
Bispecific Antibodies in Multiple Myeloma: Opportunities to Enhance Efficacy and Improve Safety

Abstract
:Simple Summary
Abstract
1. Introduction
2. Overview of Bispecific Antibodies in Myeloma
3. Improving Efficacy
4. Cyclophosphamide
5. IMiDs
6. Checkpoint Inhibitors (CPI)
7. Earlier Use within the Treatment Paradigm
8. Improving Antigen Availability
9. Safety
9.1. Infection
9.2. CRS
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowan, A.J.; Allen, C.; Barac, A.; Basaleem, H.; Bensenor, I.; Curado, M.P.; Foreman, K.; Gupta, R.; Harvey, J.; Hosgood, H.D.; et al. Global Burden of Multiple Myeloma: A Systematic Analysis for the Global Burden of Disease Study 2016. JAMA Oncol. 2018, 4, 1221–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyle, R.A.; Gertz, M.A.; Witzig, T.E.; Lust, J.A.; Lacy, M.Q.; Dispenzieri, A.; Fonseca, R.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; et al. Review of 1027 patients with newly diagnosed multiple myeloma. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2003; Volume 78, pp. 21–33. [Google Scholar] [CrossRef]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- Avet-Loiseau, H. Ultra high-risk myeloma. Hematol. Am. Soc. Hematol. Educ. Program 2010, 2010, 489–493. [Google Scholar] [CrossRef] [Green Version]
- Weinhold, N.; Ashby, C.; Rasche, L.; Chavan, S.S.; Stein, C.; Stephens, O.W.; Tytarenko, R.; Bauer, M.A.; Meissner, T.; Deshpande, S.; et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016, 128, 1735–1744. [Google Scholar] [CrossRef]
- Weinhold, N.; Heuck, C.J.; Rosenthal, A.; Thanendrarajan, S.; Stein, C.K.; Van Rhee, F.; Zangari, M.; Hoering, A.; Tian, E.; Davies, F.E.; et al. Clinical value of molecular subtyping multiple myeloma using gene expression profiling. Leukemia 2016, 30, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019, 33, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, U.H.; Cornell, R.F.; Lakshman, A.; Gahvari, Z.J.; McGehee, E.; Jagosky, M.H.; Gupta, R.; Varnado, W.; Fiala, M.A.; Chhabra, S.; et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia 2019, 33, 2266–2275. [Google Scholar] [CrossRef]
- Dahlén, E.; Veitonmäki, N.; Norlén, P. Bispecific antibodies in cancer immunotherapy. Ther. Adv. Vaccines Immunother. 2018, 6, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Guy, D.G.; Uy, G.L. Bispecific Antibodies for the Treatment of Acute Myeloid Leukemia. Curr. Hematol. Malig. Rep. 2018, 13, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Wang, Z.; Hao, M.; Li, J. Bispecific antibodies and their applications. J. Hematol. Oncol. 2015, 8, 130. [Google Scholar] [CrossRef] [Green Version]
- Przepiorka, D.; Ko, C.W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef] [Green Version]
- Swan, D.; Routledge, D.; Harrison, S. The evolving status of immunotherapies in multiple myeloma: The future role of bispecific antibodies. Br. J. Haematol. 2022, 196, 488–506. [Google Scholar] [CrossRef] [PubMed]
- Coquery, C.M.; Erickson, L.D. Regulatory roles of the tumor necrosis factor receptor BCMA. Crit. Rev. Immunol. 2012, 32, 287–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gras, M.-P.; Laâbi, Y.; Linares-Cruz, G.; Blondel, M.-O.; Rigaut, J.-P.; Brouet, J.-C.; Leca, G.; Haguenauer-Tsapis, R.; Tsapis, A. BCMAp: An integral membrane protein in the Golgi apparatus of human mature B lymphocytes. Int. Immunol. 1995, 7, 1093–1106. [Google Scholar] [CrossRef]
