


Peptidergic Systems and Cancer: Focus on Tachykinin and Calcitonin/Calcitonin Gene-Related Peptide Families




Abstract
:Simple Summary
Abstract
1. Introduction
2. Tachykinin and Calcitonin Peptide Families
2.1. Tachykinin Peptide Family
2.1.1. Genes and Products of Human Tachykinins

2.1.2. Structure of NK-2R and NK-3R
The Structure of NK-2R

The Structure of NK-3R

2.1.3. Intracellular Signaling of NK-2R and NK-3R
2.1.4. The Expression of NK-2R and NK-3R
2.2. Calcitonin/Calcitonin Gene-Related Peptide Family
2.2.1. Genes and Products of Human Calcitonin
2.2.2. Structure and Dynamics of the CGRP Receptor: A Three-Component Complex
2.2.3. Mechanisms of Signaling of the CGRP Receptor
3. Involvement of the Tachykinin and Calcitonin/Calcitonin Gene-Related Peptide Families in Cancer
3.1. Tachykinin Peptide Family
3.1.1. Breast Cancer
3.1.2. Colorectal Cancer
3.1.3. Glioma
3.1.4. Insulinoma
3.1.5. Lung Cancer
3.1.6. Medullary Thyroid Carcinoma
3.1.7. Midgut Carcinoid Tumor
3.1.8. Neuroblastoma
3.1.9. Oral Squamous Cell Carcinoma
3.1.10. Phaeochromocytoma
3.1.11. Schwannoma-Derived Cells
3.1.12. Small Bowel Neuroendocrine Tumors
3.1.13. Uterine Leiomyomata
3.2. Calcitonin/Calcitonin Gene-Related Peptide Family
3.2.1. Acute Myeloid Leukemia
Adrenomedullin
Calcitonin Gene-Related Peptide
3.2.2. Adrenocortical Tumor
Adrenomedullin
Adrenomedullin 2
Calcitonin Gene-Related Peptide
3.2.3. Bladder Cancer
Adrenomedullin
3.2.4. Breast Cancer
Adrenomedullin
Adrenomedullin 2
Calcitonin Gene-Related Peptide
3.2.5. Choriocarcinoma
Adrenomedullin
3.2.6. Colon Cancer
Adrenomedullin
Adrenomedullin 2
Amylin
Calcitonin Gene-Related Peptide
3.2.7. Cutaneous Nerve Neuromas
Calcitonin Gene-Related Peptide
3.2.8. Endometrial Cancer
Adrenomedullin
3.2.9. Ewing Sarcoma
Calcitonin Gene-Related Peptide
3.2.10. Gastric Cancer
Adrenomedullin
3.2.11. Glioma
Adrenomedullin
Adrenomedullin 2
Amylin
3.2.12. Head and Neck Squamous Cell Carcinoma
Calcitonin Gene-Related Peptide
3.2.13. Liver Cancer
Adrenomedullin
Adrenomedullin 2
Amylin
3.2.14. Lung Cancer
Adrenomedullin
Calcitonin Gene-Related Peptide
3.2.15. Melanoma
Adrenomedullin
3.2.16. Nasopharyngeal Carcinoma
Adrenomedullin
3.2.17. Neuroblastoma
Adrenomedullin
3.2.18. Neuroendocrine Tumors
Adrenomedullin
3.2.19. Oropharyngeal Squamous Cell Carcinoma
Adrenomedullin
3.2.20. Osteosarcoma
Adrenomedullin
3.2.21. Ovarian Cancer
Adrenomedullin
3.2.22. Pancreatic Cancer
Adrenomedullin
Adrenomedullin 2
Amylin
Calcitonin Gene-Related Peptide
3.2.23. Pituitary Adenoma
Adrenomedullin
Amylin
3.2.24. Prostate Cancer
Adrenomedullin
Adrenomedullin 2
Calcitonin Gene-Related Peptide
3.2.25. Renal Carcinoma
Adrenomedullin
3.2.26. Thymic Lymphomas
Amylin
3.2.27. Thyroid Cancer
Adrenomedullin 2
Amylin
Calcitonin Gene-Related Peptide
3.2.28. Uterine Cervical Carcinoma
Adrenomedullin
3.2.29. Vascular Tumors
Adrenomedullin
4. Antitumor Therapeutic Strategies Based on the Modulation of Peptidergic Systems
4.1. Tachykinin Peptide Family
4.2. Calcitonin/Calcitonin Gene-Related Peptide Family
4.2.1. Adrenomedullin
4.2.2. Adrenomedullin 2
4.2.3. Amylin
4.2.4. Calcitonin Gene-Related Peptide
5. Future Research
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Coveñas, R.; Muñoz, M. Involvement of the Substance P/Neurokinin-1 Receptor System in Cancer. Cancers 2022, 14, 3539. [Google Scholar] [CrossRef]
- Sánchez, M.L.; Coveñas, R. The Neurotensinergic System: A Target for Cancer Treatment. Curr. Med. Chem. 2022, 29, 3231–3260. [Google Scholar] [CrossRef]
- Sánchez, M.L.; Coveñas, R. The Galaninergic System: A Target for Cancer Treatment. Cancers 2022, 14, 3755. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.; Coveñas, R.; Muñoz, M. Combination Therapy of Chemotherapy or Radiotherapy and the Neurokinin-1 Receptor Antagonist Aprepitant: A New Antitumor Strategy? Curr. Med. Chem. 2023, 30, 1798–1812. [Google Scholar] [CrossRef]
- Coveñas, R.; Rodríguez, F.D.; Muñoz, M. The Neurokinin-1 Receptor: A Promising Antitumor Target. Receptors 2022, 1, 5. [Google Scholar] [CrossRef]
- Kastin, A.J. Handbook of Biologically Active Peptides, 2nd ed.; Academic Press: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Wu, Y.; Berisha, A.; Borniger, J.C. Neuropeptides in Cancer: Friend and Foe? Adv. Biol. 2022, 6, 2200111. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, T.; Liu, Q.; Cummins, S. Copy Number Alteration of Neuropeptides and Receptors in Multiple Cancers. Sci. Rep. 2017, 7, 4598. [Google Scholar] [CrossRef] [Green Version]
- Kasprzak, A.; Adamek, A. The Neuropeptide System and Colorectal Cancer Liver Metastases: Mechanisms and Management. Int. J. Mol. Sci. 2020, 21, 3494. [Google Scholar] [CrossRef]
- Colucci, R.; Blandizzi, C.; Ghisu, N.; Florio, T.; Del Tacca, M. Somatostatin Inhibits Colon Cancer Cell Growth through Cyclooxygenase-2 Downregulation: Somatostatin and Cyclooxygenase-2 in Colon Cancer. Br. J. Pharmacol. 2008, 155, 198–209. [Google Scholar] [CrossRef]
- Godlewski, J.; Kmiec, Z. Colorectal Cancer Invasion and Atrophy of the Enteric Nervous System: Potential Feedback and Impact on Cancer Progression. Int. J. Mol. Sci. 2020, 21, 3391. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Coveñas, R. Neurokinin Receptor Antagonism: A Patent Review (2014-Present). Expert Opin. Ther. Pat. 2020, 30, 527–539. [Google Scholar] [CrossRef]
- Muñoz, M.; Coveñas, R. Involvement of Substance P and the NK-1 Receptor in Human Pathology. Amino Acids 2014, 46, 1727–1750. [Google Scholar] [CrossRef]
- Ebner, K.; Sartori, S.; Singewald, N. Tachykinin Receptors as Therapeutic Targets in Stress-Related Disorders. Curr. Pharm. Des. 2009, 15, 1647–1674. [Google Scholar] [CrossRef]
- Steinhoff, M.S.; von Mentzer, B.; Geppetti, P.; Pothoulakis, C.; Bunnett, N.W. Tachykinins and Their Receptors: Contributions to Physiological Control and the Mechanisms of Disease. Physiol. Rev. 2014, 94, 265–301. [Google Scholar] [CrossRef] [Green Version]
- Coveñas, R.; Mangas, A.; Narvaez, J.A. Introduction to Neuropeptides. In Focus on Neuropeptide Research; Transworld Research Network: Trivandrum, India, 2007; pp. 1–26. [Google Scholar]
- Onaga, T. Tachykinin: Recent Developments and Novel Roles in Health and Disease. Biomol. Concepts 2014, 5, 225–243. [Google Scholar] [CrossRef]
- Bepler, G.; Rotsch, M.; Jaques, G.; Haeder, M.; Heymanns, J.; Hartogh, G.; Kiefer, P.; Havemann, K. Peptides and Growth Factors in Small Cell Lung Cancer: Production, Binding Sites, and Growth Effects. J. Cancer Res. Clin. Oncol. 1988, 114, 235–244. [Google Scholar] [CrossRef]
- Obata, K.; Shimo, T.; Okui, T.; Matsumoto, K.; Takada, H.; Takabatake, K.; Kunisada, Y.; Ibaragi, S.; Nagatsuka, H.; Sasaki, A. Tachykinin Receptor 3 Distribution in Human Oral Squamous Cell Carcinoma. Anticancer Res. 2016, 36, 6335–6342. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, M.; Coveñas, R. Neurokinin/Tachykinin Receptors. In Ecyclopedia of Molecular Pharmacology; Springer: Berlin/Heidelberg, Germany, 2021; pp. 1093–1103. [Google Scholar]
- Severini, C. The Tachykinin Peptide Family. Pharmacol. Rev. 2002, 54, 285–322. [Google Scholar] [CrossRef]
- Nässel, D.R.; Zandawala, M.; Kawada, T.; Satake, H. Tachykinins: Neuropeptides That Are Ancient, Diverse, Widespread and Functionally Pleiotropic. Front. Neurosci. 2019, 13, 1262. [Google Scholar] [CrossRef] [PubMed]
- UNIPROT. Database. Available online: https://www.uniprot.org/uniprot/P25103 (accessed on 25 October 2022).
- MacDonald, M.R.; McCourt, D.W.; Krause, J.E. Posttranslational Processing of Alpha-, Beta-, and Gamma-Preprotachykinins. Cell-Free Translation and Early Posttranslational Processing Events. J. Biol. Chem. 1988, 263, 15176–15183. [Google Scholar] [CrossRef]
- Shimizu, Y.; Matsuyama, H.; Shiina, T.; Takewaki, T.; Furness, J.B. Tachykinins and Their Functions in the Gastrointestinal Tract. Cell. Mol. Life Sci. 2008, 65, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Nawa, H.; Kotani, H.; Nakanishi, S. Tissue-Specific Generation of Two Preprotachykinin MRNAs from One Gene by Alternative RNA Splicing. Nature 1984, 312, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Nawa, H.; Doteuchi, M.; Igano, K.; Inouye, K.; Nakanishi, S. Substance K: A Novel Mammalian Tachykinin That Differs from Substance P in Its Pharmacological Profile. Life Sci. 1984, 34, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Harmar, A.J.; Armstrong, A.; Pascall, J.C.; Chapman, K.; Rosie, R.; Curtis, A.; Going, J.; Edwards, C.R.W.; Fink, G. CDNA Sequence of Human β-Preprotachykinin, the Common Precursor to Substance P and Neurokinin A. FEBS Lett. 1986, 208, 67–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kage, R.; Thim, L.; Creutzfeldt, W.; Conlon, J.M. Post-Translational Processing of Preprotachykinins. Isolation of Protachykinin-(1-37)-Peptide from Human Adrenal-Medullary Phaeochromocytoma Tissue. Biochem. J. 1988, 253, 203–207. [Google Scholar] [CrossRef] [Green Version]
- Prigge, S.T.; Mains, R.E.; Eipper, B.A.; Amzel, L.M. New Insights into Copper Monooxygenases and Peptide Amidation: Structure, Mechanism and Function. Cell. Mol. Life Sci. 2000, 57, 1236–1259. [Google Scholar] [CrossRef]
- Wong, M.; Jeng, A.Y. Posttranslational Modification of Glycine-Extended Substance P by an Alpha-Amidating Enzyme in Cultured Sensory Neurons of Dorsal Root Ganglia. J. Neurosci. Res. 1994, 37, 97–102. [Google Scholar] [CrossRef]
- Entrez-Gene. National Library of Medicine. Gene Database. Available online: https://www.ncbi.nlm.nih.gov/gene (accessed on 20 October 2022).
- Maggi, C.A. The Mammalian Tachykinin Receptors. Gen. Pharmacol. Vasc. Syst. 1995, 26, 911–944. [Google Scholar] [CrossRef]
- EXPASY. SIM Alignment Tool for Protein Sequences. Available online: https://web.expasy.org/sim/ (accessed on 20 October 2022).
- Lefkowitz, R.J. The Superfamily of Heptahelical Receptors. Nat. Cell Biol. 2000, 2, E133–E136. [Google Scholar] [CrossRef] [PubMed]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and Dynamics of GPCR Signaling Complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging Paradigms in GPCR Allostery: Implications for Drug Discovery. Nat. Rev. Drug Discov. 2013, 12, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Hilger, D. The Role of Structural Dynamics in GPCR-mediated Signaling. FEBS J. 2021, 288, 2461–2489. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zhao, T.; Yun, Y.; Xie, X. Recent Progress in Assays for GPCR Drug Discovery. Am. J. Physiol.-Cell Physiol. 2022, 323, C583–C594. [Google Scholar] [CrossRef] [PubMed]
- Schöppe, J.; Ehrenmann, J.; Klenk, C.; Rucktooa, P.; Schütz, M.; Doré, A.S.; Plückthun, A. Crystal Structures of the Human Neurokinin 1 Receptor in Complex with Clinically Used Antagonists. Nat. Commun. 2019, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentin-Hansen, L.; Frimurer, T.M.; Mokrosinski, J.; Holliday, N.D.; Schwartz, T.W. Biased Gs Versus Gq Proteins and β-Arrestin Signaling in the NK1 Receptor Determined by Interactions in the Water Hydrogen Bond Network. J. Biol. Chem. 2015, 290, 24495–24508. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Chapman, K.; Clark, L.D.; Shao, Z.; Borek, D.; Xu, Q.; Wang, J.; Rosenbaum, D.M. Crystal Structure of the Human NK 1 Tachykinin Receptor. Proc. Natl. Acad. Sci. USA 2018, 115, 13264–13269. [Google Scholar] [CrossRef] [Green Version]
- Thom, C.; Ehrenmann, J.; Vacca, S.; Waltenspühl, Y.; Schöppe, J.; Medalia, O.; Plückthun, A. Structures of Neurokinin 1 Receptor in Complex with G q and G s Proteins Reveal Substance P Binding Mode and Unique Activation Features. Sci. Adv. 2021, 7, eabk2872. [Google Scholar] [CrossRef]
- Rodríguez, F.D.; Coveñas, R. The Neurokinin-1 Receptor: Structure Dynamics and Signaling. Receptors 2022, 1, 4. [Google Scholar] [CrossRef]
- GPCR. Database. Available online: https://gpcrdb.org/protein/nk1r_human (accessed on 25 October 2022).
- KingDraw. Free Software. Available online: http://www.kingdraw.cn/en/index.html (accessed on 20 October 2022).
- Gerard, N.P.; Bao, L.; Xiao-Ping, H.; Gerard, C. Molecular Aspects of the Tachykinin Receptors. Regul. Pept. 1993, 43, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Gembitsky, D.S.; Murnin, M.; Ötvös, F.L.; Allen, J.; Murphy, R.F.; Lovas, S. Importance of the Aromatic Residue at Position 6 of [Nle10] Neurokinin A (4−10) for Binding to the NK-2 Receptor and Receptor Activation. J. Med. Chem. 1999, 42, 3004–3007. [Google Scholar] [CrossRef] [PubMed]
- Rovero, P.; Pestellini, V.; Rhaleb, N.-E.; Dion, S.; Rouissi, N.; Tousignant, C.; Télémaque, S.; Drapeau, G.; Regoli, D. Structure-Activity Studies of Neurokinin A. Neuropeptides 1989, 13, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Warner, F.J.; Mack, P.; Comis, A.; Miller, R.C.; Burcher, E. Structure–Activity Relationships of Neurokinin A (4–10) at the Human Tachykinin NK2 Receptor: The Role of Natural Residues and Their Chirality. Biochem. Pharmacol. 2001, 61, 55–60. [Google Scholar] [CrossRef]
- Regoli, D.; Boudon, A.; Fauchére, J.L. Receptors and Antagonists for Substance P and Related Peptides. Pharmacol. Rev. 1994, 45, 551–599. [Google Scholar]
- Bhogal, N.; Donnelly, D.; Findlay, J.B. The Ligand Binding Site of the Neurokinin 2 Receptor. Site-Directed Mutagenesis and Identification of Neurokinin A Binding Residues in the Human Neurokinin 2 Receptor. J. Biol. Chem. 1994, 269, 27269–27274. [Google Scholar] [CrossRef]
- Chandrashekaran, I.R.; Rao, G.S.; Cowsik, S.M. Molecular Modeling of the Peptide Agonist-Binding Site in a Neurokinin-2 Receptor. J. Chem. Inf. Model. 2009, 49, 1734–1740. [Google Scholar] [CrossRef]
- Chandrashekar, I.R.; Cowsik, S.M. Three-Dimensional Structure of the Mammalian Tachykinin Peptide Neurokinin A Bound to Lipid Micelles. Biophys. J. 2003, 85, 4002–4011. [Google Scholar] [CrossRef] [Green Version]
- Zoffmann, S.; Bertrand, S.; Do, Q.-T.; Bertrand, D.; Rognan, D.; Hibert, M.; Galzi, J.-L. Topological Analysis of the Complex Formed between Neurokinin A and the NK2 Tachykinin Receptor. J. Neurochem. 2007, 101, 506–516. [Google Scholar] [CrossRef]
- Meini, S.; Bellucci, F.; Catalani, C.; Cucchi, P.; Patacchini, R.; Rotondaro, L.; Altamura, M.; Giuliani, S.; Giolitti, A.; Maggi, C.A. Mutagenesis at the Human Tachykinin NK2 Receptor to Define the Binding Site of a Novel Class of Antagonists. Eur. J. Pharmacol. 2004, 488, 61–69. [Google Scholar] [CrossRef]
- Maillet, E.L.; Pellegrini, N.; Valant, C.; Bucher, B.; Hibert, M.; Bourguignon, J.-J.; Galzi, J.-L. A Novel, Conformation-specific Allosteric Inhibitor of the Tachykinin NK2 Receptor (NK2R) with Functionally Selective Properties. FASEB J. 2007, 21, 2124–2134. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Yuan, Q.; Zhang, H.; Yang, F.; Ling, S.; Luo, Y.; Lv, P.; Eric Xu, H.; Tian, C.; Yin, W.; et al. Structural Insights into the Activation of Neurokinin 2 Receptor by Neurokinin A. Cell Discov. 2022, 8, 72. [Google Scholar] [CrossRef]
- Harris, J.A.; Faust, B.; Gondin, A.B.; Dämgen, M.A.; Suomivuori, C.-M.; Veldhuis, N.A.; Cheng, Y.; Dror, R.O.; Thal, D.M.; Manglik, A. Selective G Protein Signaling Driven by Substance P–Neurokinin Receptor Dynamics. Nat. Chem. Biol. 2022, 18, 109–115. [Google Scholar] [CrossRef]
- AlphaFold. AlphaFold Protein Structure Database. Available online: https://alphafold.ebi.ac.uk/ (accessed on 15 October 2022).
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- PDB. Protein Data Bank (PDB). Available online: https://pdb101.rcsb.org (accessed on 25 October 2022).
- Huang, R.-R.C.; Cheung, A.H.; Mazina, K.E.; Strader, C.D.; Fong, T.M. CDNA Sequence and Heterologous Expression of the Human Neurokinin-3 Receptor. Biochem. Biophys. Res. Commun. 1992, 184, 966–972. [Google Scholar] [CrossRef]
- Mantha, A.K.; Chandrashekar, I.R.; Baquer, N.Z.; Cowsik, S.M. Three Dimensional Structure of Mammalian Tachykinin Peptide Neurokinin B Bound to Lipid Micelles. J. Biomol. Struct. Dyn. 2004, 22, 137–147. [Google Scholar] [CrossRef]
- Ganjiwale, A.D.; Rao, G.S.; Cowsik, S.M. Molecular Modeling of Neurokinin B and Tachykinin NK3 Receptor Complex. J. Chem. Inf. Model. 2011, 11, 2932–2938. [Google Scholar] [CrossRef]
- Malherbe, P.; Bissantz, C.; Marcuz, A.; Kratzeisen, C.; Zenner, M.-T.; Wettstein, J.G.; Ratni, H.; Riemer, C.; Spooren, W. Me-Talnetant and Osanetant Interact within Overlapping but Not Identical Binding Pockets in the Human Tachykinin Neurokinin 3 Receptor Transmembrane Domains. Mol. Pharmacol. 2008, 73, 1736–1750. [Google Scholar] [CrossRef] [Green Version]
- Geldenhuys, W.J.; Simmons, M.A. 3D-Quantitative Structure–Activity Relationship and Docking Studies of the Tachykinin NK3 Receptor. Bioorg. Med. Chem. Lett. 2011, 21, 7405–7411. [Google Scholar] [CrossRef]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ramnath, R.D.; Tamizhselvi, R.; Bhatia, M. Neurokinin A Engages Neurokinin-1 Receptor to Induce NF-ΚB-Dependent Gene Expression in Murine Macrophages: Implications of ERK1/2 and PI 3-Kinase/Akt Pathways. Am. J. Physiol.-Cell Physiol. 2008, 295, C679–C691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khannpnavar, B.; Mehta, V.; Qi, C.; Korkhov, V. Structure and Function of Adenylyl Cyclases, Key Enzymes in Cellular Signaling. Curr. Opin. Struct. Biol. 2020, 63, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Yang, S.; Wang, J.; Zhu, G. CAMP-PKA Cascade: An Outdated Topic for Depression? Biomed. Pharmacother. 2022, 150, 113030. [Google Scholar] [CrossRef] [PubMed]
- Sassone-Corsi, P. The Cyclic AMP Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011148. [Google Scholar] [CrossRef]
- Zhang, H.; Kong, Q.; Wang, J.; Jiang, Y.; Hua, H. Complex Roles of CAMP–PKA–CREB Signaling in Cancer. Exp. Hematol. Oncol. 2020, 9, 32. [Google Scholar] [CrossRef]
- He, S.; Li, Q.; Huang, Q.; Cheng, J. Targeting Protein Kinase C for Cancer Therapy. Cancers 2022, 14, 1104. [Google Scholar] [CrossRef]
- Maekawa, M.; Ishizaki, T.; Boku, S.; Watanabe, N.; Fujita, A.; Iwamatsu, A.; Obinata, T.; Ohashi, K.; Mizuno, K.; Narumiya, S. Signaling from Rho to the Actin Cytoskeleton Through Protein Kinases ROCK and LIM-Kinase. Science 1999, 285, 895–898. [Google Scholar] [CrossRef]
- Lee, M.-H.; Kundu, J.K.; Chae, J.-I.; Shim, J.-H. Targeting ROCK/LIMK/Cofilin Signaling Pathway in Cancer. Arch. Pharm. Res. 2019, 42, 481–491. [Google Scholar] [CrossRef]
- Kümper, S.; Mardakheh, F.K.; McCarthy, A.; Yeo, M.; Stamp, G.W.; Paul, A.; Worboys, J.; Sadok, A.; Jørgensen, C.; Guichard, S.; et al. Rho-Associated Kinase (ROCK) Function Is Essential for Cell Cycle Progression, Senescence and Tumorigenesis. Elife 2016, 5, e12203. [Google Scholar] [CrossRef]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 Biochemistry. Cardiovasc. Drugs Ther. 2009, 23, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchi, L.; Araújo, T.G.; Azevedo, F.V.P.d.V.; Mota, S.T.S.; Ávila, V.d.M.R.; Ribeiro, M.A.; Goulart, L.R. Phospholipase A2 Drives Tumorigenesis and Cancer Aggressiveness through Its Interaction with Annexin A1. Cells 2021, 10, 1472. [Google Scholar] [CrossRef] [PubMed]
- Yarla, N.S.; Bishayee, A.; Sethi, G.; Reddanna, P.; Kalle, A.M.; Dhananjaya, B.L.; Dowluru, K.S.V.G.K.; Chintala, R.; Duddukuri, G.R. Targeting Arachidonic Acid Pathway by Natural Products for Cancer Prevention and Therapy. Semin. Cancer Biol. 2016, 40–41, 48–81. [Google Scholar] [CrossRef] [PubMed]
- Hur, E.-M.; Kim, K.-T. G Protein-Coupled Receptor Signalling and Cross-Talk. Cell. Signal. 2002, 14, 397–405. [Google Scholar] [CrossRef]
- Tuluc, F.; Lai, J.P.; Kilpatrick, L.E.; Evans, D.L.; Douglas, S.D. Neurokinin 1 Receptor Isoforms and the Control of Innate Immunity. Trends Immunol. 2009, 30, 271–276. [Google Scholar] [CrossRef]
- Garcia-Recio, S.; Gascón, P. Biological and Pharmacological Aspects of the NK1-Receptor. Biomed Res. Int. 2015, 2015, 495704. [Google Scholar] [CrossRef] [Green Version]
- HPA. The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 31 October 2022).
