickins, R.A. Rerouting DOT1L inhibitors in leukemia. Blood 2020, 136, 1900–1901. [Google Scholar] [CrossRef]
- Porazzi, P.; Petruk, S.; Pagliaroli, L.; De Dominici, M.; Deming, I.D., 2nd; Puccetti, M.V.; Kushinsky, S.; Kumar, G.; Minieri, V.; Barbieri, E.; et al. Targeting Chemotherapy to Decondensed H3K27me3-Marked Chromatin of AML Cells Enhances Leukemia Suppression. Cancer Res. 2022, 82, 458–471. [Google Scholar] [CrossRef]
- Li, J.; Hlavka-Zhang, J.; Shrimp, J.H.; Piper, C.; Dupéré-Richér, D.; Roth, J.S.; Jing, D.; Casellas Román, H.L.; Troche, C.; Swaroop, A.; et al. PRC2 Inhibitors Overcome Glucocorticoid Resistance Driven by NSD2 Mutation in Pediatric Acute Lymphoblastic Leukemia. Cancer Discov. 2022, 12, 186–203. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PRC2 Component | Domain | Mutation | Paediatric/Adult | SIFT | PolyPhen-2 |
|---|---|---|---|---|---|
| EZH2 | V13A | Adult | 0.52 | 0 | |
| SBD | R25Q | Paediatric | 0 | 0.99 | |
| I146T | Adult | 0 | 0.43 | ||
| SRM | Y153C | Adult | 0 | 1 | |
| SANT1 domain | G159R | Adult | 0 | 1 | |
| D185H | Paediatric | 0 | 0.41 | ||
| E249K | Adult | 0 | 0.82 | ||
| CXC domain | H525N | Adult | 0 | 1 | |
| R561L | Adult | 0 | 1 | ||
| T568I | Adult | 0 | 0.03 | ||
| SET domain | V621M | Adult | 0.02 | 1 | |
| C642R | Adult | 0 | 1 | ||
| G643E | Both | 0 | 1 | ||
| A651V | Adult | 0.03 | 0.61 | ||
| G655R | Both | 0.01 | 1 | ||
| D659A | Adult | 0.02 | 1 | ||
| S664G | Adult | 0.01 | 1 | ||
| L669S | Adult | 0.03 | 1 | ||
| L669V | Adult | 0.03 | 1 | ||
| V674M | Adult | 0 | 1 | ||
| R679H | Both | 0 | 0.65 | ||
| R679C | Adult | 0 | 1 | ||
| K680M | Both | 0.04 | 1 | ||
| R685H | Both | 0 | 1 | ||
| R685C | Both | 0 | 1 | ||
| N688K | Adult | 0 | 1 | ||
| N688T | Adult | 0 | 1 | ||
| H689R | Adult | 0 | 1 | ||
| S690L | Both | 0 | 1 | ||
| S690P | Adult | 0 | 1 | ||
| G704S | Adult | 0.04 | 1 | ||
| post-SET domain | E740K | Both | 0.03 | 0.12 | |
| E740Q | Paediatric | 0.03 | 0.91 | ||
| SUZ12 | S51R | Adult | 0 | 0.38 | |
| ZnB domain | R103Q | Adult | 0 | 1 | |
| C2 | G163D | Adult | 0.08 | 0.98 | |
| R360L | Paediatric | 0.11 | 0 | ||
| EED | WD1 repeat | F97C | Paediatric | 0.01 | 1 |
| WD3 repeat | L196Q | Adult | 0.01 | 1 | |
| WD3 repeat | W364R | Paediatric | 0 | 1 | |
| RBBP4 | WD6 repeat | E330K | Adult | 0.05 | 0.84 |
| PRC2 Component | AML Cell Line | Type of Mutation | Protein Domain | Mutation | SIFT | PolyPhen-2 |
|---|---|---|---|---|---|---|
| EZH2 | PL21 | SNV | CXC domain | R561S | 0.03 | 1.00 |
| SKM1 | SNV | SET domain | Y641C | 0 | 0.09 | |
| OCIAML5 | SNV | R685H | 0.02 | 1.00 | ||
| P31FUJ | INS | post-SET domain | A731fs | N/A | N/A | |
| SUZ12 | KY821 | SNV | N-terminal | V68G | 0.01 | 0.00 |
| EED | GDM1 | SNV | WD40 repeat | D237E | 0.28 | 0.99 |
| JARID2 | NKM1 | SNV | EZH1/2-binding domain | D259D | 0.67 | synonymous mutation |
| PCL2 (MTF2) | MUTZ8 | SNV | Tudor domain | T65T | 1 | synonymous mutation |
| MV411 | SNV | Unknown function | Y409C | 0.14 | 0.96 | |
| LCOR (PALI1) | KG1 | SNV | G9A interaction region | E576K | 0 | 0.00 |
| P31FUJ | DEL | P588fs | N/A | N/A | ||
| SHI1 | SNV | Unknown function | G901A | 0 | 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharyya, T.; Bond, J. Polycomb Alterations in Acute Myeloid Leukaemia: From Structure to Function. Cancers 2023, 15, 1693. https://doi.org/10.3390/cancers15061693
Bhattacharyya T, Bond J. Polycomb Alterations in Acute Myeloid Leukaemia: From Structure to Function. Cancers. 2023; 15(6):1693. https://doi.org/10.3390/cancers15061693
Chicago/Turabian StyleBhattacharyya, Teerna, and Jonathan Bond. 2023. "Polycomb Alterations in Acute Myeloid Leukaemia: From Structure to Function" Cancers 15, no. 6: 1693. https://doi.org/10.3390/cancers15061693
APA StyleBhattacharyya, T., & Bond, J. (2023). Polycomb Alterations in Acute Myeloid Leukaemia: From Structure to Function. Cancers, 15(6), 1693. https://doi.org/10.3390/cancers15061693