- Laabi, Y.; Gras, M.P.; Brouet, J.C.; Berger, R.; Larsen, C.J.; Tsapis, A. The BCMA gene, preferentially expressed during B lymphoid maturation, is bidirectionally transcribed. Nucleic. Acids Res. 1994, 22, 1147–1154. [Google Scholar] [CrossRef]
- Madry, C.; Laabi, Y.; Callebaut, I.; Roussel, J.; Hatzoglou, A.; Le Coniat, M.; Mornon, J.P.; Berger, R.; Tsapis, A. The characterization of murine BCMA gene defines it as a new member of the tumor necrosis factor receptor superfamily. Int. Immunol. 1998, 10, 1693–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzoglou, A.; Roussel, J.; Bourgeade, M.F.; Rogier, E.; Madry, C.; Inoue, J.; Devergne, O.; Tsapis, A. TNF receptor family member BCMA (B cell maturation) associates with TNF receptor-associated factor (TRAF) 1, TRAF2, and TRAF3 and activates NF-kappa B, elk-1, c-Jun N-terminal kinase, and p38 mitogen-activated protein kinase. J. Immunol. 2000, 165, 1322–1330. [Google Scholar] [CrossRef] [Green Version]
- Rennert, P.; Schneider, P.; Cachero, T.G.; Thompson, J.; Trabach, L.; Hertig, S.; Holler, N.; Qian, F.; Mullen, C.; Strauch, K.; et al. A soluble form of B cell maturation antigen, a receptor for the tumor necrosis factor family member APRIL, inhibits tumor cell growth. J. Exp. Med. 2000, 192, 1677–1684. [Google Scholar] [CrossRef] [Green Version]
- Schneider, P.; MacKay, F.; Steiner, V.; Hofmann, K.; Bodmer, J.L.; Holler, N.; Ambrose, C.; Lawton, P.; Bixler, S.; Acha-Orbea, H.; et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J. Exp. Med. 1999, 189, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Ghermezi, M.; Li, M.; Vardanyan, S.; Harutyunyan, N.M.; Gottlieb, J.; Berenson, A.; Spektor, T.M.; Andreu-Vieyra, C.; Petraki, S.; Sanchez, E.; et al. Serum B-cell maturation antigen: A novel biomarker to predict outcomes for multiple myeloma patients. Haematologica 2017, 102, 785–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, E.; Li, M.; Kitto, A.; Li, J.; Wang, C.S.; Kirk, D.T.; Yellin, O.; Nichols, C.M.; Dreyer, M.P.; Ahles, C.P.; et al. Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br. J. Haematol. 2012, 158, 727–738. [Google Scholar] [CrossRef]
- Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/tecvayli (accessed on 16 January 2023).
- Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-teclistamab-cqyv-relapsed-or-refractory-multiple-myeloma (accessed on 16 January 2023).
- Moreau, P.; Garfall, A.L.; van de Donk, N.W.C.J.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Nooka, A.K.; Martin, T.; Rosinol, L.; Chari, A.; et al. Teclistamab in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2022, 387, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Bahlis, N.; Tomasson, M.; Mohty, M.; Niesvizky, R.; Nooka, A.; Manier, S.; Maisel, C.; Jethava, Y.; Martinez-Lopez, J.; Prince, H.; et al. Efficacy and Safety of Elranatamab in Patients with Relapsed/Refractory Multiple Myeloma Naïve to B-Cell Maturation Antigen (BCMA)-Directed Therapies: Results from Cohort a of the Magnetismm-3 Study. Blood 2022, 140, 391–393. [Google Scholar] [CrossRef]
- Dosani, T.; Carlsten, M.; Maric, I.; Landgren, O. The cellular immune system in myelomagenesis: NK cells and T cells in the development of MM and their uses in immunotherapies. Blood Cancer J. 2015, 5, e321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhodapkar, M.V.; Krasovsky, J.; Osman, K.; Geller, M.D. Vigorous premalignancy-specific effector T cell response in the bone marrow of patients with monoclonal gammopathy. J. Exp. Med. 2003, 198, 1753–1757. [Google Scholar] [CrossRef] [Green Version]