- Uhlen, M.; Karlsson, M.J.; Zhong, W.; Tebani, A.; Pou, C.; Mikes, J.; Lakshmikanth, T.; Forsström, B.; Edfors, F.; Odeberg, J.; et al. A Genome-Wide Transcriptomic Analysis of Protein-Coding Genes in Human Blood Cells. Science 2019, 366, eaax9198. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Blasco, V.; Pinto, F.M.; Fernández-Atucha, A.; Prados, N.; Tena-Sempere, M.; Fernández-Sánchez, M.; Candenas, L. Altered Expression of the Kisspeptin/KISS1R and Neurokinin B/NK3R Systems in Mural Granulosa and Cumulus Cells of Patients with Polycystic Ovarian Syndrome. J. Assist. Reprod. Genet. 2019, 36, 113–120. [Google Scholar] [CrossRef]
- Cañete, H.; Dorta, I.; Hernández, M.; Cejudo Roman, A.; Candenas, L.; Pinto, F.M.; Valladares, F.; Báez, D.; Montes de Oca, F.; Bello, A.R.; et al. Differentially Regulated Expression of Neurokinin B (NKB)/NK3 Receptor System in Uterine Leiomyomata. Hum. Reprod. 2013, 28, 1799–1808. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Joshi, D.D.; Hameed, M.; Qian, J.; Gascón, P.; Maloof, P.B.; Mosenthal, A.; Rameshwar, P. Increased Expression of Preprotachykinin-I and Neurokinin Receptors in Human Breast Cancer Cells: Implications for Bone Marrow Metastasis. Proc. Natl. Acad. Sci. USA 2000, 97, 388–393. [Google Scholar] [CrossRef] [Green Version]
- King, R.; Brain, S.D. CGRP. In Handbook of Biologically Active Peptides; Academic Press: Amsterdam, The Netherlands, 2013; pp. 1394–1466. [Google Scholar]
- Montuenga, L.M.; Martínez, A.; Miller, M.J.; Unsworth, E.J.; Cuttitta, F. Expression of Adrenomedullin and Its Receptor during Embryogenesis Suggests Autocrine or Paracrine Modes of Action. Endocrinology 1997, 138, 440–451. [Google Scholar] [CrossRef]
- Hinson, J.P.; Kapas, S.; Smith, D.M. Adrenomedullin, a Multifunctional Regulatory Peptide. Endocr. Rev. 2000, 21, 138–167. [Google Scholar] [CrossRef]
- Kim, W.; Moon, S.-O.; Sung, M.J.; Kim, S.H.; Lee, S.; So, J.-N.; Park, S.K. Angiogenic Role of Adrenomedullin through Activation of Akt, Mitogen-activated Protein Kinase, and Focal Adhesion Kinase in Endothelial Cells. FASEB J. 2003, 17, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Iwase, T.; Nagaya, N.; Fujii, T.; Itoh, T.; Ishibashi-Ueda, H.; Yamagishi, M.; Miyatake, K.; Matsumoto, T.; Kitamura, S.; Kangawa, K. Adrenomedullin Enhances Angiogenic Potency of Bone Marrow Transplantation in a Rat Model of Hindlimb Ischemia. Circulation 2005, 111, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Shichiri, M.; Marumo, F.; Hirata, Y. Adrenomedullin as an Autocrine/Paracrine Apoptosis Survival Factor for Rat Endothelial Cells. Endocrinology 1997, 138, 2615–2620. [Google Scholar] [CrossRef]
- Martinez, A. The Effects of Adrenomedullin Overexpression in Breast Tumor Cells. Cancer Spectr. Knowl. Environ. 2002, 94, 1226–1237. [Google Scholar] [CrossRef] [Green Version]
- Nikitenko, L.L. Adrenomedullin Is an Autocrine Regulator of Endothelial Growth in Human Endometrium. Mol. Hum. Reprod. 2000, 6, 811–819. [Google Scholar] [CrossRef]
- Oehler, M.K.; Hague, S.; Rees, M.C.; Bicknell, R. Adrenomedullin Promotes Formation of Xenografted Endometrial Tumors by Stimulation of Autocrine Growth and Angiogenesis. Oncogene 2002, 21, 2815–2821. [Google Scholar] [CrossRef] [Green Version]
- Hippenstiel, S.; Witzenrath, M.; Schmeck, B.; Hocke, A.; Krisp, M.; Krüll, M.; Seybold, J.; Seeger, W.; Rascher, W.; Schütte, H.; et al. Adrenomedullin Reduces Endothelial Hyperpermeability. Circ. Res. 2002, 91, 618–625. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Hague, S.; Manek, S.; Zhang, L.; Bicknell, R.; Rees, M.C. PCR Display Identifies Tamoxifen Induction of the Novel Angiogenic Factor Adrenomedullin by a Non Estrogenic Mechanism in the Human Endometrium. Oncogene 1998, 16, 409–415. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Han, B.; Nunobiki, O.; Kakudo, K. Adrenomedullin: A Tumor Progression Factor via Angiogenic Control. Curr. Cancer Drug Targets 2006, 6, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Ewers, M.; Mielke, M.M.; Hampel, H. Blood-Based Biomarkers of Microvascular Pathology in Alzheimer’s Disease. Exp. Gerontol. 2010, 45, 75–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucukosmanoglu, E.; Keskin, O.; Karcin, M.; Cekmen, M.; Balat, A. Plasma Adrenomedullin Levels in Children with Asthma: Any Relation with Atopic Dermatitis? Allergol. Immunopathol. 2012, 40, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Broderick, C.L.; Brooke, G.S.; DiMarchi, R.D.; Gold, G. Human and Rat Amylin Have No Effects on Insulin Secretion in Isolated Rat Pancreatic Islets. Biochem. Biophys. Res. Commun. 1991, 177, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Mather, K.J.; Paradisi, G.; Leaming, R.; Hook, G.; Steinberg, H.O.; Fineberg, N.; Hanley, R.; Baron, A.D. Role of Amylin in Insulin Secretion and Action in Humans: Antagonist Studies across the Spectrum of Insulin Sensitivity. Diabetes Metab. Res. Rev. 2002, 18, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.L.; Chen, S.; Lutz, T.A.; Parkes, D.G.; Roth, J.D. Amylin: Pharmacology, Physiology, and Clinical Potential. Pharmacol. Rev. 2015, 67, 564–600. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Kong, L.; Yang, L.; Nie, Y.Z.; Liang, Y.; Teng, C.B. Toxicity Reduction of Human Islet Amyloid Polypeptide by Trim-Away Technique in Insulinoma Cells. Yi Chuan 2020, 42, 586–598. (In Chinese) [Google Scholar]
- Muff, R.; Born, W.; Lutz, T.A.; Fischer, J.A. Biological Importance of the Peptides of the Calcitonin Family as Revealed by Disruption and Transfer of Corresponding Genes. Peptides 2004, 25, 2027–2038. [Google Scholar] [CrossRef]
- Garelja, M.L.; Hay, D.L. A Narrative Review of the Calcitonin Peptide Family and Associated Receptors as Migraine Targets: Calcitonin Gene-Related Peptide and Beyond. Headache J. Head Face Pain 2022, 62, 1093–1104. [Google Scholar] [CrossRef]
- Naot, D.; Musson, D.S.; Cornish, J. The Activity of Peptides of the Calcitonin Family in Bone. Physiol. Rev. 2019, 99, 781–805. [Google Scholar] [CrossRef]
- Hay, D.L.; Garelja, M.L.; Poyner, D.R.; Walker, C.S. Update on the Pharmacology of Calcitonin/CGRP Family of Peptides: IUPHAR Review 25: Pharmacology of the Calcitonin/CGRP Family. Br. J. Pharmacol. 2018, 175, 3–17. [Google Scholar] [CrossRef]
- Andreotti, G.; Vitale, R.M.; Avidan-Shpalter, C.; Amodeo, P.; Gazit, E.; Motta, A. Converting the Highly Amyloidogenic Human Calcitonin into a Powerful Fibril Inhibitor by Three-Dimensional Structure Homology with a Non-Amyloidogenic Analogue. J. Biol. Chem. 2011, 286, 2707–2718. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, M.G.; Mermod, J.-J.; Amara, S.G.; Swanson, L.W.; Sawchenko, P.E.; Rivier, J.; Vale, W.W.; Evans, R.M. Production of a Novel Neuropeptide Encoded by the Calcitonin Gene via Tissue-Specific RNA Processing. Nature 1983, 304, 129–135. [Google Scholar] [CrossRef]
- Amara, S.G.; Arriza, J.L.; Leff, S.E.; Swanson, L.W.; Evans, R.M.; Rosenfeld, M.G. Expression in Brain of a Messenger RNA Encoding a Novel Neuropeptide Homologous to Calcitonin Gene-Related Peptide. Science 1985, 229, 1094–1097. [Google Scholar] [CrossRef]
- Hillyard, C.J.; Abeyasekera, G.; Craig, R.; Myers, C.; Stevenson, J.; Macintyre, I. Katacalcin: A New Plasma Calcium-Lowering Hormone. Lancet 1983, 321, 846–848. [Google Scholar] [CrossRef]
- Josephs, T.M.; Belousoff, M.J.; Liang, Y.-L.; Piper, S.J.; Cao, J.; Garama, D.J.; Leach, K.; Gregory, K.J.; Christopoulos, A.; Hay, D.L.; et al. Structure and Dynamics of the CGRP Receptor in Apo and Peptide-Bound Forms. Science 2021, 372, eabf7258. [Google Scholar] [CrossRef]
- McLatchie, L.M.; Fraser, N.J.; Main, M.J.; Wise, A.; Brown, J.; Thompson, N.; Solari, R.; Lee, M.G.; Foord, S.M. RAMPs Regulate the Transport and Ligand Specificity of the Calcitonin-Receptor-like Receptor. Nature 1998, 393, 333–339. [Google Scholar] [CrossRef]
- Simms, J.; Routledge, S.; Uddin, R.; Poyner, D. The Structure of the CGRP and Related Receptors. Calcitonin Gene-Relat. Pept. CGRP Mech. 2018, 255, 23–36. [Google Scholar] [CrossRef]
- Cong, Z.; Liang, Y.-L.; Zhou, Q.; Darbalaei, S.; Zhao, F.; Feng, W.; Zhao, L.; Xu, H.E.; Yang, D.; Wang, M.-W. Structural Perspective of Class B1 GPCR Signaling. Trends Pharmacol. Sci. 2022, 43, 321–334. [Google Scholar] [CrossRef]
- Liang, Y.-L.; Khoshouei, M.; Deganutti, G.; Glukhova, A.; Koole, C.; Peat, T.S.; Radjainia, M.; Plitzko, J.M.; Baumeister, W.; Miller, L.J.; et al. Cryo-EM Structure of the Active, Gs-Protein Complexed, Human CGRP Receptor. Nature 2018, 561, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Egea, S.C.; Dickerson, I.M. Direct Interactions between Calcitonin-Like Receptor (CLR) and CGRP-Receptor Component Protein (RCP) Regulate CGRP Receptor Signaling. Endocrinology 2012, 153, 1850–1860. [Google Scholar] [CrossRef]
- Dickerson, I. Role of CGRP-Receptor Component Protein (RCP) in CLR/RAMP Function. Curr. Protein Pept. Sci. 2013, 14, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Evans, B.N.; Rosenblatt, M.I.; Mnayer, L.O.; Oliver, K.R.; Dickerson, I.M. CGRP-RCP, a Novel Protein Required for Signal Transduction at Calcitonin Gene-Related Peptide and Adrenomedullin Receptors. J. Biol. Chem. 2000, 275, 31438–31443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ter Haar, E.; Koth, C.M.; Abdul-Manan, N.; Swenson, L.; Coll, J.T.; Lippke, J.A.; Lepre, C.A.; Garcia-Guzman, M.; Moore, J.M. Crystal Structure of the Ectodomain Complex of the CGRP Receptor, a Class-B GPCR, Reveals the Site of Drug Antagonism. Structure 2010, 18, 1083–1093. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Lehto, S.G.; Zhu, D.X.D.; Sun, H.; Zhang, J.; Smith, B.P.; Immke, D.C.; Wild, K.D.; Xu, C. Pharmacologic Characterization of AMG 334, a Potent and Selective Human Monoclonal Antibody against the Calcitonin Gene-Related Peptide Receptor. J. Pharmacol. Exp. Ther. 2015, 356, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Garces, F.; Mohr, C.; Zhang, L.; Huang, C.-S.; Chen, Q.; King, C.; Xu, C.; Wang, Z. Molecular Insight into Recognition of the CGRPR Complex by Migraine Prevention Therapy Aimovig (Erenumab). Cell Rep. 2020, 30, 1714–1723.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhakta, M.; Vuong, T.; Taura, T.; Wilson, D.S.; Stratton, J.R.; Mackenzie, K.D. Migraine Therapeutics Differentially Modulate the CGRP Pathway. Cephalalgia 2021, 41, 499–514. [Google Scholar] [CrossRef]
- Klein, K.R.; Matson, B.C.; Caron, K.M. The Expanding Repertoire of Receptor Activity Modifying Protein (RAMP) Function. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 65–71. [Google Scholar] [CrossRef]
- Cottrell, G.S. CGRP Receptor Signalling Pathways. Calcitonin Gene-Relat. Pept. CGRP Mech. 2018, 255, 37–64. [Google Scholar] [CrossRef]
- Routledge, S.J.; Simms, J.; Clark, A.; Yeung, H.Y.; Wigglesworth, M.J.; Dickerson, I.M.; Kitchen, P.; Ladds, G.; Poyner, D.R. Receptor Component Protein, an Endogenous Allosteric Modulator of Family B G Protein Coupled Receptors. Biochim. Biophys. Acta BBA—Biomembr. 2020, 1862, 183174. [Google Scholar] [CrossRef] [PubMed]
- Conner, A.C.; Simms, J.; Howitt, S.G.; Wheatley, M.; Poyner, D.R. The Second Intracellular Loop of the Calcitonin Gene-Related Peptide Receptor Provides Molecular Determinants for Signal Transduction and Cell Surface Expression. J. Biol. Chem. 2006, 281, 1644–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, C.S.; Raddant, A.C.; Woolley, M.J.; Russo, A.F.; Hay, D.L. CGRP Receptor Antagonist Activity of Olcegepant Depends on the Signalling Pathway Measured. Cephalalgia 2018, 38, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Suekane, A.; Ichikawa, T.; Saito, Y.; Nakahata, S.; Morishita, K. The CGRP Receptor Antagonist MK0974 Induces EVI1 high AML Cell Apoptosis by Disrupting ERK Signaling. Anticancer Res. 2022, 42, 4743–4752. [Google Scholar] [CrossRef]
- Mancinelli, R.; Ceci, L.; Kennedy, L.; Francis, H.; Meadows, V.; Chen, L.; Carpino, G.; Kyritsi, K.; Wu, N.; Zhou, T.; et al. The Effects of Taurocholic Acid on Biliary Damage and Liver Fibrosis Are Mediated by Calcitonin-Gene-Related Peptide Signaling. Cells 2022, 11, 1591. [Google Scholar] [CrossRef]
- Yarwood, R.E.; Imlach, W.L.; Lieu, T.; Veldhuis, N.A.; Jensen, D.D.; Klein Herenbrink, C.; Aurelio, L.; Cai, Z.; Christie, M.J.; Poole, D.P.; et al. Endosomal Signaling of the Receptor for Calcitonin Gene-Related Peptide Mediates Pain Transmission. Proc. Natl. Acad. Sci. USA 2017, 114, 12309–12314. [Google Scholar] [CrossRef] [Green Version]
- Bigioni, M.; Benzo, A.; Irrissuto, C.; Maggi, C.A.; Goso, C. Role of NK-1 and NK-2 Tachykinin Receptor Antagonism on the Growth of Human Breast Carcinoma Cell Line MDA-MB-231. Anticancer Drugs 2005, 16, 1083–1089. [Google Scholar] [CrossRef]
- Nizam, E.; Erin, N. Differential Consequences of Neurokinin Receptor 1 and 2 Antagonists in Metastatic Breast Carcinoma Cells; Effects Independent of Substance P. Biomed. Pharmacother. 2018, 108, 263–270. [Google Scholar] [CrossRef]
- Gutierrez, S.; Boada, M.D. Neuropeptide-Induced Modulation of Carcinogenesis in a Metastatic Breast Cancer Cell Line (MDA-MB-231LUC+). Cancer Cell Int. 2018, 18, 216. [Google Scholar] [CrossRef]
- Fang, W.; Fu, C.; Chen, X.; Mou, X.; Liu, F.; Qian, J.; Zhao, P.; Zheng, Y.; Zheng, Y.; Deng, J.; et al. Neurokinin-2 Receptor Polymorphism Predicts Lymph Node Metastasis in Colorectal Cancer Patients. Oncol. Lett. 2015, 9, 2003–2006. [Google Scholar] [CrossRef]
- Xiang, H.; Toyoshima, Y.; Shen, W.; Wang, X.; Okada, N.; Kii, S.; Nagato, T. IFN-α/β-Mediated NK2R Expression Is Related to the Malignancy of Colon Cancer Cells. Cancer Sci. 2021, 113, 2513–2525. [Google Scholar] [CrossRef] [PubMed]
- Palma, C.; Nardelli, F.; Manzini, S.; Maggi, C.A. Substance P Activates Responses Correlated with Tumour Growth in Human Glioma Cell Lines Bearing Tachykinin NK1 Receptors. Br. J. Cancer 1999, 79, 236–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, R.; Christofides, K.; Jones, C.E. Endocytic Recycling Prevents Copper Accumulation in Astrocytoma Cells Stimulated with Copper-Bound Neurokinin B. Biochem. Biophys. Res. Commun. 2020, 523, 739–744. [Google Scholar] [CrossRef]
- McGregor, G.P.; Fehmann, C.; Hartel, R.; Voigt, K.H.; Göke, B.; Göke, R. Investigations of the Expression and Post-Translational Processing of the Preprotachykinin-I (PPT-I) Gene by Rat Pancreatic, Insulin-Producing Tumor Cell-Lines. Regul. Pept. 1993, 46, 444–446. [Google Scholar] [CrossRef] [PubMed]
- McGregor, G.P.; Hartel, R.; Fehmann, H.-C.; Lankat-Buttgereit, B.; Göke, B.; Göke, R.; Voigt, K. Characterisation of the Expression and Post-Translational Processing of the Preprotachykinin-I Gene and the Regulated Release of Tachykinins by the RINm5F Cell-Line. FEBS Lett. 1992, 312, 187–191. [Google Scholar] [CrossRef] [Green Version]
- Bishop, A.E.; Hamid, Q.A.; Adams, C.; Bretherton-Watt, D.; Jones, P.M.; Denny, P.; Stamp, G.W.H.; Hurt, R.L.; Grimelius, L.; Harmar, A.J.; et al. Expression of Tachykinins by Ileal and Lung Carcinoid Tumors Assessed by Combined in Situ Hybridization, Immunocytochemistry, and Radioimmunoassay. Cancer 1989, 63, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Norheim, I.; Wilander, E.; öberg, K.; Theodorsson-Norheim, E.; Lundqvist, M.-L.; Lindgren, P.; Bergh, J. Tachykinin Production by Carcinoid Tumours in Culture. Eur. J. Cancer Clin. Oncol. 1987, 23, 689–695. [Google Scholar] [CrossRef]
- Turner, G.B.; Johnston, B.T.; McCance, D.R.; McGinty, A.; Watson, R.G.P.; Patterson, C.C.; Ardill, J.E.S. Circulating Markers of Prognosis and Response to Treatment in Patients with Midgut Carcinoid Tumours. Gut 2006, 55, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Makridis, C.; Theodorsson, E.; Åkerström, G.; Öberg, K.; Knutson, L. Increased Intestinal Non-Substance P Tachykinin Concentrations in Malignant Midgut Carcinoid Disease: Tachykinins in Carcinoid Disease. J. Gastroenterol. Hepatol. 2002, 14, 500–507. [Google Scholar] [CrossRef]
- McGregor, G.P.; Gaedicke, G.; Voigt, K. Neurokinin-Immunoreactivity in Human Neuroblastomas: Evidence for Selective Expression of the Preprotachykinin (PPT) II Gene. FEBS Lett. 1990, 277, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Mukerji, I.; Ramkissoon, S.H.; Reddy, K.K.R.; Rameshwar, P. Autocrine Proliferation of Neuroblastoma Cells Is Partly Mediated through Neurokinin Receptors: Relevance to Bone Marrow Metastasis. J. Neurooncol. 2005, 71, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, S.; Mukai, H.; Kako, K.; Nakayama, K.; Munekata, E. Further Identification of Neurokinin Receptor Types and Mechanisms of Calcium Signaling Evoked by Neurokinins in the Murine Neuroblastoma C1300 Cell Line. J. Neurochem. 2002, 67, 1282–1292. [Google Scholar] [CrossRef] [PubMed]
- Advenier, C.; Naline, E.; Toty, L.; Bakdach, H.; Emonds-Alt, X.; Vilain, P.; Brelière, J.-C.; Fur, G.L. Effects on the Isolated Human Bronchus of SR 48968, a Potent and Selective Nonpeptide Antagonist of the Neurokinin A (NK2) Receptors. Am. Rev. Respir. Dis. 1992, 146, 1177–1181. [Google Scholar] [CrossRef]
- Obata, K.; Shimo, T.; Okui, T.; Matsumoto, K.; Takada, H.; Takabatake, K.; Kunisada, Y.; Ibaragi, S.; Yoshioka, N.; Kishimoto, K.; et al. Role of Neurokinin 3 Receptor Signaling in Oral Squamous Cell Carcinoma. Anticancer Res. 2017, 11, 6119–6123. [Google Scholar] [CrossRef]
- Kage, R.; Conlon, J.M. Neurokinin B in a Human Pheochromocytoma Measured with a Specific Radioimmunoassay. Peptides 1989, 10, 713–716. [Google Scholar] [CrossRef]
- Demitsu, T.; Murata, S.; Kakurai, M.; Kiyosawa, T.; Yaoita, H. Immunocytochemical Characterization of Malignant Schwannoma-Derived Cells in Culture. J. Dermatol. 1997, 24, 1–6. [Google Scholar] [CrossRef]
- Woltering, E.A.; Voros, B.A.; Thiagarajan, R.; Beyer, D.T.; Ramirez, R.A.; Wang, Y.-Z.; Mamikunian, G.; Boudreaux, J.P. Plasma Neurokinin A Levels Predict Survival in Well-Differentiated Neuroendocrine Tumors of the Small Bowel. Pancreas 2018, 47, 843–848. [Google Scholar] [CrossRef]
- Ardill, J.E.; Johnston, B.T.; McCance, D.R.; Eatock, M. Neurokinin A Monitoring of Response to Interferon Alpha in a Patient with an Advanced Small Bowel Neuroendocrine Tumour Uncontrolled by Somatostatin Analogue Therapy. Ann. Clin. Biochem. Int. J. Lab. Med. 2017, 54, 297–301. [Google Scholar] [CrossRef]
- Ardill, J.E.; McCance, D.R.; Stronge, W.V.; Johnston, B.T. Raised Circulating Neurokinin A Predicts Prognosis in Metastatic Small Bowel Neuroendocrine Tumours. Lowering Neurokinin A Indicates Improved Prognosis. Ann. Clin. Biochem. Int. J. Lab. Med. 2016, 53, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Ardill, J.E.S.; Armstrong, L.; Smye, M.; Doherty, R.; McCance, D.R.; Johnston, B.T. Neuroendocrine Tumours of the Small Bowel: Interpretation of Raised Circulating Chromogranin A, Urinary 5 Hydroxy Indole Acetic Acid and Circulating Neurokinin A. QJM 2016, 109, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Diebold, A.E.; Boudreaux, J.P.; Wang, Y.-Z.; Anthony, L.B.; Uhlhorn, A.P.; Ryan, P.; Mamikunian, P.; Mamikunian, G.; Woltering, E.A. Neurokinin A Levels Predict Survival in Patients with Stage IV Well Differentiated Small Bowel Neuroendocrine Neoplasms. Surgery 2012, 152, 1172–1176. [Google Scholar] [CrossRef] [PubMed]
- Balks, H.J.; Conlon, J.M.; Creutzfeldt, W.; Stöckmann, F. Effect of a Long-Acting Somatostatin Analogue (Octreotide) on Circulating Tachykinins and the Pentagastrin-Induced Carcinoid Flush. Eur. J. Clin. Pharmacol. 1989, 36, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Theodorsson-Norheim, E.; Andersson, M.; Jornvall, H.; Norheim, I.; Oberg, K.; Jacobsson, G. Isolation and Characterization of Neurokinin A, Neurokinin A(3–10) and Neurokinin A(4–10) from a Neutral Water Extract of a Metastatic Ileal Carcinoid Tumour. Eur. J. Biochem. 1987, 166, 693–698. [Google Scholar] [CrossRef] [PubMed]
- González-Santana, A.; Marrero-Hernández, S.; Dorta, I.; Hernández, M.; Pinto, F.M.; Báez, D.; Bello, A.R.; Candenas, L.; Almeida, T.A. Altered Expression of the Tachykinins Substance P/Neurokinin A/Hemokinin-1 and Their Preferred Neurokinin 1/Neurokinin 2 Receptors in Uterine Leiomyomata. Fertil. Steril. 2016, 106, 1521–1529. [Google Scholar] [CrossRef] [Green Version]
- Ogasawara, M.; Murata, J.; Ayukawa, K.; Saiki, I. Differential Effect of Intestinal Neuropeptides on Invasion and Migration of Colon Carcinoma Cells in Vitro. Cancer Lett. 1997, 119, 125–130. [Google Scholar] [CrossRef]
- Björk, J.; Nilsson, J.; Hultcrantz, R.; Johansson, C. Growth-Regulatory Effects of Sensory Neuropeptides, Epidermal Growth Factor, Insulin, and Somatostatin on the Non-Transformed Intestinal Epithelial Cell Line IEC-6 and the Colon Cancer Cell Line HT 29. Scand. J. Gastroenterol. 1993, 28, 879–884. [Google Scholar] [CrossRef]
- Heuillet, E.; Ménager, J.; Fardin, V.; Flamand, O.; Bock, M.; Garret, C.; Crespo, A.; Fallourd, A.M.; Doble, A. Characterization of a Human NK 1 Tachykinin Receptor in the Astrocytoma Cell Line U 373 MG. J. Neurochem. 1993, 60, 868–876. [Google Scholar] [CrossRef]
- Tung, W.-L.; Lee, C.-M. Effects of Tachykinins on [3H] Taurine Release from Human Astrocytoma Cells (U-373 MG). Brain Res. 1991, 549, 171–173. [Google Scholar] [CrossRef]
- Conlon, J.M.; Deacon, C.F.; Bailey, C.J.; Flatt, P.R. Effects of a Transplantable Insulinoma upon Regulatory Peptide Concentrations in the Gastrointestinal Tract of the Rat. Diabetologia 1986, 29, 334–338. [Google Scholar] [CrossRef] [Green Version]
- Hanna, F.W.F.; Ardill, J.E.S.; Johnston, C.F.; Cunningham, R.T.; Curry, W.J.; Russell, C.F.J.; Buchanan, K.D. Regulatory Peptides and Other Neuroendocrine Markers in Medullary Carcinoma of the Thyroid. J. Endocrinol. 1997, 152, 275–281. [Google Scholar] [CrossRef]
- Takahashi, K.; Satoh, F.; Hara, E.; Murakami, O.; Kumabe, T.; Tominaga, T.; Kayama, T.; Yoshimoto, T.; Shibahara, S. Production and Secretion of Adrenomedullin by Cultured Choroid Plexus Carcinoma Cells. J. Neurochem. 2002, 68, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Karpinich, N.O.; Kechele, D.O.; Espenschied, S.T.; Willcockson, H.H.; Fedoriw, Y.; Caron, K.M. Adrenomedullin Gene Dosage Correlates with Tumor and Lymph Node Lymphangiogenesis. FASEB J. 2013, 27, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, D.L.; Walker, C.S.; Poyner, D.R. Adrenomedullin and Calcitonin Gene-Related Peptide Receptors in Endocrine-Related Cancers: Opportunities and Challenges. Endocr. Relat. Cancer 2010, 18, C1–C14. [Google Scholar] [CrossRef] [PubMed]
- Zudaire, E.; Martínez, A.; Cuttitta, F. Adrenomedullin and Cancer. Regul. Pept. 2003, 112, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Nikitenko, L.L.; Fox, S.B.; Kehoe, S.; Rees, M.C.P.; Bicknell, R. Adrenomedullin and Tumour Angiogenesis. Br. J. Cancer 2006, 94, 1–7. [Google Scholar] [CrossRef]
- Cabarcas, S.M.; Mathews, L.A.; Farrar, W.L. The Cancer Stem Cell Niche-There Goes the Neighborhood? Int. J. Cancer 2011, 129, 2315–2327. [Google Scholar] [CrossRef]
- Talero, E.; Sánchez-Fidalgo, S.; Villegas, I.; de la Lastra, A.C.; Illanes, M.; Motilva, V. Role of Different Inflammatory and Tumor Biomarkers in the Development of Ulcerative Colitis-Associated Carcinogenesis. Inflamm. Bowel Dis. 2011, 17, 696–710. [Google Scholar] [CrossRef]
- Warrington, J.I.; Richards, G.O.; Wang, N. The Role of the Calcitonin Peptide Family in Prostate Cancer and Bone Metastasis. Curr. Mol. Biol. Rep. 2017, 3, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Valiya Veettil, M.; Krishna, G.; Roy, A.; Ghosh, A.; Dutta, D.; Kumar, B.; Chakraborty, S.; Anju, T.R.; Sharma-Walia, N.; Chandran, B. Kaposi’s Sarcoma-Associated Herpesvirus Infection Induces the Expression of Neuroendocrine Genes in Endothelial Cells. J. Virol. 2020, 94, e01692-19. [Google Scholar] [CrossRef]
- Gluexam, T.; Grandits, A.M.; Schlerka, A.; Nguyen, C.H.; Etzler, J.; Finkes, T.; Fuchs, M.; Scheid, C.; Heller, G.; Hackl, H.; et al. CGRP Signaling via CALCRL Increases Chemotherapy Resistance and Stem Cell Properties in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2019, 20, 5826. [Google Scholar] [CrossRef] [Green Version]
- Larrue, C.; Guiraud, N.; Mouchel, P.-L.; Dubois, M.; Farge, T.; Gotanègre, M.; Bosc, C.; Saland, E.; Nicolau-Travers, M.-L.; Sabatier, M.; et al. Adrenomedullin-CALCRL Axis Controls Relapse-Initiating Drug Tolerant Acute Myeloid Leukemia Cells. Nat. Commun. 2021, 12, 422. [Google Scholar] [CrossRef] [PubMed]
- Di Liddo, R.; Bridi, D.; Gottardi, M.; De Angeli, S.; Grandi, C.; Tasso, A.; Bertalot, T.; Martinelli, G.; Gherlinzoni, F.; Conconi, M.T. Adrenomedullin in the Growth Modulation and Differentiation of Acute Myeloid Leukemia Cells. Int. J. Oncol. 2016, 48, 1659–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonetti, G.; Angeli, D.; Petracci, E.; Fonzi, E.; Vedovato, S.; Sperotto, A.; Padella, A.; Ghetti, M.; Ferrari, A.; Robustelli, V.; et al. Adrenomedullin Expression Characterizes Leukemia Stem Cells and Associates With an Inflammatory Signature in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 684396. [Google Scholar] [CrossRef]
- Ismail, E.A.R.; El-Mogy, M.I.; Mohamed, D.S.; El-Farrash, R.A.H. Methylation Pattern of Calcitonin (CALCA) Gene in Pediatric Acute Leukemia. J. Pediatr. Hematol. Oncol. 2011, 33, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Morimoto, R.; Hirose, T.; Satoh, F.; Totsune, K. Adrenomedullin 2/Intermedin in the Hypothalamo–Pituitary–Adrenal Axis. J. Mol. Neurosci. 2011, 43, 182–192. [Google Scholar] [CrossRef]
- Mouri, T.; Takahashi, K.; Sone, M.; Murakami, O.; Ohneda, M.; Itoi, K.; Imai, Y.; Yoshinaga, K.; Sasano, N. Calcitonin Gene-Related Peptide-like Immunoreactivities in Pheochromocytomas. Peptides 1989, 10, 201–204. [Google Scholar] [CrossRef]
- Takahashi, K.; Satoh, F.; Sone, M.; Totsune, K.; Arihara, Z.; Noshiro, T.; Mouri, T.; Murakami, O. Expression of Adrenomedullin MRNA in Adrenocortical Tumors and Secretion of Adrenomedullin by Cultured Adrenocortical Carcinoma Cells. Peptides 1998, 19, 1719–1724. [Google Scholar] [CrossRef]