- Park, J.J.; Omiya, R.; Matsumura, Y.; Sakoda, Y.; Kuramasu, A.; Augustine, M.M.; Yao, S.; Tsushima, F.; Narazaki, H.; Anand, S.; et al. B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood 2010, 116, 1291–1298. [Google Scholar] [CrossRef]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Feyler, S.; Scott, G.B.; Parrish, C.; Jarmin, S.; Evans, P.; Short, M.; McKinley, K.; Selby, P.J.; Cook, G. Tumour cell generation of inducible regulatory T-cells in multiple myeloma is contact-dependent and antigen-presenting cell-independent. PLoS ONE 2012, 7, e35981. [Google Scholar] [CrossRef] [Green Version]
- Dahlhoff, J.; Manz, H.; Steinfatt, T.; Delgado-Tascon, J.; Seebacher, E.; Schneider, T.; Wilnit, A.; Mokhtari, Z.; Tabares, P.; Böckle, D.; et al. Transient regulatory T-cell targeting triggers immune control of multiple myeloma and prevents disease progression. Leukemia 2022, 36, 790–800. [Google Scholar] [CrossRef]
- Serafini, P.; Meckel, K.; Kelso, M.; Noonan, K.; Califano, J.; Koch, W.; Dolcetti, L.; Bronte, V.; Borrello, I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 2006, 203, 2691–2702. [Google Scholar] [CrossRef]
- Philipp, N.; Kazerani, M.; Nicholls, A.; Vick, B.; Wulf, J.; Straub, T.; Scheurer, M.; Muth, A.; Hänel, G.; Nixdorf, D.; et al. T-cell exhaustion induced by continuous bispecific molecule exposure is ameliorated by treatment-free intervals. Blood 2022, 140, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Ahn, S.; Lee, H.; Leblay, N.; Friedrich, M.; Maity, R.; Tilmont, R.; Barakat, E.; Raab, M.S.; Bahlis, N.J. Dysfunctional Hyper-Expanded Clonotypes and Lack of TCR Clonal Replacement Predict Resistance to T Cell Engagers in Multiple Myeloma. Blood 2022, 140, 2093–2094. [Google Scholar] [CrossRef]
- Cortes-Selva, D.; Casneuf, T.; Vishwamitra, D.; Stein, S.; Perova, T.; Skerget, S.; Ramos, E.; van Steenbergen, L.; De Maeyer, D.; Boominathan, R.; et al. Teclistamab, a B-Cell Maturation Antigen (BCMA) x CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma (RRMM): Correlative Analyses from MajesTEC-1. Blood 2022, 140, 241–243. [Google Scholar] [CrossRef]
- Naicker, S.D.; Feerick, C.L.; Lynch, K.; Swan, D.; McEllistrim, C.; Henderson, R.; Leonard, N.A.; Treacy, O.; Natoni, A.; Rigalou, A.; et al. Cyclophosphamide alters the tumor cell secretome to potentiate the anti-myeloma activity of daratumumab through augmentation of macrophage-mediated antibody dependent cellular phagocytosis. Oncoimmunology 2021, 10, 1859263. [Google Scholar] [CrossRef] [PubMed]
- Swan, D.; Henderson, R.; McEllistrim, C.; Naicker, S.D.; Quinn, J.; Cahill, M.R.; Mykytiv, V.; Lenihan, E.; Mulvaney, E.; Nolan, M.; et al. CyBorD-DARA in Newly Diagnosed Transplant-Eligible Multiple Myeloma: Results from the 16-BCNI-001/CTRIAL-IE 16-02 Study Show High Rates of MRD Negativity at End of Treatment. Clin. Lymphoma Myeloma Leuk. 2022, 22, 847–852. [Google Scholar] [CrossRef]
- Arnold, H.; Bourseaux, F.; Brock, N. Chemotherapeutic action of a cyclic nitrogen mustard phosphamide ester (B 518-ASTA) in experimental tumours of the rat. Nature 1958, 181, 931. [Google Scholar] [CrossRef]
- Swan, D.; Gurney, M.; Krawczyk, J.; Ryan, A.E.; O’Dwyer, M. Beyond DNA Damage: Exploring the Immunomodulatory Effects of Cyclophosphamide in Multiple Myeloma. Hemasphere 2020, 4, e350. [Google Scholar] [CrossRef] [PubMed]
- Rollinghoff, M.; Starzinski-Powitz, A.; Pfizenmaier, K.; Wagner, H. Cyclophosphamide-sensitive T lymphocytes suppress the in vivo generation of antigen-specific cytotoxic T lymphocytes. J. Exp. Med. 1977, 145, 455–459. [Google Scholar] [CrossRef]