- Zeng, Z.-P.; Liu, D.-M.; Li, H.-Z.; Fan, X.-R.; Liu, G.-Q.; Yan, W.-G.; Tong, A.-L.; Zheng, X. Expression and Effect of Adrenomedullin in Pheochromocytoma. Ann. N. Y. Acad. Sci. 2006, 1073, 270–276. [Google Scholar] [CrossRef]
- Liu, D.-M.; Zeng, Z.-P.; Li, H.-Z.; Fan, X.-R.; Liu, G.-Q.; Yan, W.-G.; Tong, A.-L.; Zheng, X. Expression of Adrenomedullin and Its Receptor MRNA in the Tissues of Normal Adrenal Medulla and Pheochromocytoma. Zhonggou Yi Xue Ke Yuan Xue Bao Acta Acad. Med. Sin. 2005, 4, 452–456. [Google Scholar]
- Morimoto, R.; Satoh, F.; Murakami, O.; Hirose, T.; Totsune, K.; Imai, Y.; Arai, Y.; Suzuki, T.; Sasano, H.; Ito, S.; et al. Expression of Adrenomedullin 2/Intermedin in Human Adrenal Tumors and Attached Non-Neoplastic Adrenal Tissues. J. Endocrinol. 2008, 198, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Zhang, X.; Li, F.; Zhao, Y.; Guo, Y.; Yang, R. RNA Interference Targeting Adrenomedullin Induces Apoptosis and Reduces the Growth of Human Bladder Urothelial Cell Carcinoma. Med. Oncol. 2013, 30, 616. [Google Scholar] [CrossRef] [PubMed]
- Oehler, M.K.; Fischer, D.C.; Orlowska-Volk, M.; Herrle, F.; Kieback, D.G.; Rees, M.C.P.; Bicknell, R. Tissue and Plasma Expression of the Angiogenic Peptide Adrenomedullin in Breast Cancer. Br. J. Cancer 2003, 89, 1927–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siclari, V.A.; Mohammad, K.S.; Tompkins, D.R.; Davis, H.; McKenna, C.R.; Peng, X.; Wessner, L.L.; Niewolna, M.; Guise, T.A.; Suvannasankha, A.; et al. Tumor-Expressed Adrenomedullin Accelerates Breast Cancer Bone Metastasis. Breast Cancer Res. 2014, 16, 458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.-L.; Chen, S.-L.; Huang, Y.-H.; Yang, X.; Wang, C.-H.; He, J.-H.; Yun, J.-P.; Luo, R.-Z. Adrenomedullin Inhibits Tumor Metastasis and Is Associated with Good Prognosis in Triple-Negative Breast Cancer Patients. Am. J. Transl. Res. 2020, 12, 773–786. [Google Scholar] [PubMed]
- Benyahia, Z.; Dussault, N.; Cayol, M.; Sigaud, R.; Berenguer-Daizé, C.; Delfino, C.; Tounsi, A.; Garcia, S.; Martin, P.-M.; Mabrouk, K.; et al. Stromal Fibroblasts Present in Breast Carcinomas Promote Tumor Growth and Angiogenesis through Adrenomedullin Secretion. Oncotarget 2017, 8, 15744–15762. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.-M.; Zhong, J.-B.; Wang, H.-Y.; Yu, X.-F.; Li, Z.-Q. The Prognostic Value of Intermedin in Patients with Breast Cancer. Dis. Mark. 2015, 2015, 862158. [Google Scholar] [CrossRef]
- Kong, L.; Xiong, Y.; Wang, D.; Huang, L.; Li, M.; Feng, Z.; Zhou, Y.; Zhang, H.; Liu, F.; Xiao, F.; et al. Intermedin (Adrenomedullin 2) Promotes Breast Cancer Metastasis via Src/c-Myc-Mediated Ribosome Production and Protein Translation. Breast Cancer Res. Treat. 2022, 195, 91–103. [Google Scholar] [CrossRef]
- Papantoniou, V.; Tsiouris, S.; Sotiropoulou, M.; Valsamaki, P.; Koutsikos, J.; Ptohis, N.; Dimitrakakis, C.; Sotiropoulou, E.; Melissinou, M.; Nakopoulou, L.; et al. The Potential Role of Calcitonin Gene-Related Peptide (CGRP) in Breast Carcinogenesis and Its Correlation With 99mTc-(V)DMSA Scintimammography. Am. J. Clin. Oncol. 2007, 30, 420–427. [Google Scholar] [CrossRef]
- Zhao, H.; Ning, L.; Wang, Z.; Li, H.; Qiao, D.; Yao, Y.; Qin, H. Calcitonin Gene-Related Peptide Inhibits Osteolytic Factors Induced by Osteoblast In Co-Culture System with Breast Cancer. Cell Biochem. Biophys. 2014, 70, 1097–1104. [Google Scholar] [CrossRef]
- Moriyama, T.; Otani, T.; Maruo, T. Expression of Adrenomedullin by Human Placental Cytotrophoblasts and Choriocarcinoma JAr Cells. J. Clin. Endocrinol. Metab. 2001, 86, 3958–3961. [Google Scholar] [CrossRef]
- Uemura, M.; Yamamoto, H.; Takemasa, I.; Mimori, K.; Mizushima, T.; Ikeda, M.; Sekimoto, M.; Doki, Y.; Mori, M. Hypoxia-Inducible Adrenomedullin in Colorectal Cancer. Anticancer Res. 2011, 31, 507–514. [Google Scholar] [PubMed]
- Nouguerède, E.; Berenguer, C.; Garcia, S.; Bennani, B.; Delfino, C.; Nanni, I.; Dahan, L.; Gasmi, M.; Seitz, J.; Martin, P.; et al. Expression of Adrenomedullin in Human Colorectal Tumors and Its Role in Cell Growth and Invasion in Vitro and in Xenograft Growth in Vivo. Cancer Med. 2013, 2, 196–207. [Google Scholar] [CrossRef]
- Hikosaka, T.; Tsuruda, T.; Nagata, S.; Kuwasako, K.; Tsuchiya, K.; Hoshiko, S.; Inatsu, H.; Chijiiwa, K.; Kitamura, K. Adrenomedullin Production Is Increased in Colorectal Adenocarcinomas; Its Relation to Matrix Metalloproteinase-9. Peptides 2011, 32, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Park, W.-D.; Lee, S.; Park, J.-H. Adrenomedullin Is Involved in the Progression of Colonic Adenocarcinoma. Mol. Med. Rep. 2012, 6, 1030–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Gala, M.; Yamamoto, M.; Pino, M.S.; Kikuchi, H.; Shue, D.S.; Shirasawa, S.; Austin, T.R.; Lynch, M.P.; Rueda, B.R.; et al. Adrenomedullin Is a Therapeutic Target in Colorectal Cancer: K-Ras Regulates Adrenomedullin in Hypoxia. Int. J. Cancer 2014, 134, 2041–2050. [Google Scholar] [CrossRef] [Green Version]
- Ochoa-Callejero, L.; García-Sanmartín, J.; Martínez-Herrero, S.; Rubio-Mediavilla, S.; Narro-Íñiguez, J.; Martínez, A. Small Molecules Related to Adrenomedullin Reduce Tumor Burden in a Mouse Model of Colitis-Associated Colon Cancer. Sci. Rep. 2017, 7, 17488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, D.M.; Leah, J.D.; Zimmermann, M. The Localization and Release of Substance P and Calcitonin Gene-Related Peptide at Nerve Fibre Endings in Rat Cutaneous Nerve Neuroma. Brain Res. 1989, 503, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.J.; Chitcholtan, K.; Dann, J.M.; Guilford, P.; Harris, G.; Lewis, L.K.; Nagase, J.; Welkamp, A.A.W.; Zwerus, R.; Sykes, P.H. Adrenomedullin Interacts with VEGF in Endometrial Cancer and Has Varied Modulation in Tumours of Different Grades. Gynecol. Oncol. 2012, 125, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Oehler, M.K.; Norbury, C.; Hague, S.; Rees, M.C.; Bicknell, R. Adrenomedullin Inhibits Hypoxic Cell Death by Upregulation of Bcl-2 in Endometrial Cancer Cells: A Possible Promotion Mechanism for Tumour Growth. Oncogene 2001, 20, 2937–2945. [Google Scholar] [CrossRef] [Green Version]
- Nunobiki, O.; Nakamura, M.; Taniguchi, E.; Utsunomiya, H.; Mori, I.; Tsubota, Y.; Mabuchi, Y.; Kakudo, K. Adrenomedullin, Bcl-2 and Microvessel Density in Normal, Hyperplastic and Neoplastic Endometrium. Pathol. Int. 2009, 59, 530–536. [Google Scholar] [CrossRef]
- Dallmayer, M.; Li, J.; Ohmura, S.; Alba Rubio, R.; Baldauf, M.C.; Hölting, T.L.B.; Musa, J.; Knott, M.M.L.; Stein, S.; Cidre-Aranaz, F.; et al. Targeting the CALCB/RAMP1 Axis Inhibits Growth of Ewing Sarcoma. Cell Death Dis. 2019, 10, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Y.; Peng, L.; Wang, Q.; Chen, N.; Teng, Y.; Wang, T.; Mao, F.; Zhang, J.; Cheng, P.; Liu, Y.; et al. Degranulation of Mast Cells Induced by Gastric Cancer-Derived Adrenomedullin Prompts Gastric Cancer Progression. Cell Death Dis. 2018, 9, 1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, A.P.; Serrano, J.; Amorim, M.A.; Pozo-Rodrigalvarez, A.; Martinez-Murillo, R. Adrenomedullin and Nitric Oxide: Implications for the Etiology and Treatment of Primary Brain Tumors. CNS Neurol. Disord. Drug Targets 2011, 10, 820–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metellus, P.; Voutsinos-Porche, B.; Nanni-Metellus, I.; Colin, C.; Fina, F.; Berenguer, C.; Dussault, N.; Boudouresque, F.; Loundou, A.; Intagliata, D.; et al. Adrenomedullin Expression and Regulation in Human Glioblastoma, Cultured Human Glioblastoma Cell Lines and Pilocytic Astrocytoma. Eur. J. Cancer 2011, 47, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Ouafik, L.; Sauze, S.; Boudouresque, F.; Chinot, O.; Delfino, C.; Fina, F.; Vuaroqueaux, V.; Dussert, C.; Palmari, J.; Dufour, H.; et al. Neutralization of Adrenomedullin Inhibits the Growth of Human Glioblastoma Cell Lines in Vitro and Suppresses Tumor Xenograft Growth in Vivo. Am. J. Pathol. 2002, 160, 1279–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudouresque, F.; Berthois, Y.; Martin, P.-M.; Figarella-Branger, D.; Chinot, O.; Ouafik, L. Role of Adrenomedullin in Glioblastomas Growth. Bull Cancer 2005, 4, 317–326. [Google Scholar]
- Ouafik, L.; Berenguer-Daize, C.; Berthois, Y. Adrenomedullin Promotes Cell Cycle Transit and Up-Regulates Cyclin D1 Protein Level in Human Glioblastoma Cells through the Activation of c-Jun/JNK/AP-1 Signal Transduction Pathway. Cell. Signal. 2009, 21, 597–608. [Google Scholar] [CrossRef]
- Lim, S.Y.; Ahn, S.-H.; Park, H.; Lee, J.; Choi, K.; Choi, C.; Choi, J.H.; Park, E.-M.; Choi, Y.-H. Transcriptional Regulation of Adrenomedullin by Oncostatin M in Human Astroglioma Cells: Implications for Tumor Invasion and Migration. Sci. Rep. 2014, 4, 6444. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Cheng, M.; Hu, J.; Liu, L.; Liu, P.; Chen, L.; Cao, D.; Tang, J. MiR-1297 Sensitizes Glioma Cells to Temozolomide (TMZ) Treatment through Targeting Adrenomedullin (ADM). J. Transl. Med. 2022, 20, 443. [Google Scholar] [CrossRef]
- Huang, L.; Wang, D.; Feng, Z.; Zhao, H.; Xiao, F.; Wei, Y.; Zhang, H.; Li, H.; Kong, L.; Li, M.; et al. Inhibition of Intermedin (Adrenomedullin 2) Suppresses the Growth of Glioblastoma and Increases the Antitumor Activity of Temozolomide. Mol. Cancer Ther. 2021, 20, 284–295. [Google Scholar] [CrossRef]
- Gitter, B.D.; Cox, L.M.; Carlson, C.D.; May, P.C. Human Amylin Stimulates Inflammatory Cytokine Secretion from Human Glioma Cells. Neuroimmunomodulation 2000, 7, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, M.; Liu, Z.; Wang, X.; Ji, T. The Neuropeptide Calcitonin Gene-Related Peptide Links Perineural Invasion with Lymph Node Metastasis in Oral Squamous Cell Carcinoma. BMC Cancer 2021, 21, 1254. [Google Scholar] [CrossRef]
- Zhang, Y.; Lin, C.; Wang, X.; Ji, T. Calcitonin Gene-related Peptide: A Promising Bridge between Cancer Development and Cancer-associated Pain in Oral Squamous Cell Carcinoma (Review). Oncol. Lett. 2020, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Park, S.C.; Yoon, J.-H.; Lee, J.-H.; Yu, S.J.; Myung, S.J.; Kim, W.; Gwak, G.-Y.; Lee, S.-H.; Lee, S.-M.; Jang, J.J.; et al. Hypoxia-Inducible Adrenomedullin Accelerates Hepatocellular Carcinoma Cell Growth. Cancer Lett. 2008, 271, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Nakata, T.; Seki, N.; Miwa, S.; Kobayashi, A.; Soeda, J.; Nimura, Y.; Kawasaki, S.; Miyagawa, S. Identification of Genes Associated with Multiple Nodules in Hepatocellular Carcinoma Using CDNA Microarray: Multicentric Occurrence or Intrahepatic Metastasis? Hepatogastroenterology 2008, 55, 865–872. [Google Scholar]
- Zhou, C.; Zheng, Y.; Li, L.; Zhai, W.; Li, R.; Liang, Z.; Zhao, L. Adrenomedullin Promotes Intrahepatic Cholangiocellular Carcinoma Metastasis and Invasion by Inducing Epithelial-Mesenchymal Transition. Oncol. Rep. 2015, 34, 610–616. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Yang, R.; Zhang, X.; Liu, A.; Zhao, Y.; Guo, Y. Silencing of Hypoxia-inducible Adrenomedullin Using RNA Interference Attenuates Hepatocellular Carcinoma Cell Growth in Vivo. Mol. Med. Rep. 2014, 10, 1295–1302. [Google Scholar] [CrossRef]
- Qu, Z.; Jiang, Y.; Xu, M.; Lu, M.Z.; Zhou, B.; Ding, Y. Correlation of Adrenomedullin with the Erythropoietin Receptor and Microvessel Density in Hepatocellular Carcinoma. Arch. Med. Sci. 2015, 5, 978–981. [Google Scholar] [CrossRef]
- Guo, X.; Schmitz, J.C.; Kenney, B.C.; Uchio, E.M.; Kulkarni, S.; Cha, C.H. Intermedin Is Overexpressed in Hepatocellular Carcinoma and Regulates Cell Proliferation and Survival. Cancer Sci. 2012, 103, 1474–1480. [Google Scholar] [CrossRef]
- Shang, H.; Hao, Z.; Fu, X.; Hua, X.; Ma, Z.; Ai, F.; Feng, Z.; Wang, K.; Li, W.; Li, B. Intermedin Promotes Hepatocellular Carcinoma Cell Proliferation through the Classical Wnt Signaling Pathway. Oncol. Lett. 2018, 15, 5966–5970. [Google Scholar] [CrossRef]
- Xiao, F.; Li, H.; Feng, Z.; Huang, L.; Kong, L.; Li, M.; Wang, D.; Liu, F.; Zhu, Z.; Wei, Y.; et al. Intermedin Facilitates Hepatocellular Carcinoma Cell Survival and Invasion via ERK1/2-EGR1/DDIT3 Signaling Cascade. Sci. Rep. 2021, 11, 488. [Google Scholar] [CrossRef]
- Sheriff, S.; Fischer, J.E.; Balasubramaniam, A. Characterization of Amylin Binding Sites in a Human Hepatoblastoma Cell Line. Peptides 1992, 13, 1193–1199. [Google Scholar] [CrossRef]
- Buyukberber, S.; Sari, I.; Camci, C.; Buyukberber, N.M.; Sevinc, A.; Turk, H.M. Adrenomedullin Expression Does Not Correlate with Survival in Lung Cancer. Med. Oncol. 2007, 24, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Portal-Nuñez, S.; Shankavaram, U.T.; Rao, M.; Datrice, N.; Atay, S.; Aparicio, M.; Camphausen, K.A.; Fernández-Salguero, P.M.; Chang, H.; Lin, P.; et al. Aryl Hydrocarbon Receptor-Induced Adrenomedullin Mediates Cigarette Smoke Carcinogenicity in Humans and Mice. Cancer Res. 2012, 72, 5790–5800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edbrooke, M.R.; Parker, D.; McVey, J.H.; Riley, J.H.; Sorenson, G.D.; Pettengill, O.S.; Craig, R.K. Expression of the Human Calcitonin/CGRP Gene in Lung and Thyroid Carcinoma. EMBO J. 1985, 4, 715–724. [Google Scholar] [CrossRef]