- Zhao, J.; Cao, Y.; Lei, Z.; Yang, Z.; Zhang, B.; Huang, B. Selective depletion of CD4+CD25+Foxp3+ regulatory T cells by low-dose cyclophosphamide is explained by reduced intracellular ATP levels. Cancer Res. 2010, 70, 4850–4858. [Google Scholar] [CrossRef] [Green Version]
- Heylmann, D.; Bauer, M.; Becker, H.; van Gool, S.; Bacher, N.; Steinbrink, K.; Kaina, B. Human CD4+CD25+ regulatory T cells are sensitive to low dose cyclophosphamide: Implications for the immune response. PLoS ONE 2013, 8, e83384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavoni, G.; Sistigu, A.; Valentini, M.; Mattei, F.; Sestili, P.; Spadaro, F.; Sanchez, M.; Lorenzi, S.; D’Urso, M.T.; Belardelli, F.; et al. Cyclophosphamide Synergizes with Type I Interferons through Systemic Dendritic Cell Reactivation and Induction of Immunogenic Tumor Apoptosis. Cancer Res. 2011, 71, 768–778. [Google Scholar] [CrossRef] [Green Version]
- Bryant, C.; Suen, H.; Brown, R.; Yang, S.; Favaloro, J.; Aklilu, E.; Gibson, J.; Ho, P.J.; Iland, H.; Fromm, P.; et al. Long-term survival in multiple myeloma is associated with a distinct immunological profile, which includes proliferative cytotoxic T-cell clones and a favourable Treg/Th17 balance. Blood Cancer J. 2013, 3, e148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meermeier, E.W.; Welsh, S.J.; Sharik, M.E.; Du, M.T.; Garbitt, V.M.; Riggs, D.L.; Shi, C.X.; Stein, C.K.; Bergsagel, M.; Chau, B.; et al. Tumor burden limits bispecific antibody efficacy through T cell exhaustion averted by concurrent cytotoxic therapy. Blood Cancer Discov. 2021, 2, 354–369. [Google Scholar] [CrossRef] [PubMed]
- Kronke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Krämer, I.; Engelhardt, M.; Fichtner, S.; Neuber, B.; Medenhoff, S.; Bertsch, U.; Hillengass, J.; Raab, M.-S.; Hose, D.; Ho, A.D.; et al. Lenalidomide enhances myeloma-specific T-cell responses in vivo and in vitro. Oncoimmunology 2016, 5, e1139662. [Google Scholar] [CrossRef] [Green Version]
- Alsina, M.; Becker, P.S.; Zhong, X.; Adams, A.; Hari, P.; Rowley, S.; Stadtmauer, E.A.; Vesole, D.H.; Logan, B.; Weisdorf, D.; et al. Lenalidomide maintenance for high-risk multiple myeloma after allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transpl. 2014, 20, 1183–1189. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.J.; Minnema, M.C.; Lee, H.C.; Spencer, A.; Kapoor, P.; Madduri, D.; Larsen, J.; Ailawadhi, S.; Kaufman, J.L.; Raab, M.S.; et al. A Phase 1 First in Human (FIH) Study of AMG 701, an Anti-B-Cell Maturation Antigen (BCMA) Half-Life Extended (HLE) BiTE® (bispecific T-cell engager) Molecule, in Relapsed/Refractory (RR) Multiple Myeloma (MM). Blood 2020, 136, 28–29. [Google Scholar] [CrossRef]
- Cho, S.F.; Lin, L.; Xing, L.; Li, Y.; Wen, K.; Yu, T.; Hsieh, P.A.; Munshi, N.; Wahl, J.; Matthes, K.; et al. The immunomodulatory drugs lenalidomide and pomalidomide enhance the potency of AMG 701 in multiple myeloma preclinical models. Blood Adv. 2020, 4, 4195–4207. [Google Scholar] [CrossRef]
- Richardson, P.G.; Trudel, S.; Quach, H.; Popat, R.; Lonial, S.; Orlowski, R.Z.; Kim, K.; Mateos, M.-V.; Pawlyn, C.; Ramasamy, K.; et al. Mezigdomide (CC-92480), a Potent, Novel Cereblon E3 Ligase Modulator (CELMoD), Combined with Dexamethasone (DEX) in Patients (pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Preliminary Results from the Dose-Expansion Phase of the CC-92480-MM-001 Trial. Blood 2022, 140, 1366–1368. [Google Scholar] [CrossRef]
- Lonial, S.; Popat, R.; Hulin, C.; Jagannath, S.; Oriol, A.; Richardson, P.G.; Facon, T.; Weisel, K.; Larsen, J.T.; Minnema, M.C.; et al. Iberdomide plus dexamethasone in heavily pretreated late-line relapsed or refractory multiple myeloma (CC-220-MM-001): A multicentre, multicohort, open-label, phase 1/2 trial. Lancet Haematol 2022, 9, e822–e832. [Google Scholar] [CrossRef]
- Seckinger, A.; Delgado, J.A.; Moser, S.; Moreno, L.; Neuber, B.; Grab, A.; Lipp, S.; Merino, J.; Prosper, F.; Emde, M.; et al. Target Expression, Generation, Preclinical Activity, and Pharmacokinetics of the BCMA-T Cell Bispecific Antibody EM801 for Multiple Myeloma Treatment. Cancer Cell 2017, 31, 396–410. [Google Scholar] [CrossRef] [Green Version]
- Paiva, B.; Gaffney, B.; Burnett, K.; Castiglioni, P.; Angelo, M.; Pierce, D.W.; Boss, I.W. Synergistic Antitumor Activity of Alnuctamab (ALNUC.; BMS-986349; CC-93269), a BCMA 2 + 1 T Cell Engager (TCE), and Celmod Agents in Multiple Myeloma (MM) Preclinical Models. Blood 2022, 140 (Suppl. 1), 7054–7055. [Google Scholar] [CrossRef]
- Searle, E.; Quach, H.; Wong, S.W.; Costa, L.J.; Hulin, C.; Janowski, W.; Berdeja, J.; Anguille, S.; Matous, J.V.; Touzeau, C.; et al. Teclistamab in Combination with Subcutaneous Daratumumab and Lenalidomide in Patients with Multiple Myeloma: Results from One Cohort of MajesTEC-2, a Phase1b, Multicohort Study. Blood 2022, 140 (Suppl. 1), 394–396. [Google Scholar] [CrossRef]
- Wang, L.; Wang, H.; Chen, H.; Wang, W.D.; Chen, X.Q.; Geng, Q.R.; Xia, Z.J.; Lu, Y. Serum levels of soluble programmed death ligand 1 predict treatment response and progression free survival in multiple myeloma. Oncotarget 2015, 6, 41228–41236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feucht, J.; Kayser, S.; Gorodezki, D.; Hamieh, M.; Döring, M.; Blaeschke, F.; Schlegel, P.; Bösmüller, H.; Quintanilla-Fend, L.; Ebinger, M.; et al. T-cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 on leukemic blasts. Oncotarget 2016, 7, 76902–76919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhnke, T.; Krupka, C.; Tischer, J.; Knösel, T.; Subklewe, M. Increase of PD-L1 expressing B-precursor ALL cells in a patient resistant to the CD19/CD3-bispecific T cell engager antibody blinatumomab. J. Hematol. Oncol. 2015, 8, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Stagg, N.J.; Johnston, J.; Harris, M.J.; Menzies, S.A.; DiCara, D.; Clark, V.; Hristopoulos, M.; Cook, R.; Slaga, D.; et al. Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing. Cancer Cell 2017, 31, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Krönke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 775–783. [Google Scholar] [CrossRef]
- Verkleij, C.; Broekmans, M.; Wong, A.; Zweegman, S.; Verona, R.; Adams, H.; Mutis, T.; van de Donk, N. Mechanisms of Resistance and Determinants of Response of the GPRC5D-Targeting T-Cell Redirecting Bispecific Antibody JNJ-7564 in Multiple Myeloma. Blood 2020, 136, 8–9. [Google Scholar] [CrossRef]
- Badros, A.; Hyjek, E.; Ma, N.; Lesokhin, A.; Dogan, A.; Rapoport, A.P.; Kocoglu, M.; Lederer, E.; Philip, S.; Milliron, T.; et al. Pembrolizumab, pomalidomide, and low-dose dexamethasone for relapsed/refractory multiple myeloma. Blood 2017, 130, 1189–1197. [Google Scholar] [CrossRef] [Green Version]
- Usmani, S.Z.; Schjesvold, F.; Rocafiguera, A.O.; Karlin, L.; Rifkin, R.M.; Yimer, H.A.; LeBlanc, R.; Takezako, N.; McCroskey, R.D.; Suzuki, K.; et al. A phase 3 randomized study of pembrolizumab (pembro) plus lenalidomide (len) and low-dose dexamethasone (Rd) versus Rd for newly diagnosed and treatment-naive multiple myeloma (MM): KEYNOTE-185. J. Clin. Oncol. 2018, 36, 8010. [Google Scholar] [CrossRef]