- Schifter, S.; Johannsen, L.; Bunker, C.; Brickell, P.; Bork, E.; Lindeberg, H.; Faber, J. Calcitonin Gene-Related Peptide in Small Cell Lung Carcinomas. Clin. Endocrinol. 1993, 39, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Huang, Y.; Bong, R.; Ding, Y.; Song, N.; Wang, X.; Song, X.; Luo, Y. Tumor-Associated Macrophages Promote Angiogenesis and Melanoma Growth via Adrenomedullin in a Paracrine and Autocrine Manner. Clin. Cancer Res. 2011, 17, 7230–7239. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-L.; Huang, S.-X.; Lin, S.; Chai, L. Plasma Adrenomedullin Levels and Nasopharyngeal Carcinoma Prognosis. Clin. Chim. Acta 2015, 440, 172–176. [Google Scholar] [CrossRef]
- Dötsch, J.; Harmjanz, A.; Christiansen, H.; Hänze, J.; Lampert, F.; Rascher, W. Gene Expression of Neuronal Nitric Oxide Synthase and Adrenomedullin in Human Neuroblastoma Using Real-Time PCR. Int. J. Cancer 2000, 88, 172–175. [Google Scholar] [CrossRef]
- Zimmermann, U.; Fischer, J.A.; Muff, R. Adrenomedullin and Calcitonin Gene-Related Peptide Interact with the Same Receptor in Cultured Human Neuroblastoma SK-N-MC Cells. Peptides 1995, 16, 421–424. [Google Scholar] [CrossRef]
- Pavel, M.E.; Hoppe, S.; Papadopoulos, T.; Linder, V.; Mohr, B.; Hahn, E.G.; Lohmann, T.; Schuppan, D. Adrenomedullin Is a Novel Marker of Tumor Progression in Neuroendocrine Carcinomas. Horm. Metab. Res. 2006, 38, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.d.L.; Peterle, G.T.; dos Santos, M.; Trivilin, L.O.; Mendes, S.O.; de Oliveira, M.M.; dos Santos, J.G.; Stur, E.; Agostini, L.P.; Couto, C.V.M.d.S.; et al. JMJD1A, H3K9me1, H3K9me2 and ADM Expression as Prognostic Markers in Oral and Oropharyngeal Squamous Cell Carcinoma. PLoS ONE 2018, 13, e0194884. [Google Scholar] [CrossRef]
- Dai, X.; Ma, W.; He, X.J.; Jha, R.K. Elevated Expression of Adrenomedullin Is Correlated with Prognosis and Disease Severity in Osteosarcoma. Med. Oncol. 2013, 30, 347. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-Y.; Hao, C.-P.; Ling, M.; Guo, C.-H.; Ma, W. Hypoxia-Induced Apoptosis Is Blocked by Adrenomedullin via Upregulation of Bcl-2 in Human Osteosarcoma Cells. Oncol. Rep. 2015, 34, 787–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacalone, P.-L.; Vuaroqueaux, V.; Daurés, J.-P.; Houafic, L.; Martin, P.-M.; Laffargue, F.; Maudelonde, T. Expression of Adrenomedullin in Human Ovaries, Ovarian Cysts and Cancers. Eur. J. Obstet. Gynecol. Reprod. Biol. 2003, 110, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Deng, B.; Zhang, S.; Miao, Y.; Han, Z.; Zhang, X.; Wen, F.; Zhang, Y. Adrenomedullin Expression in Epithelial Ovarian Cancers and Promotes HO8910 Cell Migration Associated with Upregulating Integrin A5β1 and Phosphorylating FAK and Paxillin. J. Exp. Clin. Cancer Res. 2012, 31, 19. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Hong, L.; Liao, M.; Guo, G. Expression and Clinical Significance of Focal Adhesion Kinase and Adrenomedullin in Epithelial Ovarian Cancer. Oncol. Lett. 2015, 10, 1003–1007. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y. Effect of Inhibition of the Adrenomedullin Gene on the Growth and Chemosensitivity of Ovarian Cancer Cells. Oncol. Rep. 2012, 27, 1461–1466. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yang, X.; Ma, J.; Pang, X.; Dong, M. Adrenomedullin Promotes Angiogenesis in Epithelial Ovarian Cancer through Upregulating Hypoxiainducible Factor-1α and Vascular Endothelial Growth Factor. Sci. Rep. 2017, 16, 40524. [Google Scholar] [CrossRef]
- Hata, K. Expression of the Adrenomedullin Gene in Epithelial Ovarian Cancer. Mol. Hum. Reprod. 2000, 6, 867–872. [Google Scholar] [CrossRef] [Green Version]
- Keleg, S.; Kayed, H.; Jiang, X.; Penzel, R.; Giese, T.; Büchler, M.W.; Friess, H.; Kleeff, J. Adrenomedullin Is Induced by Hypoxia and Enhances Pancreatic Cancer Cell Invasion. Int. J. Cancer 2007, 121, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Chen, J.; Wang, J.; Okada, F.; Sugiyama, T.; Kobayashi, T.; Shindo, M.; Higashino, F.; Katoh, H.; Asaka, M.; et al. Adrenomedullin Antagonist Suppresses in Vivo Growth of Human Pancreatic Cancer Cells in SCID Mice by Suppressing Angiogenesis. Oncogene 2003, 22, 1238–1242. [Google Scholar] [CrossRef] [Green Version]
- Aggarwai, G.; Ramachandran, V.; Javeed, N.; Arumugam, T.; Dutta, S. Adrenomedullin Is Up-Regulated in Patients With Pancreatic Cancer and Causes Insulin Resistance in β Cells and Mice. Gastroenterology 2012, 6, 1510–1517. [Google Scholar] [CrossRef] [Green Version]
- Letizia, C.; Tamburrano, G.; Alo, P.; Paoloni, A.; Caliumi, C.; Marinoni, E.; di Iorio, R.; d’Erasmo, E. Adrenomedullin, a New Peptide, in Patients with Insulinoma. Eur. J. Endocrinol. 2001, 144, 517–520. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, V.; Arumugam, T.; Hwang, R.F.; Greenson, J.K.; Simeone, D.M.; Logsdon, C.D. Adrenomedullin Is Expressed in Pancreatic Cancer and Stimulates Cell Proliferation and Invasion in an Autocrine Manner via the Adrenomedullin Receptor, ADMR. Cancer Res. 2007, 67, 2666–2675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, K.; Tanaka, M.; Kamiyoshi, A.; Sakurai, T.; Ichikawa-Shindo, Y.; Kawate, H.; Cui, N.; Wei, Y.; Tanaka, M.; Kakihara, S.; et al. Deficiency of the Adrenomedullin-RAMP3 System Suppresses Metastasis through the Modification of Cancer-Associated Fibroblasts. Oncogene 2020, 39, 1914–1930. [Google Scholar] [CrossRef]
- Xu, M.; Qi, F.; Zhang, S.; Ma, X.; Wang, S.; Wang, C.; Fu, Y.; Luo, Y. Adrenomedullin Promotes the Growth of Pancreatic Ductal Adenocarcinoma through Recruitment of Myelomonocytic Cells. Oncotarget 2016, 7, 55043–55056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollander, L.L.; Guo, X.; Salem, R.R.; Cha, C.H. The Novel Tumor Angiogenic Factor, Adrenomedullin-2, Predicts Survival in Pancreatic Adenocarcinoma. J. Surg. Res. 2015, 197, 219–224. [Google Scholar] [CrossRef]
- Mäkimattila, S.; Hietaniemi, K.; Kiviluoto, T.; Timonen, T.; Järvien, H. In Vivo Glucose-Stimulated Amylin Secretion Is Increased in Nondiabetic Patients with Pancreatic Cancer. Metabolism 2001, 50, 1036–1042. [Google Scholar] [CrossRef]
- Gao, F.; Liu, G.; Wang, J.; Huang, S.; Ding, F.; Lian, W.; Lv, X.; Guo, Y.; Fan, X.; Zhang, S.; et al. Methylation of CALCA and CALCB in Pancreatic Ductal Adenocarcinoma. Oxid. Med. Cell. Longev. 2021, 2021, 2088345. [Google Scholar] [CrossRef]
- Ramachandran, V.; Arumugam, T.; Langley, R.; Hwang, R.F.; Vivas-Mejia, P.; Sood, A.K.; Lopez-Berestein, G.; Logsdon, C.D. The ADMR Receptor Mediates the Effects of Adrenomedullin on Pancreatic Cancer Cells and on Cells of the Tumor Microenvironment. PLoS ONE 2009, 4, e7502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, T. Immunocytochemical Staining for Islet Amyloid Polypeptide in Pancreatic Endocrine Tumors. Islets 2011, 3, 344–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardero, M.; Kovacs, K.; Scheithauer, B.; Rotondo, F.; Salehi, F.; Lloyd, R. Adrenomedullin Expression in Pituitary Adenomas and Nontumoral Adenohypophyses. Histol. Histopathol. 2007, 23, 11–17. [Google Scholar] [CrossRef]
- Rocchi, P.; Boudouresque, F.; Zamora, A.J.; Muracciole, X.; Lechevallier, E.; Martin, P.M.; Ouafik, L. Expression of Adrenomedullin and Peptide Amidation Activity in Human Prostate Cancer and in Human Prostate Cancer Cell Lines. Cancer Res. 2001, 3, 1196–1206. [Google Scholar]
- Berenguer-Daizé, C.; Boudouresque, F.; Bastide, C.; Tounsi, A.; Benyahia, Z.; Acunzo, J.; Dussault, N.; Delfino, C.; Baeza, N.; Daniel, L.; et al. Adrenomedullin Blockade Suppresses Growth of Human Hormone–Independent Prostate Tumor Xenograft in Mice. Clin. Cancer Res. 2013, 19, 6138–6150. [Google Scholar] [CrossRef] [Green Version]
- Calvo, A.; Abasolo, I.; Jiménez, N.; Wang, Z.; Montuenga, L. Adrenomedullin and Proadrenomedullin N-Terminal 20 Peptide in the Normal Prostate and in Prostate Carcinoma: AM and PAMP in the Prostate. Microsc. Res. Tech. 2002, 57, 98–104. [Google Scholar] [CrossRef]
- Abasolo, I.; Wang, Z.; Montuenga, L.M.; Calvo, A. Adrenomedullin Inhibits Prostate Cancer Cell Proliferation through a CAMP-Independent Autocrine Mechanism. Biochem. Biophys. Res. Commun. 2004, 322, 878–886. [Google Scholar] [CrossRef]
- Abasolo, I.; Montuenga, L.M.; Calvo, A. Adrenomedullin Prevents Apoptosis in Prostate Cancer Cells. Regul. Pept. 2006, 133, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Oulidi, A.; Bokhobza, A.; Gkika, D.; Vanden Abeele, F.; Lehen’kyi, V.; Ouafik, L.; Mauroy, B.; Prevarskaya, N. TRPV2 Mediates Adrenomedullin Stimulation of Prostate and Urothelial Cancer Cell Adhesion, Migration and Invasion. PLoS ONE 2013, 8, e64885. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-L.; Wang, Y.-Y.; He, H.-D.; Xie, X.; Yu, Z.-J.; Pan, Y.-M. Association of Plasma Intermedin Levels with Progression and Metastasis in Men after Radical Prostatectomy for Localized Prostatic Cancer. Cancer Biomark. 2015, 15, 799–805. [Google Scholar] [CrossRef]
- Suzuki, K.; Kobayashi, Y.; Morita, T. Serum Calcitonin Gene-Related Peptide Levels in Untreated Prostate Cancer Patients: CGRP Level in Untreated Prostate Cancer. Int. J. Urol. 2006, 13, 781–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Kobayashi, Y.; Morita, T. Significance of Serum Calcitonin Gene-Related Peptide Levels in Prostate Cancer Patients Receiving Hormonal Therapy. Urol. Int. 2009, 82, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Sheng, D.; Shao, Y.; Zhang, Q.; Peng, Y. Neuronal Calcitonin Gene-Related Peptide Promotes Prostate Tumor Growth in the Bone Microenvironment. Peptides 2021, 135, 170423. [Google Scholar] [CrossRef] [PubMed]
- Michelsen, J.; Thiesson, H.; Walter, S.; Ottosen, P.D.; Skøtt, O.; Jensen, B.L. Tissue Expression and Plasma Levels of Adrenomedullin in Renal Cancer Patients. Clin. Sci. 2006, 111, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Nikitenko, L.L.; Leek, R.; Henderson, S.; Pillay, N.; Turley, H.; Generali, D.; Gunningham, S.; Morrin, H.R.; Pellagatti, A.; Rees, M.C.P.; et al. The G-Protein–Coupled Receptor CLR Is Upregulated in an Autocrine Loop with Adrenomedullin in Clear Cell Renal Cell Carcinoma and Associated with Poor Prognosis. Clin. Cancer Res. 2013, 19, 5740–5748. [Google Scholar] [CrossRef] [Green Version]
- Fujita, Y.; Mimata, H.; Nasu, N.; Nomura, T.; Nomura, Y.; Nakagawa, M. Involvement of Adrenomedullin Induced by Hypoxia in Angiogenesis in Human Renal Cell Carcinoma. Int. J. Urol. 2002, 9, 285–295. [Google Scholar] [CrossRef]
- Venkatanarayan, A.; Raulji, P.; Norton, W.; Chakravarti, D.; Coarfa, C.; Su, X.; Sandur, S.K.; Ramirez, M.S.; Lee, J.; Kingsley, C.V.; et al. IAPP-Driven Metabolic Reprogramming Induces Regression of P53-Deficient Tumours in Vivo. Nature 2015, 517, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.T.; Lim, M.A.; Lee, S.E.; Kim, H.J.; Koh, H.Y.; Lee, J.H.; Jun, S.M.; Kim, J.M.; Kim, K.H.; Shin, H.S.; et al. Adrenomedullin2 Stimulates Progression of Thyroid Cancer in Mice and Humans under Nutrient Excess Conditions. J. Pathol. 2022, 258, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Alevizaki, M.; Grigorakis, S.; Tseleni-Balafouta, S.; Alevizaki, C.; Philippou, G.; Anastasiou, E.; Souvatzoglou, A. High Plasma Amylin/Islet Amyloid Polypeptide Levels in Patients with Residual Medullary Thyroid Carcinoma after Total Thyroidectomy. Eur. J. Endocrinol. 2001, 145, 585–589. [Google Scholar] [CrossRef] [Green Version]
- Haller-Brem, S.; Muff, R.; Petermann, J.B.; Born, W.; Roos, B.A.; Fischer, J.A. Role of Cytosolic Free Calcium Concentration in the Secretion of Calcitonin Gene-Related Peptide and Calcitonin from Medullary Thyroid Carcinoma Cells. Endocrinology 1987, 121, 1272–1277. [Google Scholar] [CrossRef]
- Pacini, F.; Fugazzola, L.; Basolo, F.; Elisei, R.; Pinchera, A. Expression of Calcitonin Gene-Related Peptide in Medullary Thyroid Cancer. J. Endocrinol. Investig. 1992, 15, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Takeuchi, S.; Otani, T.; Maruo, T. Implications of Adrenomedullin Expression in the Invasion of Squamous Cell Carcinoma of the Uterine Cervix. Int. J. Clin. Oncol. 2001, 6, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Takeuchi, S.; Ohara, N.; Maruo, T. Paradoxically Abundant Expression of Bcl-2 and Adrenomedullin in Invasive Cervical Squamous Carcinoma. Int. J. Clin. Oncol. 2003, 8, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yoshinoya, A.; Murakami, O.; Totsune, K.; Shibahara, S. Production and Secretion of Two Vasoactive Peptides, Adrenomedullin and Endothelin-1, by Cultured Human Adrenocortical Carcinoma Cells. Peptides 2000, 21, 251–256. [Google Scholar] [CrossRef]