- Mateos, M.V.; Blacklock, H.; Schjesvold, F.; Rocafiguera, A.O.; Simpson, D.; George, A.; Goldschmidt, H.; Larocca, A.; Sherbenou, D.W.; Avivi, I.; et al. A phase 3 randomized study of pembrolizumab (Pembro) plus pomalidomide (Pom) and dexamethasone (Dex) for relapsed/refractory multiple myeloma (RRMM): KEYNOTE-183. J. Clin. Oncol. 2018, 36, 8021. [Google Scholar] [CrossRef]
- Chung, A.; Huang, C.-Y.; Martin, T.; Wolf, J.L.; Wong, S.W.; Shah, N. Phase II Study of Pembrolizumab in Multiple Myeloma Patients Relapsing after or Refractory to Anti-BCMA CAR-T Therapies. Blood 2022, 140 (Suppl. 1), 12651–12652. [Google Scholar] [CrossRef]
- Chong, E.A.; Alanio, C.; Svoboda, J.; Nasta, S.D.; Landsburg, D.J.; Lacey, S.F.; Ruella, M.; Bhattacharyya, S.; Wherry, E.J.; Schuster, S.J. Pembrolizumab for B-cell lymphomas relapsing after or refractory to CD19-directed CAR T-cell therapy. Blood 2022, 139, 1026–1038. [Google Scholar] [CrossRef]
- Vrohlings, M.; Müller, J.; Jungmichel, S.; Senn, D.; Howald, A.B.; Schleier, T.; Scheifele, F.; Wendelspiess, S.; Richle, P.; Merten, H.; et al. Preclinical Assessment of CDR101—A BCMAxCD3xPD-L1 Trispecific Antibody with Superior Anti-Tumor Efficacy. Blood 2021, 138, 1583. [Google Scholar] [CrossRef]
- Danhof, S.; Schreder, M.; Knop, S.; Rasche, L.; Strifler, S.; Löffler, C.; Gogishvili, T.; Einsele, H.; Hudecek, M. Expression of programmed death-1 on lymphocytes in myeloma patients is lowered during lenalidomide maintenance. Haematologica 2018, 103, e126–e129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Pruteanu, I.; Cohen, A.D.; Garfall, A.L.; Milone, M.C.; Tian, L.; Gonzalez, V.E.; Gill, S.; Frey, N.V.; Barrett, D.M.; et al. Identification and Validation of Predictive Biomarkers to CD19- and BCMA-Specific CAR T-Cell Responses in CAR T-Cell Precursors. Blood 2019, 134, 622. [Google Scholar] [CrossRef]
- Dancy, E.; Garfall, A.L.; Cohen, A.D.; Fraietta, J.A.; Davis, M.; Levine, B.L.; Siegel, D.L.; Stadtmauer, E.A.; Vogl, D.T.; Waxman, A.; et al. Clinical Predictors of T Cell Fitness for CAR T Cell Manufacturing and Efficacy in Multiple Myeloma. Blood 2018, 132 (Suppl. 1), 1886. [Google Scholar] [CrossRef]
- Chung, D.J.; Pronschinske, K.B.; Shyer, J.A.; Wu, V.; Hassoun, H.; Landau, H.; Lendvai, N.; Lesokhin, A.M.; Koehne, G.; Giralt, S.A.; et al. Immune Reconstitution after Autologous Stem Cell Transplantation for Multiple Myeloma. Biol. Blood Marrow Transplant. 2014, 20, S60–S62. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Yu, T.; Lin, L.; Xing, L.; Cho, S.-F.; Wen, K.; Aardalen, K.; Oka, A.; Lam, J.; Daley, M.; et al. γ-secretase inhibitors augment efficacy of BCMA-targeting bispecific antibodies against multiple myeloma cells without impairing T-cell activation and differentiation. Blood Cancer J. 2022, 12, 118. [Google Scholar] [CrossRef]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.R.; et al. γ-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef] [PubMed]
- Samur, M.K.; Fulciniti, M.; Aktas Samur, A.; Bazarbachi, A.H.; Tai, Y.-T.; Prabhala, R.; Alonso, A.; Sperling, A.S.; Campbell, T.; Petrocca, F.; et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat. Commun. 2021, 12, 868. [Google Scholar] [CrossRef] [PubMed]
- Da Vià, M.C.; Dietrich, O.; Truger, M.; Arampatzi, P.; Duell, J.; Heidemeier, A.; Zhou, X.; Danhof, S.; Kraus, S.; Chatterjee, M.; et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat. Med. 2021, 27, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Truger, M.S.; Duell, J.; Zhou, X.; Heimeshoff, L.; Ruckdeschel, A.; John, M.; Riedel, A.; Hüper, S.; Peter, J.; Walter, W.; et al. Single- and double-hit events in genes encoding immune targets before and after T cell-engaging antibody therapy in MM. Blood Adv. 2021, 5, 3794–3798. [Google Scholar] [CrossRef] [PubMed]
- Atamaniuk, J.; Gleiss, A.; Porpaczy, E.; Kainz, B.; Grunt, T.W.; Raderer, M.; Hilgarth, B.; Drach, J.; Ludwig, H.; Gisslinger, H.; et al. Overexpression of G protein-coupled receptor 5D in the bone marrow is associated with poor prognosis in patients with multiple myeloma. Eur. J. Clin. Investig. 2012, 42, 953–960. [Google Scholar] [CrossRef]
- Franco, A.; Damdinsuren, B.; Ise, T.; Dement-Brown, J.; Li, H.; Nagata, S.; Tolnay, M. Human Fc receptor-like 5 binds intact IgG via mechanisms distinct from those of Fc receptors. J. Immunol. 2013, 190, 5739–5746. [Google Scholar] [CrossRef] [Green Version]
- An, G.; Jiang, H.; Acharya, C.; Zhong, M.Y.; Cai, T.; Yang, G.; Song, Z.; Theilhaber, J.; Adrian, F.; Tai, Y.-T.; et al. SAR 650984, a Therapeutic Anti-CD38 Monoclonal Antibody, Blocks CD38-CD31 Interaction in Multiple Myeloma. Blood 2014, 124, 4729. [Google Scholar] [CrossRef]
- Chari, A.; Minnema, M.C.; Berdeja, J.G.; Oriol, A.; van de Donk, N.W.C.J.; Rodríguez-Otero, P.; Askari, E.; Mateos, M.-V.; Costa, L.J.; Caers, J.; et al. Talquetamab, a T-Cell–Redirecting GPRC5D Bispecific Antibody for Multiple Myeloma. N. Engl. J. Med. 2022, 387, 2232–2244. [Google Scholar] [CrossRef]
- Mailankody, S.; Devlin, S.M.; Landa, J.; Nath, K.; Diamonte, C.; Carstens, E.J.; Russo, D.; Auclair, R.; Fitzgerald, L.; Cadzin, B.; et al. GPRC5D-Targeted CAR T Cells for Myeloma. N. Engl. J. Med. 2022, 387, 1196–1206. [Google Scholar] [CrossRef]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11, eaau7746. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Richter, J.; Trudel, S.; Cohen, A.D.; Spencer, A.; Forsberg, P.A.; Laubach, J.P.; Thomas, S.K.; Bahlis, N.J.; Costa, L.J.; et al. Enduring Responses after 1-Year, Fixed-Duration Cevostamab Therapy in Patients with Relapsed/Refractory Multiple Myeloma: Early Experience from a Phase I Study. Blood 2022, 140 (Suppl. 1), 4415–4417. [Google Scholar] [CrossRef]
- Pihlgren, M.; Hall, O.; Carretero, L.; Estoppey, C.; Drake, A.; Pais, D.; Loyau, J.; Berret, J.; Gruber, I.; Suere, P.; et al. ISB 2001, a First-in-Class Trispecific BCMA and CD38 T Cell Engager Designed to Overcome Mechanisms of Escape from Treatments for Multiple Myeloma By Targeting Two Antigens. Blood 2022, 140 (Suppl. 1), 858–859. [Google Scholar] [CrossRef]
- Stefano, S.; Grandclement, C.; Dehilly, E.; Panagopoulou, M.; Martini, E.; Castillo, R.; Suere, P.; Pouleau, B.; Estoppey, C.; Frei, J.; et al. ISB 1442, a First-in-Class CD38 and CD47 Bispecific Antibody Innate Cell Modulator for the Treatment of Relapsed Refractory Multiple Myeloma. Blood 2021, 138 (Suppl. 1), 73. [Google Scholar] [CrossRef]
- Perumal, D.; Imai, N.; Laganà, A.; Finnigan, J.; Melnekoff, D.; Leshchenko, V.V.; Solovyov, A.; Madduri, D.; Chari, A.; Cho, H.J.; et al. Mutation-derived Neoantigen-specific T-cell Responses in Multiple Myeloma. Clin. Cancer Res. 2020, 26, 450–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodysh, J.; Marron, T.; Rubinsteyn, A.; O’Donnell, T.; Finnigan, J.; Blazquez, A.; Saxena, M.; Meseck, M.; Friedlander, P.; Bhardwaj, N. Abstract CT173: PGV-001: A phase I trial of a multipeptide personalized neoantigen vaccine in the adjuvant setting. Cancer Res. 2020, 80 (Suppl. 16), CT173. [Google Scholar] [CrossRef]