- Thouënnon, E.; Pierre, A.; Tanguy, Y.; Guillemot, J.; Manecka, D.-L.; Guérin, M.; Ouafik, L.; Muresan, M.; Klein, M.; Bertherat, J.; et al. Expression of Trophic Amidated Peptides and Their Receptors in Benign and Malignant Pheochromocytomas: High Expression of Adrenomedullin RDC1 Receptor and Implication in Tumoral Cell Survival. Endocr. Relat. Cancer 2010, 17, 637–651. [Google Scholar] [CrossRef]
- Letizia, C.; De Toma, G.; Caliumi, C.; Cerci, S.; Massa, R.; Di Iorio, R.; Alo, P.; Marinoni, E.; Diacinti, D.; D’Erasmo, E. Plasma Adrenomedullin Concentrations in Patients with Adrenal Pheochromocytoma. Horm. Metab. Res. 2001, 33, 290–294. [Google Scholar] [CrossRef]
- Cotesta, D.; Caliumi, C.; Alò, P.; Petramala, L.; Reale, M.G.; Masciangelo, R.; Signore, A.; Cianci, R.; D’Erasmo, E.; Letizia, C. High Plasma Levels of Human Chromogranin a and Adrenomedullin in Patients with Pheochromocytoma. Tumori J. 2005, 91, 53–58. [Google Scholar] [CrossRef]
- Kobayashi, H.; Itoh, S.; Yanagita, T.; Yokoo, H.; Sugano, T.; Wada, A. Expression of Adrenomedullin and Proadrenomedullin N-Terminal 20 Peptide in PC12 Cells after Exposure to Nerve Growth Factor. Neuroscience 2004, 125, 973–980. [Google Scholar] [CrossRef]
- Brekhman, V.; Lugassie, J.; Zaffryar-Eilot, S.; Sabo, E.; Kessler, O.; Smith, V.; Golding, H.; Neufeld, G. Receptor Activity Modifying Protein-3 Mediates the Protumorigenic Activity of Lysyl Oxidase-like Protein-2. FASEB J. 2011, 25, 55–65. [Google Scholar] [CrossRef]
- Paré, M.; Darini, C.Y.; Yao, X.; Chignon-Sicard, B.; Rekima, S.; Lachambre, S.; Virolle, V.; Aguilar-Mahecha, A.; Basik, M.; Dani, C.; et al. Breast Cancer Mammospheres Secrete Adrenomedullin to Induce Lipolysis and Browning of Adjacent Adipocytes. BMC Cancer 2020, 20, 784. [Google Scholar] [CrossRef]
- Nakayama, M.; Takahashi, K.; Murakami, O.; Shirato, K.; Shibahara, S. Induction of Adrenomedullin by Hypoxia and Cobalt Chloride in Human Colorectal Carcinoma Cells. Biochem. Biophys. Res. Commun. 1998, 243, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Eissele, R.; Neuhaus, C.; Trautmann, M.E.; Funk, A.; Arnold, R.; Höfler, H. Immunoreactivity and Expression of Amylin in Gastroenteropancreatic Endocrine Tumors. Am. J. Phatologie 1993, 1, 283–291. [Google Scholar]
- Godlewski, J.; Kaleczyc, J. Somatostatin, Substance P and Calcitonin Gene-Related Peptide-Positive Intramural Nerve Structures of the Human Large Intestine Affected by Carcinoma. Folia Histochem. Cytobiol. 2010, 48, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Bozkurt, K.K.; Yalçın, Y.; Erdemoğlu, E.; Tatar, B.; Erdemoğlu, E.; Çerçi, S.S.; Çiriş, İ.M.; Başpınar, Ş.; Uğuz, A.; Kapucuoğlu, N. The Role of Immunohistochemical Adrenomedullin and Bcl-2 Expression in Development of Type-1 Endometrial Adenocarcinoma. Pathol.—Res. Pract. 2016, 212, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Cerci, S.S.; Yalçın, Y.; Bozkurt, K.K.; Erdemoğlu, E.; Tatar, B. Hypoxia-Inducible Factor-1α, Adrenomedullin and Bcl-2 Although Expected Are Not Related to Increased Uptake of Fluorine-18-Fluorodeoxyglucose in Endometrial Cancer. Hell. J. Nuc. Med. 2015, 18, 228–232. [Google Scholar]
- Höppener, J.W.M.; Steenbergh, P.H.; Slebos, R.J.C.; Visser, A.; Lips, C.J.M.; Jansz, H.S.; Bechet, J.M.; Lenoir, G.M.; Born, W.; Haller-Brem, S.; et al. Expression of the Second Calcitonin/Calcitonin Gene-Related Peptide Gene in Ewing Sarcoma Cell Lines. J. Clin. Endocrinol. Metab. 1987, 64, 809–817. [Google Scholar] [CrossRef]
- Qiao, F.; Fang, J.; Xu, J.; Zhao, W.; Ni, Y.; Akuo, B.A.; Zhang, W.; Liu, Y.; Ding, F.; Li, G.; et al. The Role of Adrenomedullin in the Pathogenesis of Gastric Cancer. Oncotarget 2017, 8, 88464–88474. [Google Scholar] [CrossRef] [Green Version]
- Benes, L. The Immunohistochemical Expression of Calcitonin Receptor-like Receptor (CRLR) in Human Gliomas. J. Clin. Pathol. 2004, 57, 172–176. [Google Scholar] [CrossRef] [Green Version]
- Kitamuro, T.; Takahashi, K.; Nakayama, M.; Murakami, O.; Hida, W.; Shirato, K.; Shibahara, S. Induction of Adrenomedullin During Hypoxia in Cultured Human Glioblastoma Cells. J. Neurochem. 2002, 75, 1826–1833. [Google Scholar] [CrossRef]
- Takahashi, K.; Satoh, F.; Hara, E.; Sone, M.; Murakami, O.; Kayama, T.; Yoshimoto, T.; Shibahara, S. Production and Secretion of Adrenomedullin From Glial Cell Tumors and Its Effects on CAMP Production. Peptides 1997, 18, 1117–1124. [Google Scholar] [CrossRef]
- Takahashi, K.; Udono-Fujimori, R.; Totsune, K.; Murakami, O.; Shibahara, S. Suppression of Cytokine-Induced Expression of Adrenomedullin and Endothelin-1 by Dexamethasone in T98G Human Glioblastoma Cells. Peptides 2003, 24, 1053–1062. [Google Scholar] [CrossRef]
- McIlvried, L.A.; Atherton, M.A.; Horan, N.L.; Goch, T.N.; Scheff, N.N. Sensory Neurotransmitter Calcitonin Gene-Related Peptide Modulates Tumor Growth and Lymphocyte Infiltration in Oral Squamous Cell Carcinoma. Adv. Biol. 2022, 6, e2200019. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, L.-J.; Xiao, F.; Wei, Y.; Ke, W.; Xin, H.-B. Intermedin: A Novel Regulator for Vascular Remodeling and Tumor Vessel Normalization by Regulating Vascular Endothelial-Cadherin and Extracellular Signal–Regulated Kinase. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2721–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, H.; Hao, Z.Q.; Fu, X.B.; Hua, X.D.; Ma, Z.H.; Ai, F.L.; Feng, Z.Q.; Wang, K.; Li, W.X.; Li, B. Intermedin Promotes Hepatic Carcinoma Cell Proliferation through Upregulation of miR-155. Int. J. Clin. Exp. Pahotlogy 2018, 11, 3961–3968. [Google Scholar]
- Greillier, L.; Tounsi, A.; Berenguer-Daizé, C.; Dussault, N.; Delfino, C.; Benyahia, Z.; Cayol, M.; Mabrouk, K.; Garcia, S.; Martin, P.-M.; et al. Functional Analysis of the Adrenomedullin Pathway in Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2016, 11, 94–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, A.; Miller, M.J.; Unsworth, E.J.; Siegfried, J.M.; Cuttitta, F. Expression of Adrenomedullin in Normal Human Lung and in Pulmonary Tumors. Endocrinology 1995, 136, 4099–4105. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, Y. Immunohistochemical Analysis of Calcitonin and Calcitonin Gene-Related Peptide in Human Lung. Hum. Pathol. 1989, 20, 896–902. [Google Scholar] [CrossRef]
- MartÍnez, A.; Elsasser, T.H.; Muro-Cacho, C.; Moody, T.W.; Miller, M.J.; Macri, C.J.; Cuttitta, F. Expression of Adrenomedullin and Its Receptor in Normal and Malignant Human Skin: A Potential Pluripotent Role in the Integument. Endocrinology 1997, 138, 5597–5604. [Google Scholar] [CrossRef]
- Kitamuro, T.; Takahashi, K.; Totsune, K.; Nakayama, M.; Murakami, O.; Hida, W.; Shirato, K.; Shibahara, S. Differential Expression of Adrenomedullin and Its Receptor Component, Receptor Activity Modifying Protein (RAMP) 2 during Hypoxia in Cultured Human Neuroblastoma Cells. Peptides 2001, 22, 1795–1801. [Google Scholar] [CrossRef]
- Xu, Y.; Krukoff, T.L. Adrenomedullin Stimulates Nitric Oxide Release from SK-N-SH Human Neuroblastoma Cells by Modulating Intracellular Calcium Mobilization. Endocrinology 2005, 146, 2295–2305. [Google Scholar] [CrossRef] [Green Version]
- Deville, J.-L.; Bartoli, C.; Berenguer, C.; Fernandez-Sauze, S.; Kaafarani, I.; Delfino, C.; Fina, F.; Salas, S.; Muracciole, X.; Mancini, J.; et al. Expression and Role of Adrenomedullin in Renal Tumors and Value of Its MRNA Levels as Prognostic Factor in Clear-Cell Renal Carcinoma. Int. J. Cancer 2009, 125, 2307–2315. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Wang, Y.; Ma, W.; Han, X.; He, X.; Dai, X. Downregulation of Adrenomedullin Leads to the Inhibition of the Tumorigenesis via VEGF Pathway in Human and Nude Mice Osteosarcoma Models. Arch. Med. Res. 2019, 50, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Giacalone, P.-L.; Daurés, J.-P.; Ouafik, L.; Martin, P.-M.; Laffargue, F.; Maudelonde, T. Steroids and Adrenomedullin Growth Patterns in Human Ovarian Cancer Cells: Estrogenic-Regulation Assay. Gynecol. Oncol. 2003, 91, 651–656. [Google Scholar] [CrossRef]
- Liu, J.; Bützow, R.; Hydén-Granskog, C.; Voutilainen, R. Expression of Adrenomedullin in Human Ovaries, Ovarian Sex Cord–Stromal Tumors and Cultured Granulosa–Luteal Cells. Gynecol. Endocrinol. 2009, 25, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, S.; Shang, H.; Pang, X.; Zhao, Y. Basic Fibroblast Growth Factor Upregulates Adrenomedullin Expression in Ovarian Epithelial Carcinoma Cells via JNK-AP-1 Pathway. Regul. Pept. 2009, 157, 44–50. [Google Scholar] [CrossRef]
- Pang, X.; Shang, H.; Deng, B.; Wen, F.; Zhang, Y. The Interaction of Adrenomedullin and Macrophages Induces Ovarian Cancer Cell Migration via Activation of RhoA Signaling Pathway. Int. J. Mol. Sci. 2013, 14, 2774–2787. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Chen, P.; Pang, X.; Hu, Y.; Zhang, Y. Adrenomedullin Up-Regulates the Expression of Vascular Endothelial Growth Factor in Epithelial Ovarian Carcinoma Cells via JNK/AP-1 Pathway. Int. J. Gynecol. Cancer 2015, 25, 953–960. [Google Scholar] [CrossRef] [Green Version]
- Javeed, N.; Sagar, G.; Dutta, S.K.; Smyrk, T.C.; Lau, J.S.; Bhattacharya, S.; Truty, M.; Petersen, G.M.; Kaufman, R.J.; Chari, S.T.; et al. Pancreatic Cancer–Derived Exosomes Cause Paraneoplastic β-Cell Dysfunction. Clin. Cancer Res. 2015, 21, 1722–1733. [Google Scholar] [CrossRef] [Green Version]
- Sagar, G.; Sah, R.P.; Javeed, N.; Dutta, S.K.; Smyrk, T.C.; Lau, J.S.; Giorgadze, N.; Tchkonia, T.; Kirkland, J.L.; Chari, S.T.; et al. Pathogenesis of Pancreatic Cancer Exosome-Induced Lipolysis in Adipose Tissue. Gut 2016, 65, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, A.; Srivastava, S.K.; Singh, S.; Tyagi, N.; Arora, S.; Carter, J.E.; Khushman, M.; Singh, A.P. MYB Promotes Desmoplasia in Pancreatic Cancer through Direct Transcriptional Up-Regulation and Cooperative Action of Sonic Hedgehog and Adrenomedullin. J. Biol. Chem. 2016, 291, 16263–16270. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Flatt, P.R.; Permert, J.; Adrian, T.E. Pancreatic Cancer Cells Selectively Stimulate Islet β Cells to Secrete Amylin. Gastroenterology 1998, 114, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Stridsberg, M.; Eriksson, B.; Lundqvist, G.; Skogseid, B.; Wilander, E.; Öberg, K. Islet Amyloid Polypeptide (IAPP) in Patients with Neuroendocrine Tumours. Regul. Pept. 1995, 55, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, F.; Vidal, S.; Bell, D.; Horvath, E.; Kovacs, K.; Scheithauer, B.W.; Lloyd, R.V. Immunohistochemical Localization of Amylin in Human Pancreas, Thyroid, Pituitary and Their Tumors. Acta Histochem. 2003, 105, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Brand, R.E.; Ding, X.-Z.; Young, C.M.; Adrian, T.E. The Specificity of Amylin for the Diagnosis of Pancreatic Adenocarcinoma. Int. J. Gastrointest. Cancer 2002, 31, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Chari, S.T.; Klee, G.G.; Miller, L.J.; Raimondo, M.; DiMagno, E.P. Islet Amyloid Polypeptide Is Not a Satisfactory Marker for Detecting Pancreatic Cancer. Gastroenterology 2001, 121, 640–645. [Google Scholar] [CrossRef]
- Garnett, S.; de Bruyns, A.; Provencher-Tom, V.; Dutchak, K.; Shu, R.; Dankort, D. Metabolic Regulator IAPP (Amylin) Is Required for BRAF and RAS Oncogene-Induced Senescence. Mol. Cancer Res. 2021, 19, 874–885. [Google Scholar] [CrossRef]
- kim, H.S.; Joo, S.S.; Yoo, J.-M. The Correlation between Amylin and Insulin by Treatment with 2-Deoxy-D-Glucose and/or Mannose in Rat Insulinoma INS-1E Cells. J. Physiol. Pharmacol. 2022, 72, 102–123. [Google Scholar] [CrossRef]
- Williams, A.J.K.; Coates, P.J.; Lowe, D.G.; McLEAN, C.; Gale, E.A.M. Immunochemical Investigation of Insulinomas for Islet Amyloid Polypeptide and Insulin: Evidence for Differential Synthesis and Storage. Histopathology 1992, 21, 215–223. [Google Scholar] [CrossRef]
- Oskarsson, M.E.; Singh, K.; Wang, J.; Vlodavsky, I.; Li, J.; Westermark, G.T. Heparan Sulfate Proteoglycans Are Important for Islet Amyloid Formation and Islet Amyloid Polypeptide-Induced Apoptosis. J. Biol. Chem. 2015, 290, 15121–15132. [Google Scholar] [CrossRef] [Green Version]
- Magruder, J.T.; Elahi, D.; Andersen, D.K. Diabetes and Pancreatic Cancer: Chicken or Egg? Pancreas 2011, 40, 339–351. [Google Scholar] [CrossRef]
- Bretherton-Watt, D.; Ghatei, M.A.; Bloom, S.E.; Williams, S.; Bloom, S.R. Islet Amyloid Polypeptide-like Immunoreactivity in Human Tissue and Endocrine Tumors. J. Clin. Endocrinol. Metab. 1993, 76, 1072–1074. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sun, Y.; Liu, W.; Mai, Y. Silibinin Ameliorates Amylin-Induced Pancreatic β-Cell Apoptosis Partly via Upregulation of GLP-1R/PKA Pathway. Mol. Cell Biochem. 2019, 1–2, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, J.; MacGibbon, G.; Dragunow, M.; Cooper, G.J.S. Increased Expression and Activation of C-Jun Contributes to Human Amylin-Induced Apoptosis in Pancreatic Islet β-Cells. J. Mol. Biol. 2002, 324, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Abasolo, I.; Yang, L.; Haleem, R.; Xiao, W.; Pio, R.; Cuttitta, F.; Montuenga, L.M.; Kozlowski, J.M.; Calvo, A.; Wang, Z. Overexpression of Adrenomedullin Gene Markedly Inhibits Proliferation of PC3 Prostate Cancer Cells in Vitro and in Vivo. Mol. Cell. Endocrinol. 2003, 199, 179–187. [Google Scholar] [CrossRef]
- Gonzalez-Moreno, O.; Calvo, A.; Joshi, B.H.; Abasolo, I.; Leland, P.; Wang, Z.; Montuenga, L.; Puri, R.K.; Green, J.E. Gene Expression Profiling Identifies IL-13 Receptor ?2 Chain as a Therapeutic Target in Prostate Tumor Cells Overexpressing Adrenomedullin. Int. J. Cancer 2005, 114, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.H.; Leland, P.; Calvo, A.; Green, J.E.; Puri, R.K. Human Adrenomedullin Up-Regulates Interleukin-13 Receptor A2 Chain in Prostate Cancer In Vitro and In Vivo: A Novel Approach to Sensitize Prostate Cancer to Anticancer Therapy. Cancer Res. 2008, 68, 9311–9317. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, N.; Abasolo, I.; Jongsma, J.; Calvo, A.; Garayoa, M.; van der Kwast, T.H.; van Steenbrugge, G.J.; Montuenga, L.M. Androgen-Independent Expression of Adrenomedullin and Peptidylglycine Alphaamidating Monooxygenase in Human Prostatic Carcinoma. Mol. Carcinog. 2003, 38, 14–24. [Google Scholar] [CrossRef]
- Berenguer, C.; Boudouresque, F.; Dussert, C.; Daniel, L.; Muracciole, X.; Grino, M.; Rossi, D.; Mabrouk, K.; Figarella-Branger, D.; Martin, P.-M.; et al. Adrenomedullin, an Autocrine/Paracrine Factor Induced by Androgen Withdrawal, Stimulates ‘Neuroendocrine Phenotype’ in LNCaP Prostate Tumor Cells. Oncogene 2008, 27, 506–518. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.S.; Gao, L.; Bledsoe, G.; Chao, L.; Chao, J. Intermedin Is a New Angiogenic Growth Factor. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H1040–H1047. [Google Scholar] [CrossRef] [Green Version]
- Nagakawa, O.; Ogasawara, M.; Fujii, H.; Murakami, K.; Murata, J.; Fuse, H.; Saiki, I. Effect of Prostatic Neuropeptides on Invasion and Migration of PC-3 Prostate Cancer Cells. Cancer Lett. 1998, 133, 27–33. [Google Scholar] [CrossRef]
- Di Sant’Agnese, P.A. Neuroendocrine Differentiation in Carcinoma of the Prostate. Diagnostic, Prognostic, and Therapeutic Implications. Cancer 1992, 70, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Hagner, S.; Stahl, U.; Grimm, T.; Sturzl, M.; Lang, R.E. Expression of Calcitonin Receptor-like Receptor in Human Vascular Tumours. J. Clin. Pathol. 2006, 59, 1104–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stables, J.M.; Beresford, I.J.M.; Arkinstall, S.; Ireland, S.J.; Walsh, D.M.; Seale, P.W.; Ward, P.; Hagan, R.M. GR138676, a Novel Peptidic Tachykinin Antagonist Which Is Potent at NK3 Receptors. Neuropeptides 1994, 27, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, S.; Wang, S.; Shi, L.; Wang, C.; Zhang, J.; Gao, Y.; Li, G.; Qi, Y.; An, X.; et al. Targeting Neurokinin-3 Receptor: A Novel Anti-Angiogenesis Strategy for Cancer Treatment. Oncotarget 2017, 8, 40713–40723. [Google Scholar] [CrossRef] [Green Version]
- Kitani, M.; Sakata, J.; Asada, Y.; Kitamura, K.; Eto, T. Distribution and Expression of Adrenomedullin in Human Gastrointestinal Tissue. Ann. Clin. Biochem. Int. J. Lab. Med. 1998, 35, 643–648. [Google Scholar] [CrossRef] [Green Version]
- Marutsuka, K.; Nawa, Y.; Asada, Y.; Hara, S.; Kitamura, K.; Eto, T.; Sumiyoshi, A. Adrenomedullin and Proadrenomudullin N-Terminal 20 Peptide (PAMP) Are Present in Human Colonic Epithelia and Exert an Antimicrobial Effect. Exp. Physiol. 2001, 86, 543–545. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.J.; Martínez, A.; Unsworth, E.J.; Thiele, C.J.; Moody, T.W.; Elsasser, T.; Cuttitta, F. Androgen-Independent Expression of Adrenomedullin and Peptidylglycine Alphaamidating Monooxygenase in Human Prostatic Carcinoma. J. Biol. Chem. 1996, 271, 23345–23351. [Google Scholar] [CrossRef] [Green Version]
- Kaafarani, I.; Fernandez-Sauze, S.; Berenguer, C.; Chinot, O.; Delfino, C.; Dussert, C.; Metellus, P.; Boudouresque, F.; Mabrouk, K.; Grisoli, F.; et al. Targeting Adrenomedullin Receptors with Systemic Delivery of Neutralizing Antibodies Inhibits Tumor Angiogenesis and Suppresses Growth of Human Tumor Xenografts in Mice. FASEB J. 2009, 23, 3424–3435. [Google Scholar] [CrossRef]
- Martínez, A.; Weaver, C.; López, J.; Bhathena, S.J.; Elsasser, T.H.; Miller, M.J.; Moody, T.W.; Unsworth, E.J.; Cuttitta, F. Regulation of Insulin Secretion and Blood Glucose Metabolism by Adrenomedullin. Endocrinology 1996, 137, 2626–2632. [Google Scholar] [CrossRef] [Green Version]
- Vázquez, R.; Riveiro, M.E.; Berenguer-Daizé, C.; O’Kane, A.; Gormley, J.; Touzelet, O.; Rezai, K.; Bekradda, M.; Ouafik, L. Targeting Adrenomedullin in Oncology: A Feasible Strategy With Potential as Much More Than an Alternative Anti-Angiogenic Therapy. Front. Oncol. 2021, 10, 589218. [Google Scholar] [CrossRef]
- Larráyoz, I.M.; Martínez-Herrero, S.; García-Sanmartín, J.; Ochoa-Callejero, L.; Martínez, A. Adrenomedullin and Tumour Microenvironment. J. Transl. Med. 2014, 12, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, S.; Hirata, Y.; Iwasaki, H.; Sato, K.; Watanabe, T.X.; Inui, T.; Nakajima, K.; Sakakibara, S.; Marumo, F. Structure-Activity Relationship of Adrenomedullin, a Novel Vasodilatory Peptide, in Cultured Rat Vascular Smooth Muscle Cells. Endocrinology 1994, 135, 2454–2458. [Google Scholar] [CrossRef] [PubMed]
- Avgoustou, P.; Jailani, A.B.A.; Zirimwabagabo, J.-O.; Tozer, M.J.; Gibson, K.R.; Glossop, P.A.; Mills, J.E.J.; Porter, R.A.; Blaney, P.; Bungay, P.J.; et al. Discovery of a First-in-Class Potent Small Molecule Antagonist against the Adrenomedullin-2 Receptor. ACS Pharmacol. Transl. Sci. 2020, 3, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Zirimwabagabo, J.-O.; Jailani, A.B.A.; Avgoustou, P.; Tozer, M.J.; Gibson, K.R.; Glossop, P.A.; Mills, J.E.J.; Porter, R.A.; Blaney, P.; Wang, N.; et al. Discovery of a First-In-Class Small Molecule Antagonist against the Adrenomedullin-2 Receptor: Structure–Activity Relationships and Optimization. J. Med. Chem. 2021, 64, 3299–3319. [Google Scholar] [CrossRef]
- Jailani, A.B.A.; Bigos, K.J.A.; Avgoustou, P.; Egan, J.L.; Hathway, R.A.; Skerry, T.M.; Richards, G.O. Targeting the Adrenomedullin-2 Receptor for the Discovery and Development of Novel Anti-Cancer Agents. Expert Opin. Drug Discov. 2022, 17, 839–848. [Google Scholar] [CrossRef]
- Miseki, T.; Kawakami, H.; Natsuizaka, M.; Darmanin, S.; Cui, H.Y.; Chen, J.; Fu, Q.; Okada, F.; Shindo, M.; Higashino, F.; et al. Suppression of Tumor Growth by Intra-Muscular Transfer of Naked DNA Encoding Adrenomedullin Antagonist. Cancer Gene Ther. 2007, 14, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, K.; Hida, K.; Hida, Y.; Muraki, C.; Ohga, N. Adrenomedullin Antagonist Suppresses Tumor Formation in Renal Cell Carcinoma through Inhibitory Effects on Tumor Endothelial Cells and Endothelial Progenitor Mobilization. Int. J. Oncol. 2010, 36, 1379–1386. [Google Scholar] [CrossRef]
- Gao, Y.; Li, J.; Qiao, N.; Meng, Q.; Zhang, M.; Wang, X.; Jia, J.; Yang, S.; Qu, C.; Li, W.; et al. Adrenomedullin Blockade Suppresses Sunitinib-Resistant Renal Cell Carcinoma Growth by Targeting the ERK/MAPK Pathway. Oncotarget 2016, 7, 63374–63387. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Koyama, T.; Sakurai, T.; Kamiyoshi, A.; Ichikawa-Shindo, Y.; Kawate, H.; Liu, T.; Xian, X.; Imai, A.; Zhai, L.; et al. The Endothelial Adrenomedullin-RAMP2 System Regulates Vascular Integrity and Suppresses Tumour Metastasis. Cardiovasc. Res. 2016, 111, 398–409. [Google Scholar] [CrossRef] [Green Version]
- Zudaire, E.; Martínez, A.; Garayoa, M.; Pío, R.; Kaur, G.; Woolhiser, M.R.; Metcalfe, D.D.; Hook, W.A.; Siraganian, R.P.; Guise, T.A.; et al. Adrenomedullin Is a Cross-Talk Molecule That Regulates Tumor and Mast Cell Function during Human Carcinogenesis. Am. J. Pathol. 2006, 168, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Herrero, S.; Martínez, A. Cancer Protection Elicited by a Single Nucleotide Polymorphism Close to the Adrenomedullin Gene. J. Clin. Endocrinol. Metab. 2013, 98, E807–E810. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.L.; Hsu, S.Y.T. Development of Chimeric and Bifunctional Antagonists for CLR/RAMP Receptors. PLoS ONE 2019, 14, e0216996. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xiao, F.; Kong, L.; Wang, D.; Li, H.; Wei, Y.; Tan, C.; Zhao, H.; Zhang, T.; Cao, G.; et al. Intermedin Enlarges the Vascular Lumen by Inducing the Quiescent Endothelial Cell Proliferation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 398–413. [Google Scholar] [CrossRef] [PubMed]
- Al-Keilani, M.; Alsmadi, D.; Darweesh, R.; Alzoubi, K. Pramlintide, an Antidiabetic, Is Antineoplastic in Colorectal Cancer and Synergizes with Conventional Chemotherapy. Clin. Pharmacol. Adv. Appl. 2018, 10, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Chiba, T.; Yamaguchi, A.; Yamatani, T.; Nakamura, A.; Morishita, T.; Inui, T.; Fukase, M.; Noda, T.; Fujita, T. Calcitonin Gene-Related Peptide Receptor Antagonist Human CGRP-(8-37). Am. J. Physiol.-Endocrinol. Metab. 1989, 256, E331–E335. [Google Scholar] [CrossRef]
- Balood, M.; Ahmadi, M.; Eichwald, T.; Ahmadi, A.; Majdoubi, A.; Roversi, K.; Roversi, K.; Lucido, C.T.; Restaino, A.C.; Huang, S.; et al. Nociceptor Neurons Affect Cancer Immunosurveillance. Nature 2022, 611, 405–412. [Google Scholar] [CrossRef]
- Pío, R.; Martínez, A.; Unsworth, E.J.; Kowalak, J.A.; Bengoechea, J.A.; Zipfel, P.F.; Elsasser, T.H.; Cuttitta, F. Complement Factor H Is a Serum-Binding Protein for Adrenomedullin, and the Resulting Complex Modulates the Bioactivities of Both Partners. J. Biol. Chem. 2001, 276, 12292–12300. [Google Scholar] [CrossRef] [Green Version]
- Chirgwin, J.M.; Mohammad, K.S.; Guise, T.A. Tumor-Bone Cellular Interactions in Skeletal Metastases. J. Musculoeskelet Neuronal Interact. 2004, 4, 308–318. [Google Scholar]
- Majkowska-Pilip, A.; Halik, P.K.; Gniazdowska, E. The Significance of NK1 Receptor Ligands and Their Application in Targeted Radionuclide Tumour Therapy. Pharmaceutics 2019, 11, 443. [Google Scholar] [CrossRef] [Green Version]
- Merck Sharp & Dohme Corp. Radiolabeled CGRP Antagonists. WO2011014383A1 (2011), 3 February 2011. [Google Scholar]
- Tang, B.; Yong, X.; Xie, R.; Li, Q.-W.; Yang, S.-M. Vasoactive Intestinal Peptide Receptor-Based Imaging and Treatment of Tumors. Int. J. Oncol. 2014, 44, 1023–1031. [Google Scholar] [CrossRef] [Green Version]














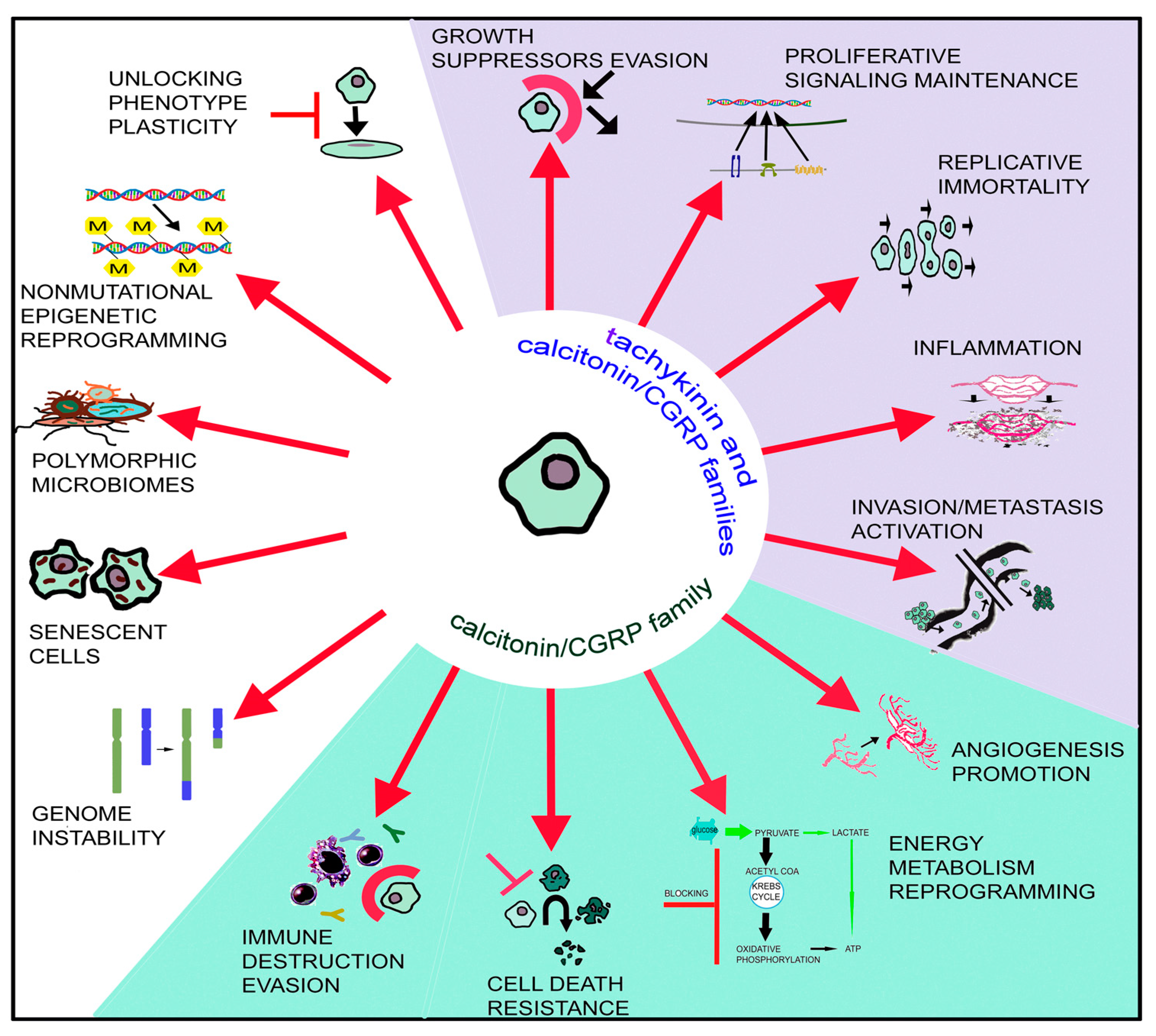{kind=link}
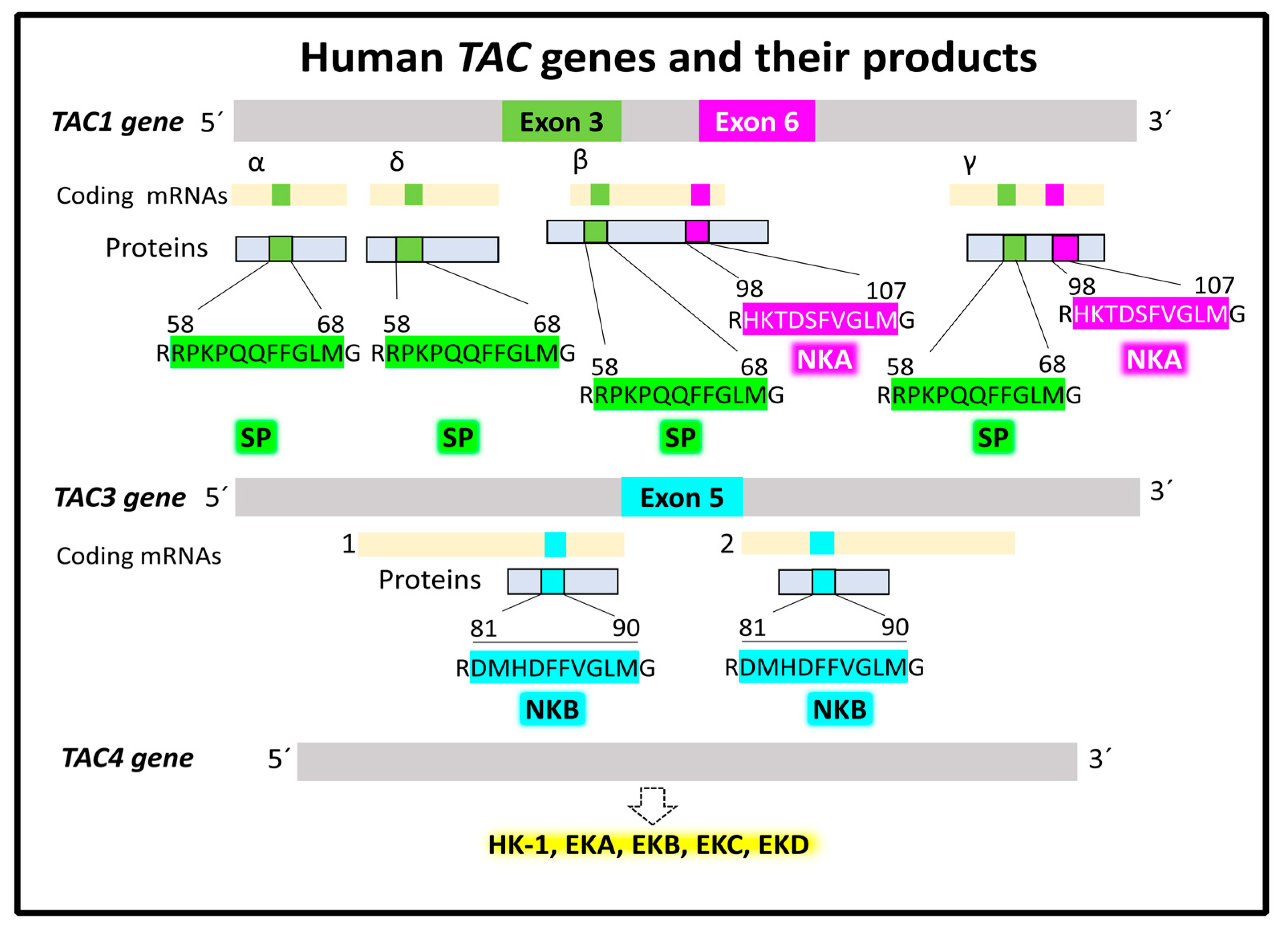{kind=link}
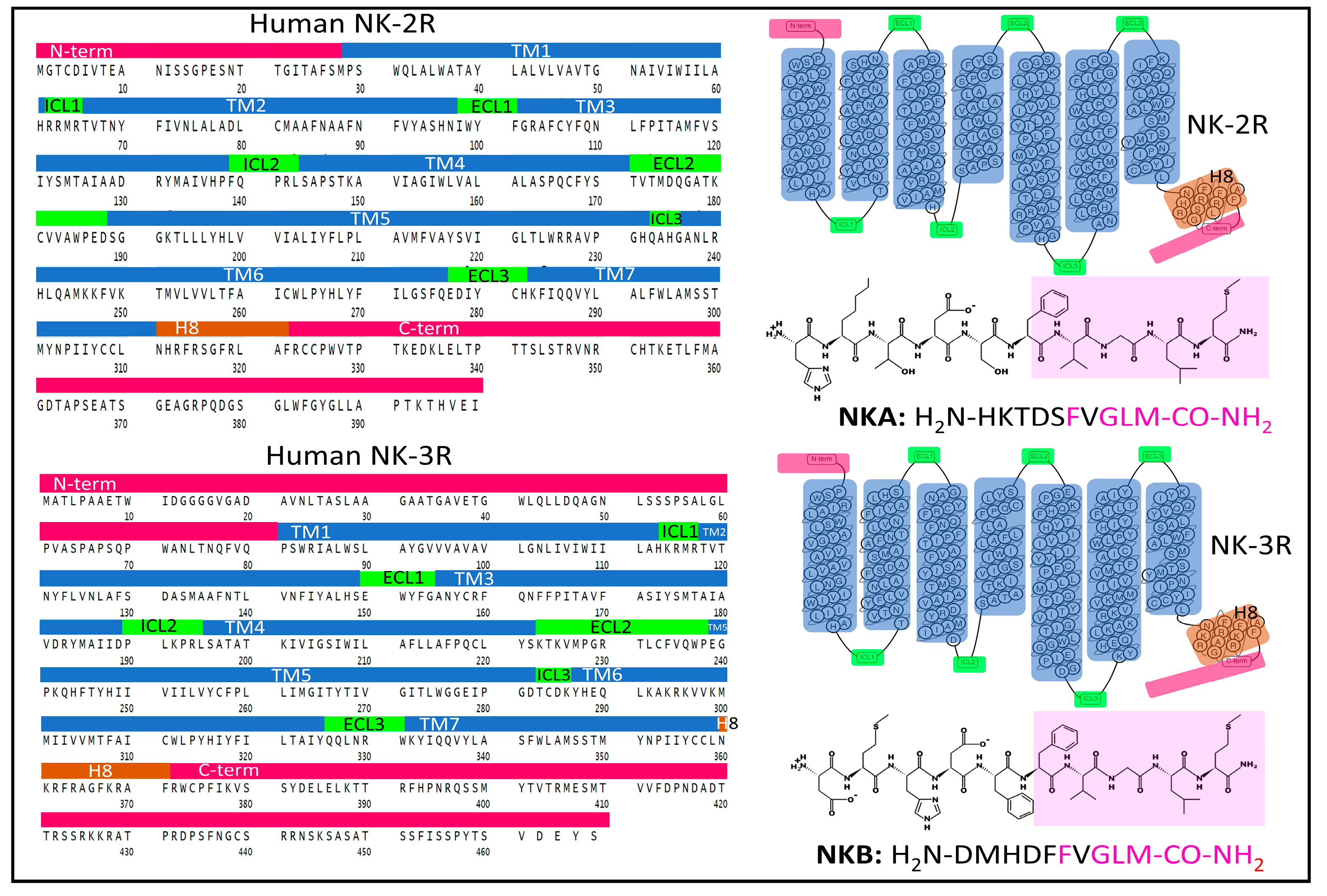{kind=link}
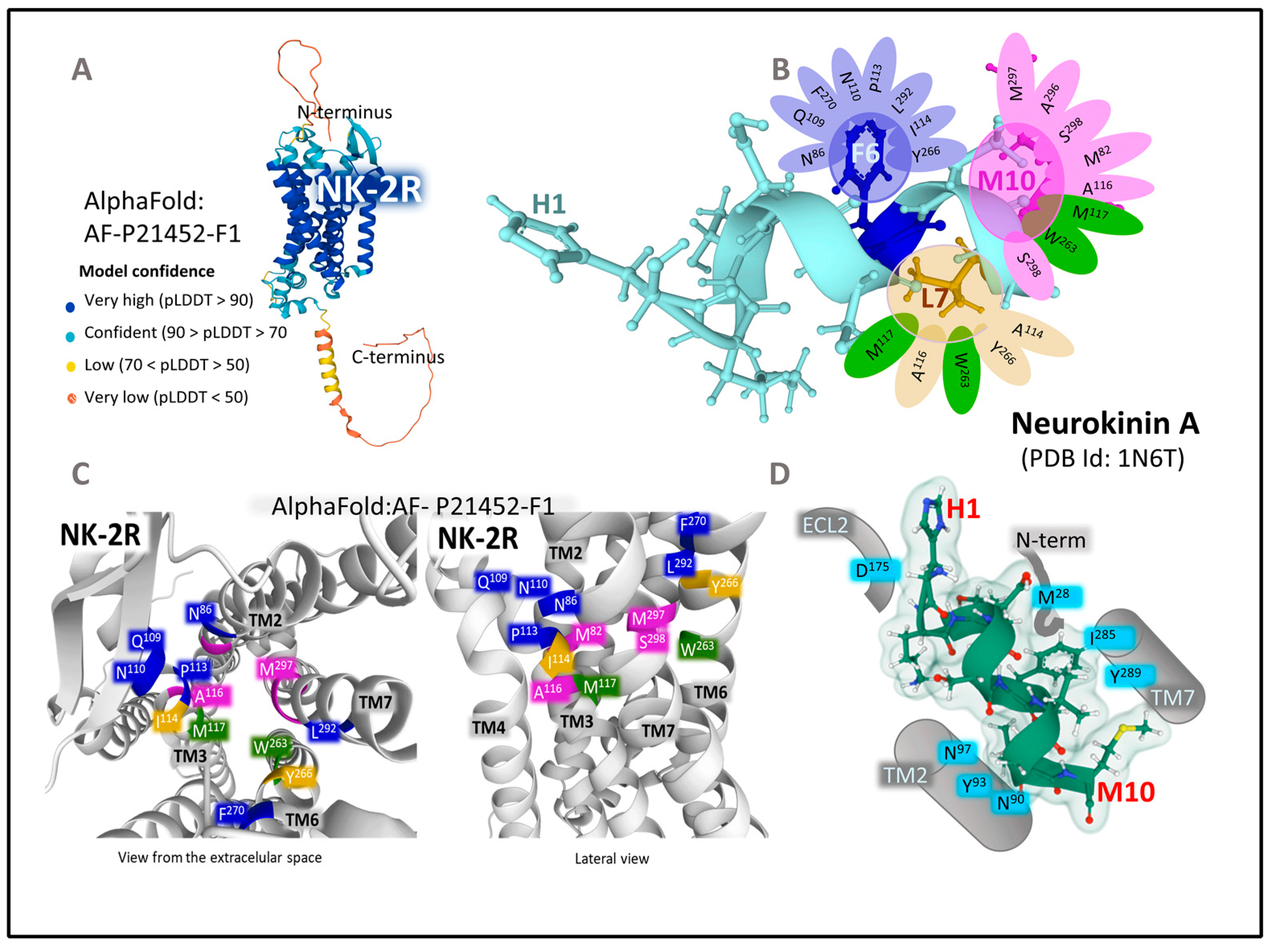{kind=link}
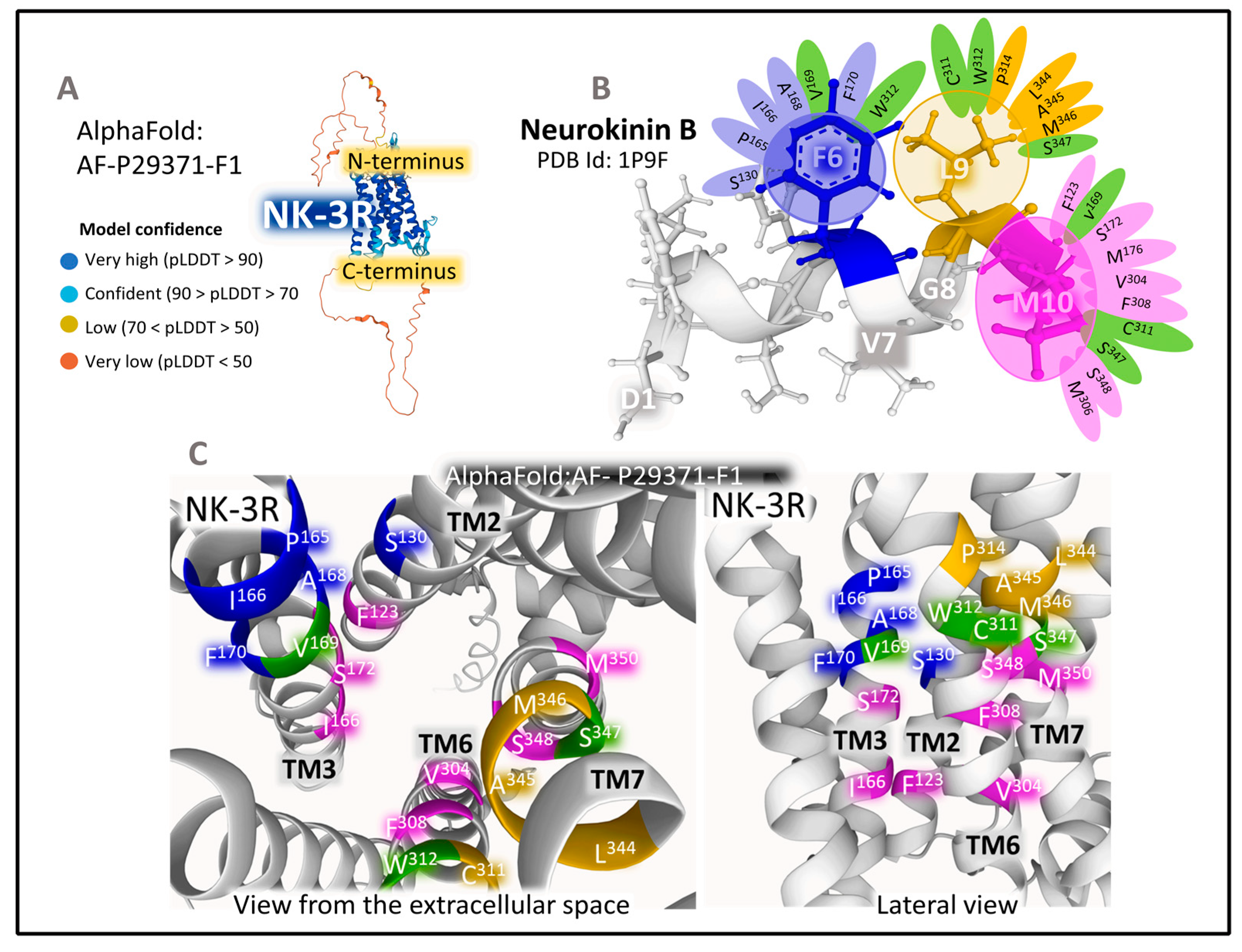{kind=link}
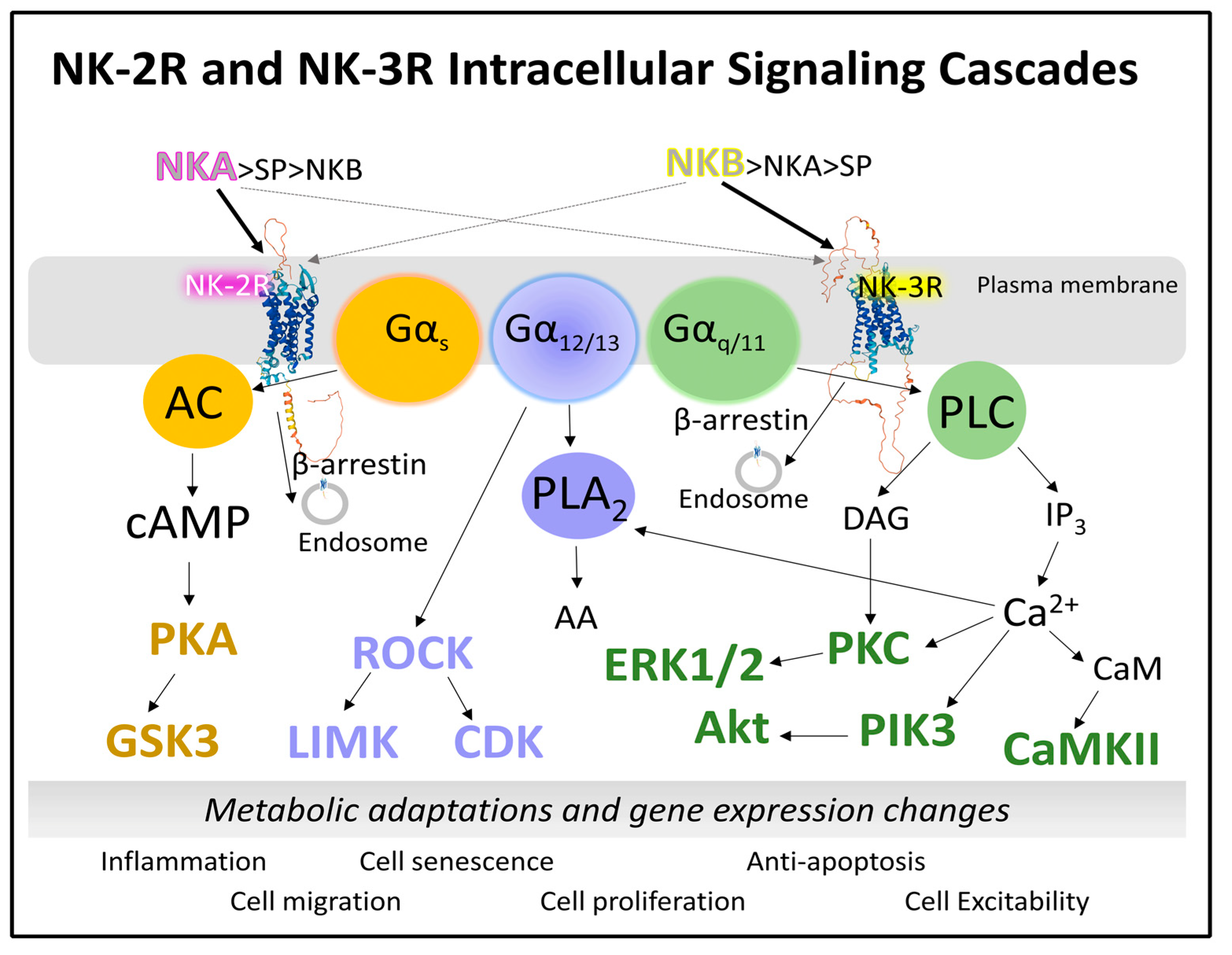{kind=link}
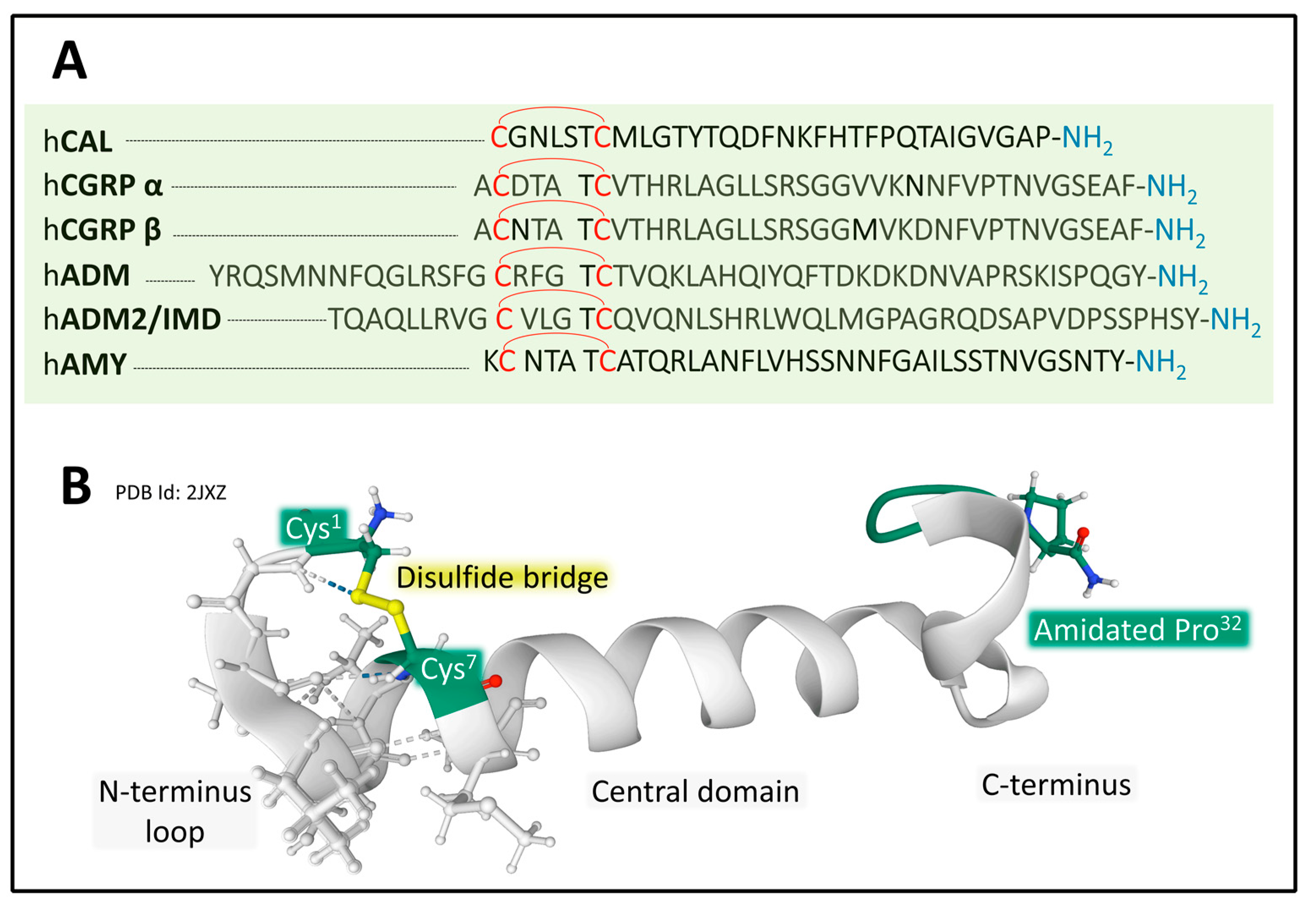{kind=link}
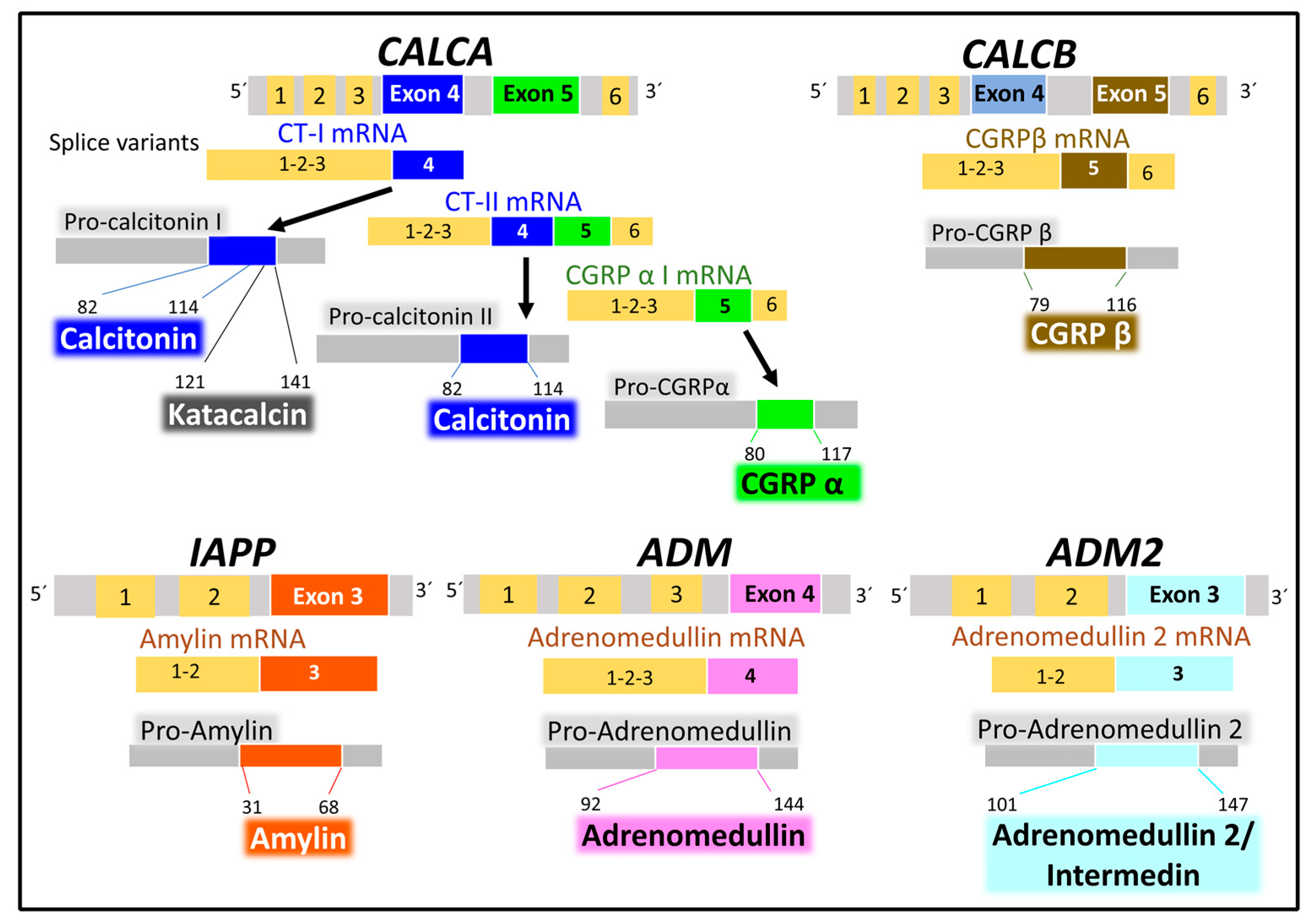{kind=link}
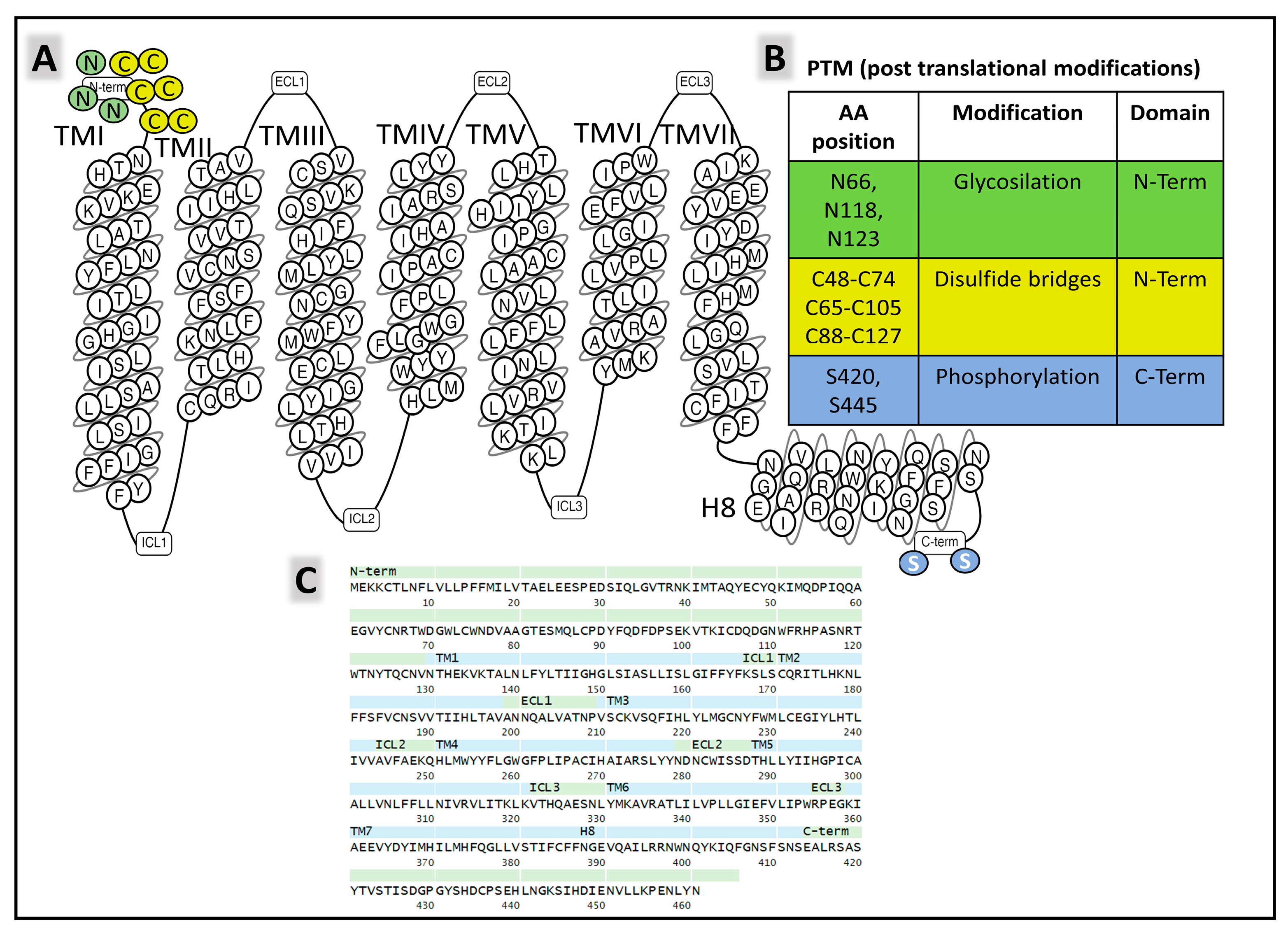{kind=link}
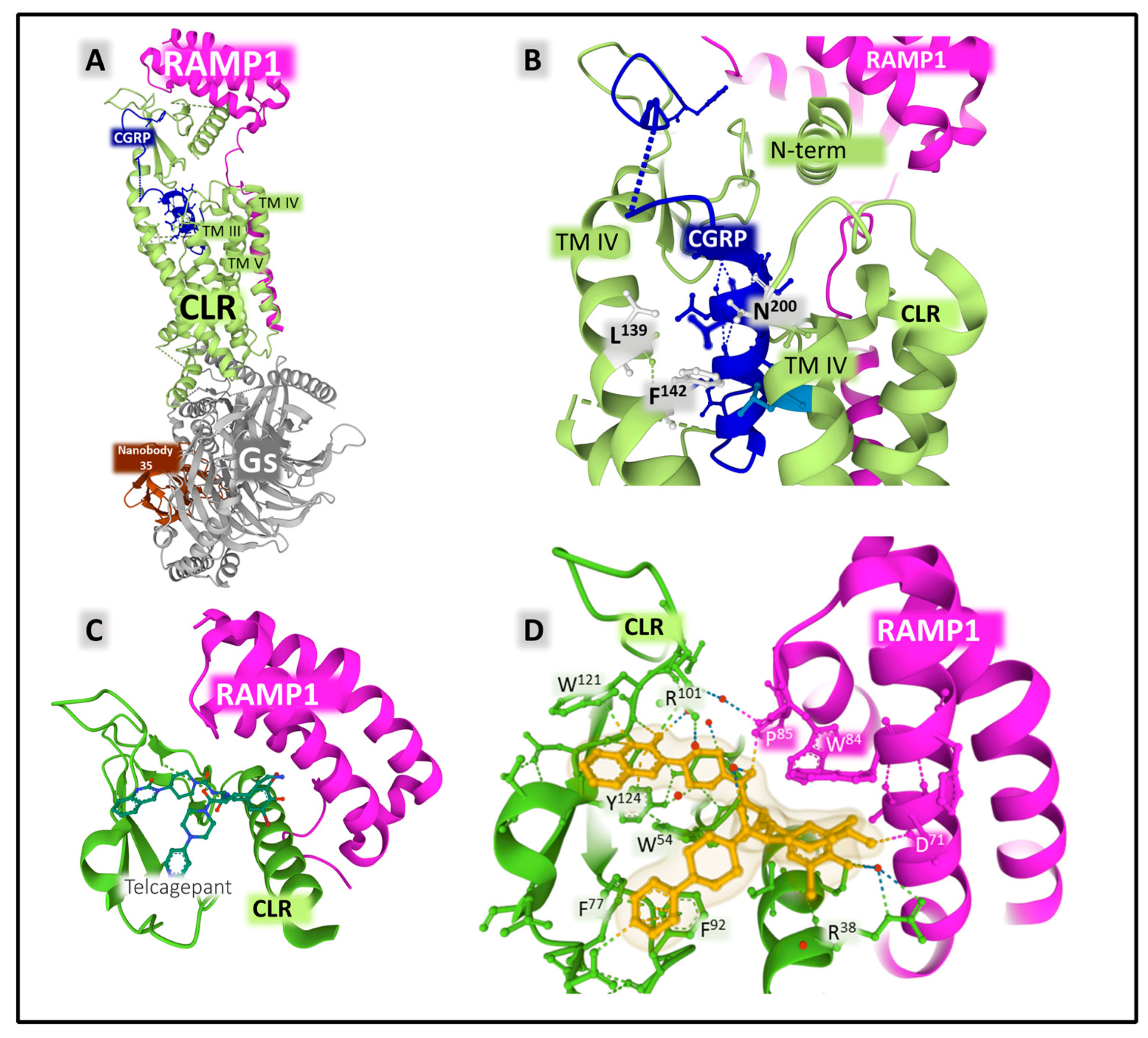{kind=link}
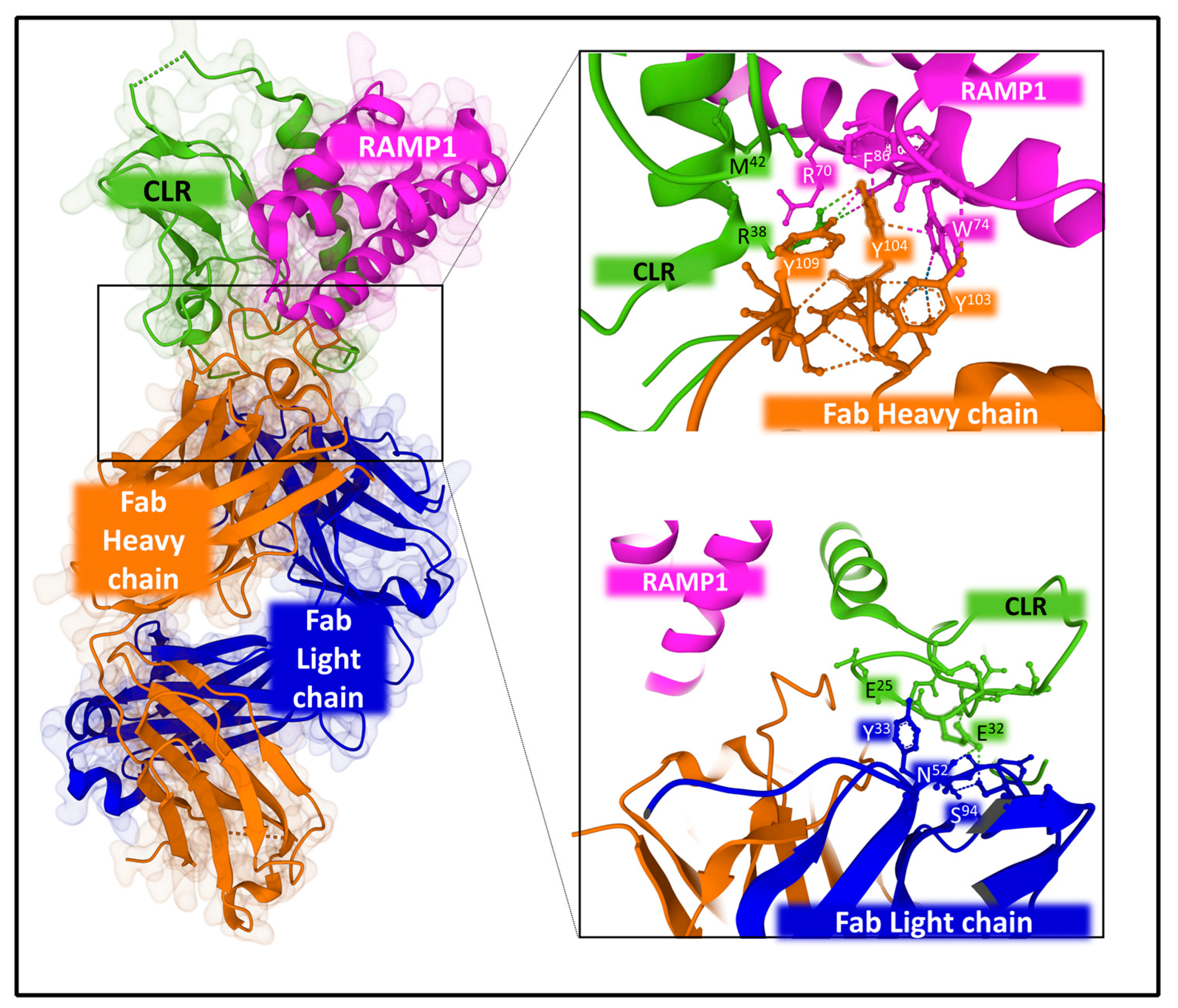{kind=link}
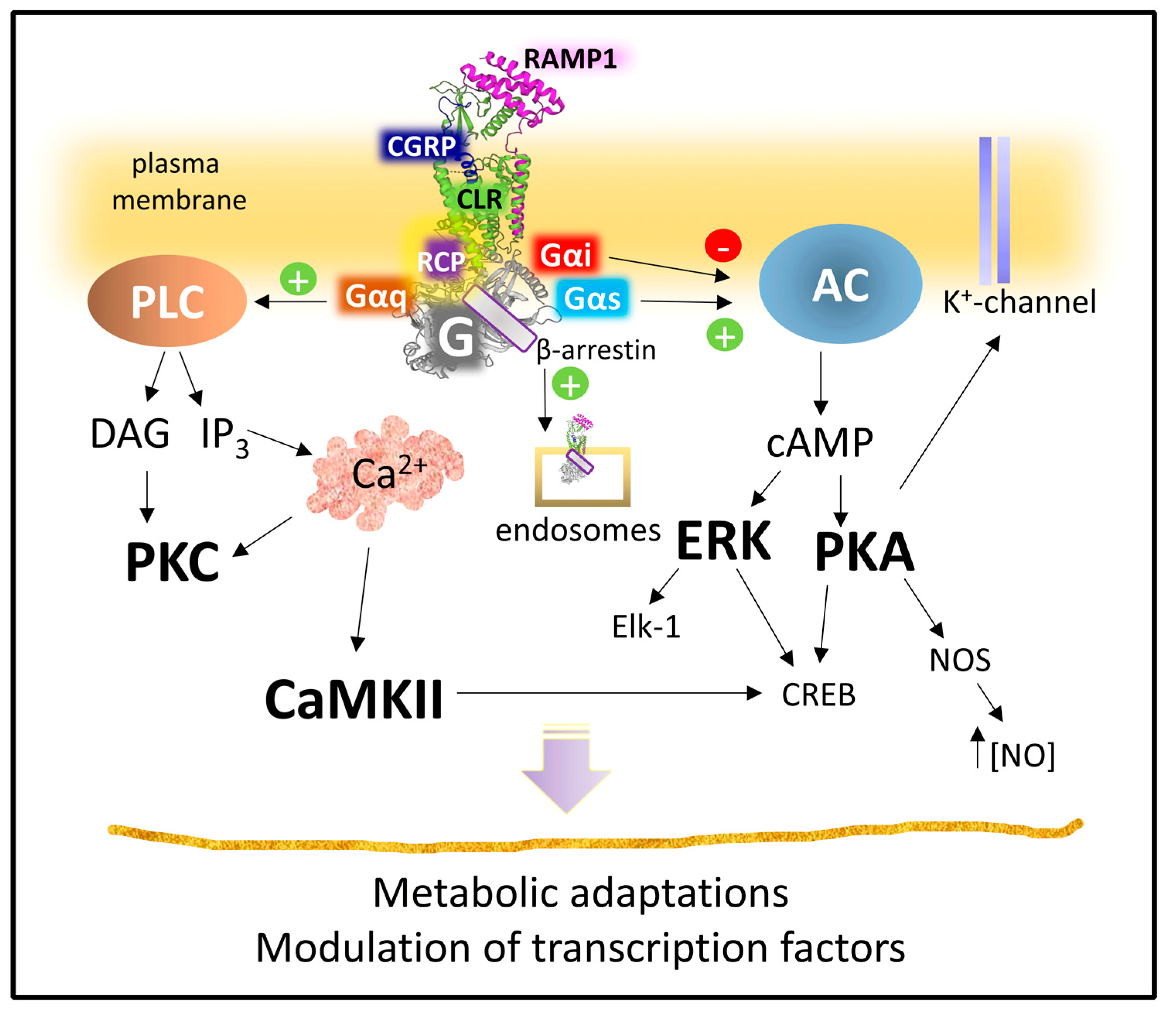{kind=link}
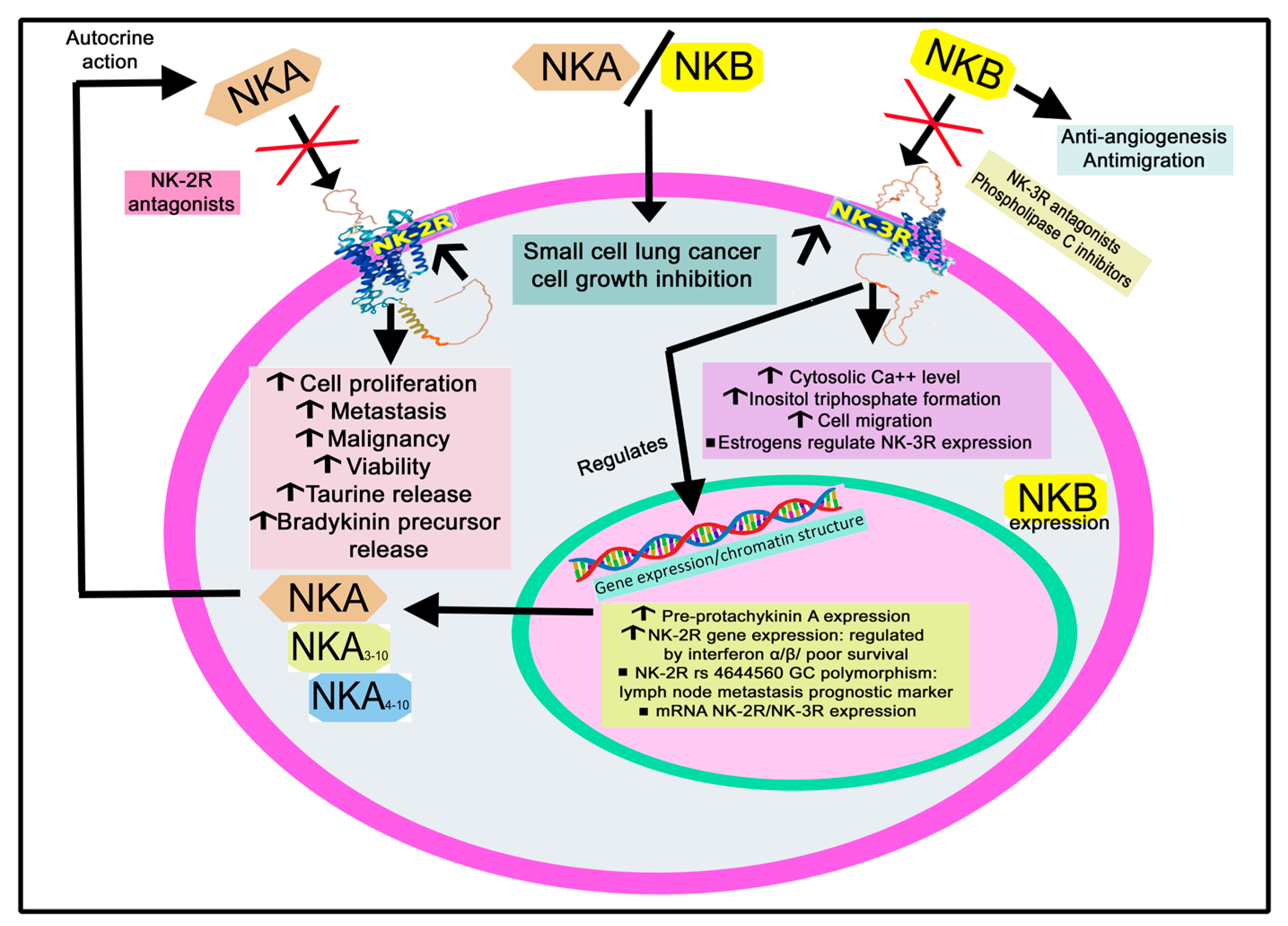{kind=link}
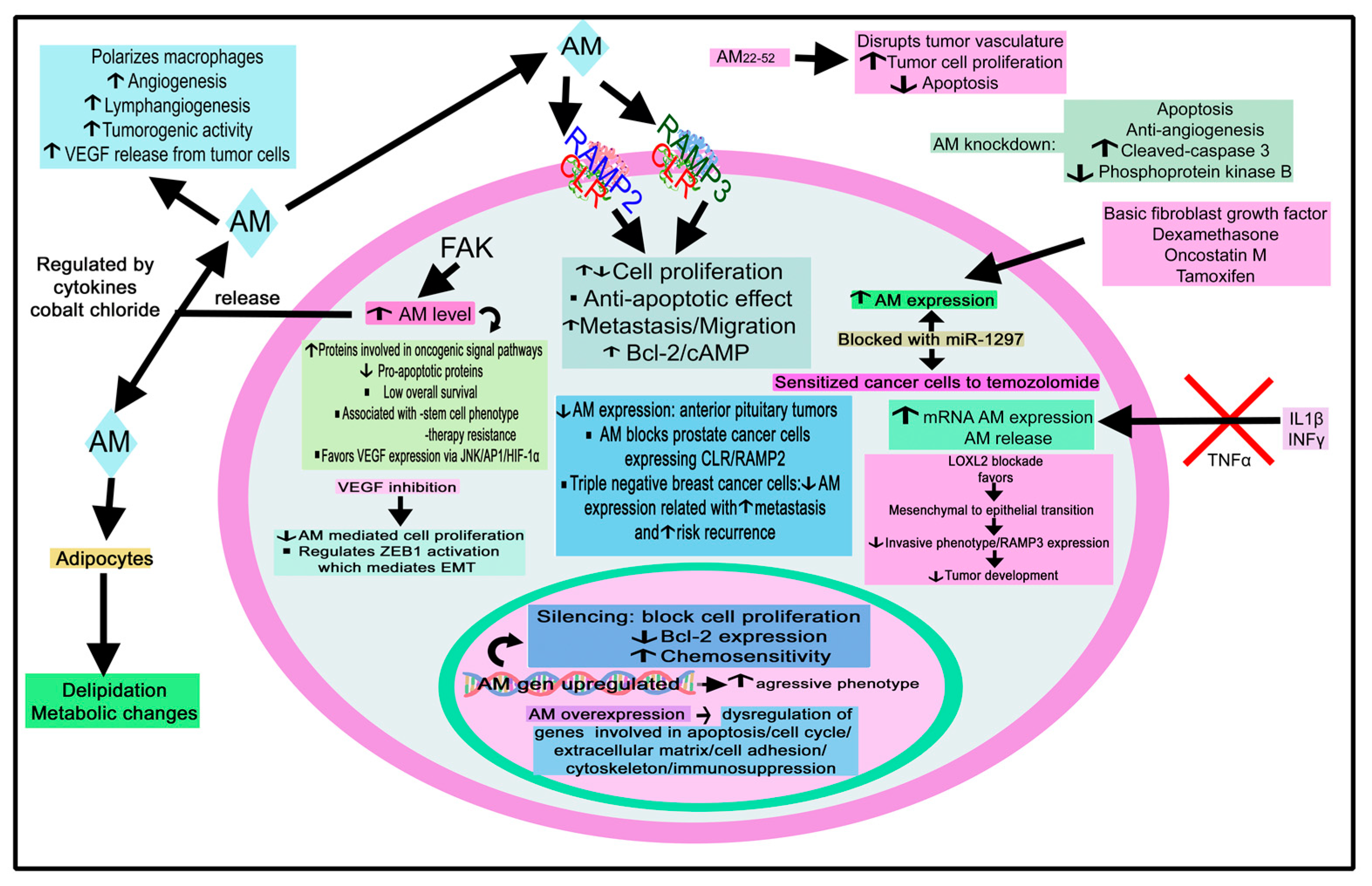{kind=link}
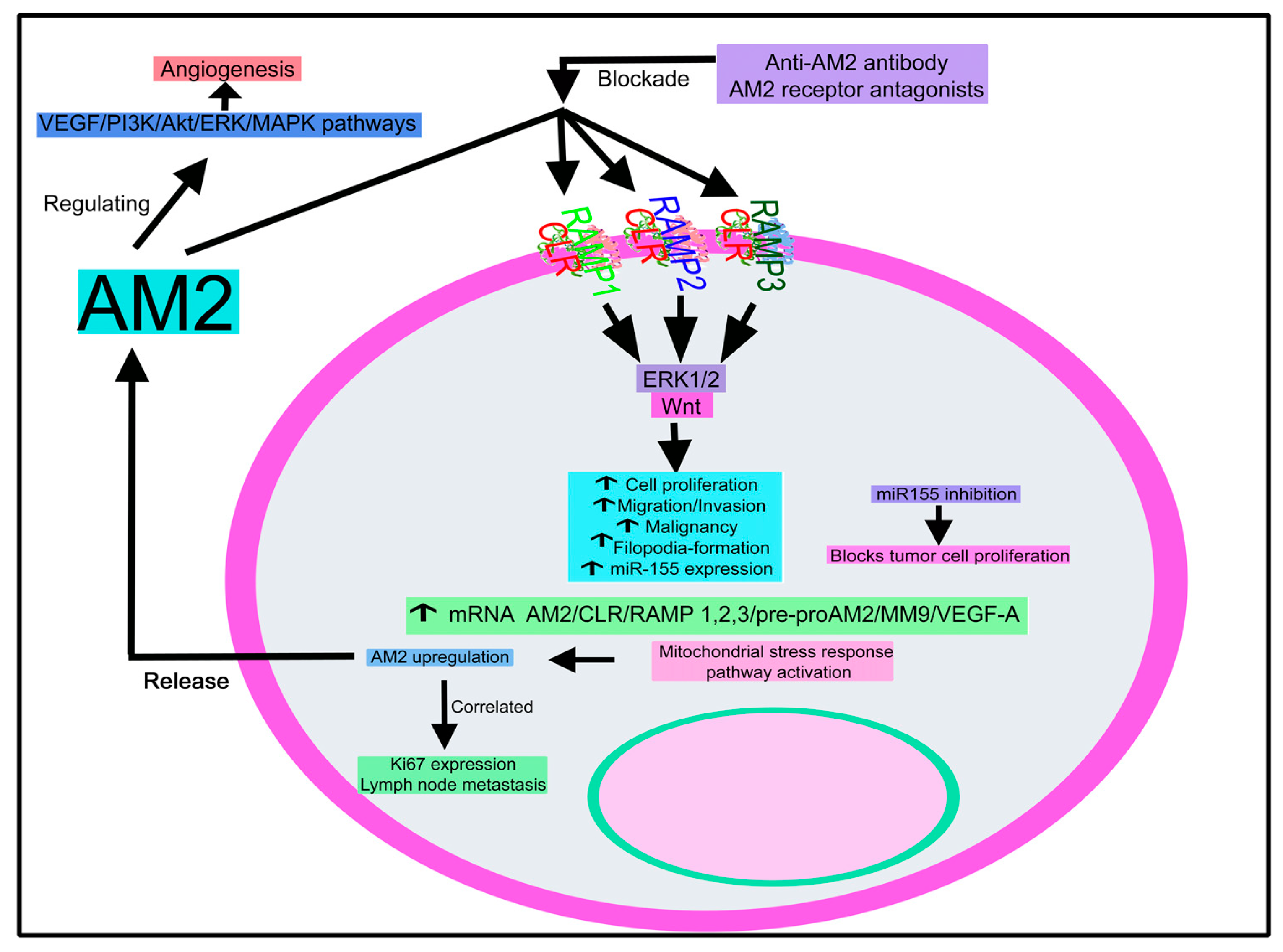{kind=link}
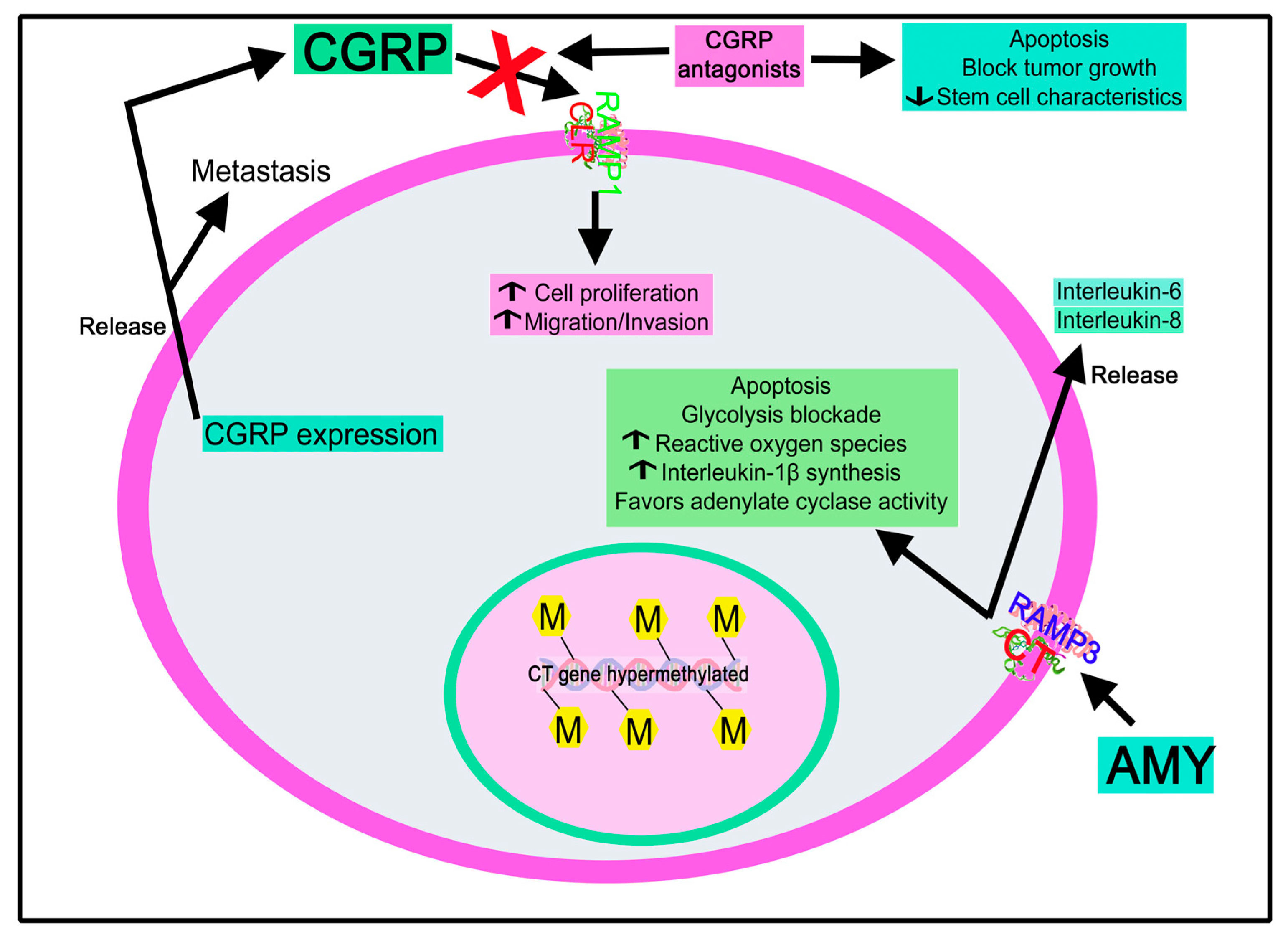{kind=link}
| Tumor | NKA/NKB | References |
|---|---|---|
| Breast cancer | Malignant biopsies/cancer cells: increased pre-protachykinin A expression. Metastatic cancer cells: NK-2R overexpression; NKA increases aggressiveness and NK-2R expression. NKA favors cell proliferation. NK-2R mediates cancer cell proliferation but not in normal cells. NK-2R antagonists block tumor cell proliferation. | [92,139,140,141] |
| Colorectal cancer | High NK-2R gene expression relates to poor survival. NK-2R rs4644560 GC polymorphism alone or combined with NK-1R rs10198644 GC: a prognostic marker for lymph node metastasis. NK-2R overexpression: increases tumorigenesis and metastatic colonization. NKA increases viability and tumor cell proliferation. NK-2R antagonists decrease tumor cell proliferation. | [142,143] |
| Glioma | NKA promotes tumor cell proliferation through NK-1R. Astrocytoma: NK-3R expression. | [144,145] |
| Insulinoma | Cancer cells release NKA and express the pre-protachykinin A gene. | [146,147] |
| Lung cancer | mRNA pre-protachykinin/NKA expression. NKA/NKB blocks tumor cell growth. | [20,148] |
| Midgut carcinoid tumor | High plasma NKA level: worse survival. Plasma NKA level: a biomarker of prognosis. Pre-pro-tachykinin I mRNA/NKA expression. NKA/NKB increased luminal content. | [148,149,150,151] |
| Neuroblastoma | mRNA NK-2R/NK-3R expression. NKB but not NKA expression. NK-2R/NK-3R: activated independently by NKA. NK-2R mediates cell proliferation. NKA, via NK-3R, increases Ca++ cytosolic level through phospholipase C activation; this action is blocked with SR-48968. | [152,153,154,155] |
| Oral squamous cell carcinoma | High NK-3R expression. NKB expression was not observed. B-222,200 inhibits tumorigenesis | [21,156] |
| Phaeochromocytoma | Human phaeochromocytoma extract: NKA expression. NKB was observed in one of 10 phaeochromocytomas. | [31,157] |
| Schwannoma-derived cells | NKA expression. | [158] |
| Small bowel neuroendocrine tumors | Plasma NKA level: biomarker. High levels are associated with poor prognosis. Improved prognosis by lowering plasma NKA level. Ileal metastatic carcinoid: circulating NKA increase. NKA, NKA3–10, and NKA4–10 expression. | [159,160,161,162,163,164,165] |
| Uterine leiomyomata | NK-2R mRNA upregulation. High NKB/NK-3R expression. NK-3R activation: nuclear translocation affecting gene expression/chromatin structure. | [91,166] |
| Tumor | AM/AM2/AMY/CGRP | References |
|---|---|---|
| Acute myeloid leukemia | AM, CLR, RAMP 2/3 expression. A high AM level is associated with low overall survival/disease-free survival. AM/AM22–52 regulates cell growth. AM expression associated with genes related to immunosuppression resistance. AM correlates with adverse outcomes. Targeting calcitonin receptor-like receptors prevents relapse. Calcitonin gene methylation pattern: independent prognostic factor. High calcitonin receptor expression: poor prognosis and correlates with chemotherapy resistance. Olcegepant decreases leukemic burden/stem cell characteristics. MK0974 promotes apoptosis. | [136,182,183,184,185,186] |
| Adrenocortical tumor | AM is synthesized/released from adrenocortical tumors and phaeochromocytomas. Phaeochromocytomas: higher AM/receptor mRNA expression. Phaeochromocytomas and adenomas: AM2 expression. AM blocks phaeochromocytoma cell proliferation. AM level as a biomarker. Adrenal tumors: AM2, CLR, RAMP1/RAMP2/RAMP3 mRNA expression. Phaeochromocytomas: high CGRP tissue level. | [187,188,189,190,191,192] |
| Bladder cancer | High AM level. AM knockdown promotes apoptosis. AM knockdown/cisplatin combination decreases tumor growth. | [193] |
| Breast cancer | AM expression associated with axillary lymph node metastasis. RAMP3: involved in metastasis. Tumor cells overexpressing AM: potential angiogenic increase/less apoptotic mechanisms. Tumor cells expressing/releasing AM: favor cell proliferation, breast cancer bone metastasis, and angiogenesis. AM is a tumor survival factor. Triple-negative breast cancer samples: AM expression decreased; this low expression is related to poor prognosis and increased risk of recurrence/metastasis. AM22–52 disrupts tumor vasculature, decreases tumor cell proliferation, and induces apoptosis. Plasma AM2 level associated with poor patient outcomes. AM2 expression increased: correlated with Ki67 expression/lymph node metastasis. AM2 promotes cancer cells’ growth, migration, and invasion; these actions were blocked with anti-AM2 antibodies. CGRP expression increased. CGRP is involved in metastasis. | [99,194,195,196,197,198,199,200,201] |
| Choriocarcinoma | AM mRNA expression. | [202] |
| Colon cancer | CLR and RAMP2/3 expressions. High levels of AM, CLR, and RAMP2/3 are correlated with lymph nodes and distant metastasis. High AM level is related to low disease-free survival. Higher AM level and AM mRNA expression. AM promotes tumor cell proliferation/invasion and antiapoptotic effect. AM level associated with clinical survival rate/cancer stage. Knockdown AM promotes apoptosis/blocks angiogenesis. AM expression is associated with vascular endothelial growth factor and hypoxia-inducible factor-1α. AM positive modulator (145425) decreases the number of tumors. Higher mRNA pre-proAM, pre-proAM2, CLR, RAMP2/3, MMP-9, and VEGF-A expression. Positive correlation between MMP-9 gene expression and pre-proAM, but not pre-proAM2. | [179,203,204,205,206,207,208] |
| Cutaneous nerve neuromas | Saphenous nerve neuromas: CGRP release from nerve fibers. | [209] |
| Endometrial cancer | AM favors angiogenesis/tumor growth and blocks tumor cell death. AM level increases from normal, simple, or complex hyperplasia with or without atypia to grade 1 adenocarcinoma. | [101,210,211,212] |
| Ewing sarcoma | CGRP expression. CGRP promotes the proliferation of cancer cells. | [213] |
| Gastric cancer | Tumor-derived AM promotes mast cell degranulation, as well as favors tumor cell proliferation, and the apoptosis blockade in cancer cells. | [214] |
| Glioma | AM favors mitogenesis in tumor cells and exerts angiogenic/antiapoptotic effects. AM mRNA is associated with tumor type and grade. c-Jun/JNK pathway is involved in the growth regulatory activity mediated by AM. AM expression is upregulated in temozolomide-resistant glioma samples: miR-1297 targets AM, blocks its expression, and sensitizes tumor cells to temozolomide treatment. AM2 expression is increased and correlated with higher-grade gliomas. AM2 increases the invasive capacity of tumor cells and improves tumor blood. AMY promotes the release of inflammatory cytokines from tumor cells. The signal transducer and STAT-3 in astroglioma control AM expression. AM promotes the migration of astroglioma cells. | [215,216,217,218,219,220,221,222,223] |
| Head and neck squamous cell carcinoma | Peripheral nerve terminals release CGRP, which exerts a paracrine action on tumor cells. CGRP links perineural invasion and lymph node metastasis. Pre-operative plasma CGRP level: lymph node metastasis predictor. | [224,225] |
| Liver cancer | CLR and RAMP2/3 expression. High AM level is related to increased intrahepatic metastasis. Higher AM mRNA levels in tumor tissues than in adjacent nontumor tissues. AM mediates the epithelial–mesenchymal transition and promotes tumor cell growth. Knockdown AM expression promotes apoptotic mechanisms and, combined with cisplatin, decreases tumor growth. Microvessel density and mRNA AM/erythropoietin receptor levels are higher in hepatocellular carcinoma than in nontumor tissues: both levels are correlated with tumor metastasis, pathological differentiation, and capsule invasion. AM is associated with N-cadherin intensity, vascular invasion, and poor prognosis. AM level as a prognostic factor. High AM2 mRNA expression even in early stages. AM2 increases tumor cell proliferation and survival, and is involved in angiogenesis: AM217–47 blocked this proliferation. AMY binding site expression. | [226,227,228,229,230,231,232,233,234] |
| Lung cancer | AM expression does not correlate with survival, cancer stage, or tumor differentiation. AM contributes to the carcinogenicity of tobacco-activated aryl hydrocarbon receptor products. CGRP gene expression. | [235,236,237,238] |
| Melanoma | Tumor-associated macrophages, through AM, favor melanoma growth and angiogenesis. The number of tumor-associated macrophages (express/release AM) in the tumor microenvironment is correlated with poor prognosis. Tumor-associated macrophages favor the migration of endothelial cells and increase tumor cell growth. | [239] |
| Nasopharyngeal carcinoma | AM level as a biomarker for predicting prognosis. | [240] |
| Neuroblastoma | AM receptor expression. AM mRNA expression is associated with tumor differentiation. | [241,242] |
| Neuroendocrine tumors | High plasma and tissue AM expression: predictive factors for tumor progression and worsened prognosis. | [243] |
| Oropharyngeal squamous cell carcinoma | Jumonji domain-containing 1A, H3K9me1/2, and AM expression: predictor markers for progression and prognosis. JMJD1A gene target AM favors tumorigenesis/cell growth. | [244] |
| Osteosarcoma | AM expression is associated with metastasis degree/malignancy. AM overexpression. AM exerts an antiapoptotic effect. | [245,246] |
| Ovarian cancer | High AM level is related to tumor stage. AM gene is correlated with histological grade, lymph node metastasis, and prognosis but not with disease stage, histological subtype, residual tumor mass after initial surgery, and patient’s age at diagnosis. Tumor cells express AM mRNA for both ligand/receptor. AM is involved in tumor progression/cell migration. AM gene silencing blocks cell proliferation and increases tumor cell chemosensitivity. Cancer patients with high AM expression show larger residual size of tumors, shorter disease-free/overall survival time, and higher metastasis incidence. AM as biomarker to evaluate prognosis/malignant potential. AM is correlated with expressions of HIF-1α, VEGF, or microvessel density. AM favors angiogenesis. | [247,248,249,250,251,252] |
| Pancreatic cancer | High AM levels are associated with disease-free survival decrease. Higher AM plasma level. AM level is higher in cancer patients with diabetes than those without diabetes. Insulinoma: circulating AM increased. AM and its receptor are expressed. AM is involved in tumor cell proliferation, migration, invasion, metastasis, and angiogenesis. AM mRNA/protein expressions are increased. AM as a tumor marker. AM receptor silencing blocks AM-induced cell growth and invasion. AM antagonists decrease tumor cell growth/blood vessel diameter. Selective RAMP2 activation/RAMP3 inhibition: tumor metastasis suppression. AM2 as tumor angiogenic factor. AM2 level: poorer survival predictor. AM2 is a biomarker predicting survival. Insulinomas express AMY. Plasma AMY levels are increased in nondiabetic patients with cancer; this level is low in patients with diabetes. CALCA (αCGRP) and CALCB (βCGRP) methylation increases. | [253,254,255,256,257,258,259,260,261,262,263,264] |
| Pituitary adenoma | AM expression is decreased in anterior pituitary tumors. | [265] |
| Prostate cancer | AM is expressed in prostate carcinomas. A high AM level is associated with a high Gleason score. Plasma AM2 level is associated with Gleason’s score, tumor node metastasis, and 5 year metastasis. AM overexpression inhibits tumor cell growth and dysregulates genes involved in apoptosis, cell cycle, extracellular matrix, cell adhesion, and cytoskeleton. AM promotes human prostate growth via the AM2 receptor (CLR/RAMP3) subtype. AM blocks apoptosis in specific tumor cells. AM, upon androgen ablation, is involved in hormone-independent tumor growth, lymphangiogenesis, and neoangiogenesis. AM promotes cancer cell migration and invasion. AM2 is involved in cancer cell migration/angiogenesis. Higher plasma AM2 level in patients with prostate cancer. Patients with a Gleason score ≥7, unconfined organ, seminal vesicle invasion, tumor node metastasis stage T2, positive lymph node, or extra-prostatic extension show high AM2. AM2 as a prognostic, predictive biomarker for 5 year metastasis and 5 year progression. CGRP increases the invasive/migratory capacity of tumor cells. CGRP serum level correlates with cancer progression. Higher serum CGRP level is associated with higher histological grade/clinical stages. CGRP receptor mediates metastasis. CGRP promotes tumor growth which is blocked with CGRP antagonists. | [180,266,267,268,269,270,271,272,273,274,275] |
| Renal carcinoma | AM, CLR, and RAMP2/3 expression. High CLR level is associated with high tumor grade. AM and AM mRNA expression. AM mRNA expression is correlated with VEGF-A mRNA. AM, via CLR/RAMP2 and CLR/RAMP3 receptors, promotes cell proliferation, migration, and invasion. High AM mRNA level is related to increased risk of relapse. | [276,277,278] |
| Thymic lymphomas | Pramlintide promotes tumor regression. AMY blocks glycolysis, promotes reactive oxygen species formation, and induces apoptosis. | [279] |
| Thyroid cancer | AM2 expression is increased in obese patients with thyroid cancer, showing locoregional recurrence, a high prevalence of lymph node metastasis, and larger tumor size. High circulating AM2/tumor cell AM2 expression levels are associated with aggressive pathological parameters. AM2 as a biomarker for predicting thyroid tumor progression. Medullary thyroid carcinoma: high AMY levels. Plasma AMY/insulin levels are correlated in medullary thyroid carcinoma. CGRP gene expression. Plasma CGRP level marker for medullary thyroid carcinoma. | [237,280,281,282,283] |
| Uterine cervical carcinoma | High AM and AM mRNA expression. AM is involved in promoting malignant progression and in selecting carcinoma cells resistant to apoptosis. | [284,285] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez, M.L.; Rodríguez, F.D.; Coveñas, R. Peptidergic Systems and Cancer: Focus on Tachykinin and Calcitonin/Calcitonin Gene-Related Peptide Families. Cancers 2023, 15, 1694. https://doi.org/10.3390/cancers15061694
Sánchez ML, Rodríguez FD, Coveñas R. Peptidergic Systems and Cancer: Focus on Tachykinin and Calcitonin/Calcitonin Gene-Related Peptide Families. Cancers. 2023; 15(6):1694. https://doi.org/10.3390/cancers15061694
Chicago/Turabian StyleSánchez, Manuel Lisardo, Francisco D. Rodríguez, and Rafael Coveñas. 2023. "Peptidergic Systems and Cancer: Focus on Tachykinin and Calcitonin/Calcitonin Gene-Related Peptide Families" Cancers 15, no. 6: 1694. https://doi.org/10.3390/cancers15061694
APA StyleSánchez, M. L., Rodríguez, F. D., & Coveñas, R. (2023). Peptidergic Systems and Cancer: Focus on Tachykinin and Calcitonin/Calcitonin Gene-Related Peptide Families. Cancers, 15(6), 1694. https://doi.org/10.3390/cancers15061694