- Mazahreh, F.; Mazahreh, L.; Schinke, C.; Thanendrarajan, S.; Zangari, M.; Shaughnessy, J.D., Jr.; Zhan, F.; van Rhee, F.; Al Hadidi, S. Risk of Infections Associated with the Use of Bispecific Antibodies in Multiple Myeloma: A Pooled Analysis. Blood 2022, 140 (Suppl. 1), 4378. [Google Scholar] [CrossRef]
- Lancman, G.; Parsa, K.; Rodriguez, C.; Richter, J.; Cho, H.J.; Parekh, S.; Richard, S.; Rossi, A.; Sanchez, L.; Thibaud, S.; et al. Infections and Severe Hypogammaglobulinemia in Multiple Myeloma Patients Treated with Anti-BCMA Bispecific Antibodies. Blood 2022, 140 (Suppl. 1), 10073–10074. [Google Scholar] [CrossRef]
- Mohan, M.; Nagavally, S.; Dhakal, B.; Radhakrishnan, S.V.; Chhabra, S.; D’Souza, A.; Hari, P. Risk of infections with B-cell maturation antigen-directed immunotherapy in multiple myeloma. Blood Adv. 2022, 6, 2466–2470. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.W.; Bar, N.; Paris, L.; Hofmeister, C.C.; Hansson, M.; Santoro, A.; Mateos, M.-V.; Rodríguez-Otero, P.; Lund, J.; Encinas, C.; et al. Alnuctamab (ALNUC; BMS-986349; CC-93269), a B-Cell Maturation Antigen (BCMA) x CD3 T-Cell Engager (TCE), in Patients (pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Results from a Phase 1 First-in-Human Clinical Study. Blood 2022, 140 (Suppl. 1), 400–402. [Google Scholar] [CrossRef]
- D’Souza, A.; Shah, N.; Rodriguez, C.; Voorhees, P.M.; Weisel, K.; Bueno, O.F.; Pothacamury, R.K.; Freise, K.J.; Yue, S.; Ross, J.A.; et al. A Phase I First-in-Human Study of ABBV-383, a B-Cell Maturation Antigen × CD3 Bispecific T-Cell Redirecting Antibody, in Patients With Relapsed/Refractory Multiple Myeloma. J. Clin. Oncol. 2022, 40, 3576–3586. [Google Scholar] [CrossRef]
- Bumma, N.; Richter, J.; Brayer, J.; Zonder, J.A.; Dhodapkar, M.; Shah, M.R.; Hoffman, J.E.; Mawad, R.; Maly, J.J.; Lentzsch, S.; et al. Updated Safety and Efficacy of REGN5458, a BCMAxCD3 Bispecific Antibody, Treatment for Relapsed/Refractory Multiple Myeloma: A Phase 1/2 First-in-Human Study. Blood 2022, 140, 10140–10141. [Google Scholar] [CrossRef]
- Elmeliegy, M.; Viqueira, A.; Vandendries, E.; Hickman, A.; Hibma, J.; Lon, H.-K.; Piscitelli, J.; Soltantabar, P.; Wang, D.; Jiang, S.; et al. Dose Optimization to Mitigate the Risk of CRS with Elranatamab in Multiple Myeloma. Blood 2022, 140 (Suppl. 1), 7174–7175. [Google Scholar] [CrossRef]
- Foureau, D.M.; Bhutani, M.; Robinson, M.; Guo, F.; Pham, D.; Buelow, B.; Steuerwald, N.; Rigby, K.; Tjaden, E.; Leonidas, M.; et al. Ex vivo efficacy of BCMA-bispecific antibody TNB-383B in relapsed/refractory multiple myeloma. EJHaem 2020, 1, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraju, D.V.; Alapa, M.; O’Donohue, A.; La Motte-Mohs, R.; Boss, I.W.; Hagner, P.; Pourdehnad, M.; Burgess, M.R.; Pierceall, W.E.; Thakurta, A. Suppression of Myeloid Cell-Derived Proinflammatory Cytokines with Celmod Agents: Implications for CRS with T-Cell Engagers (TCEs). Blood 2022, 140 (Suppl. 1), 7070–7071. [Google Scholar] [CrossRef]
- Molony, R.D.; Funk, T.; Trabucco, G.; Corcoran, E.; Ruddy, D.; Varadarajan, M.; Elliot, G.; Piquet, M.; Lam, J.; Meyer, M.J.; et al. CRISPR screening identifies T cell-intrinsic regulators of CD3-bispecific antibody responses. Front. Immunol. 2022, 13, 909979. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
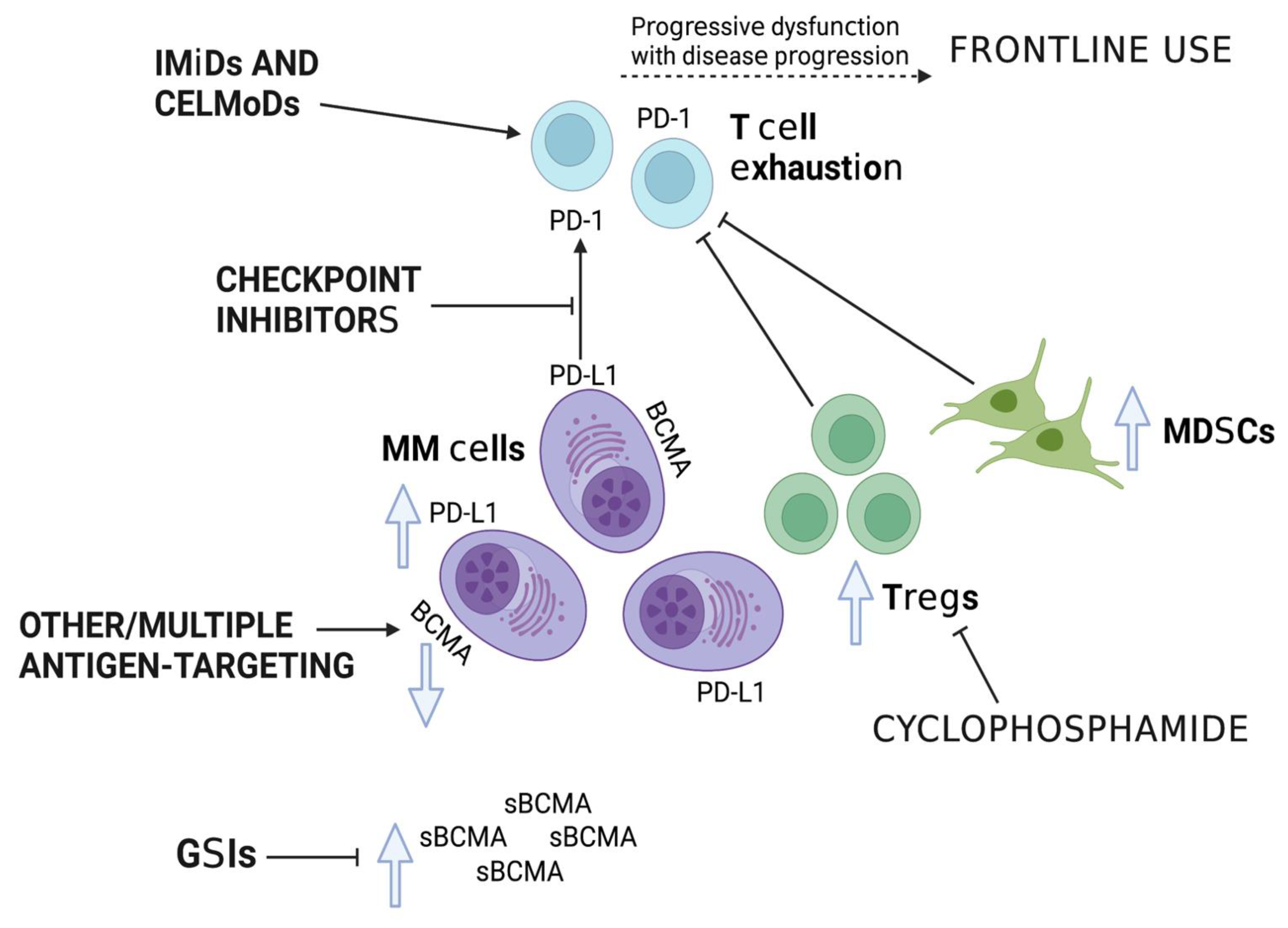{kind=link}
| Bispecific Antibody | Clinical Trials Identifier | Antibody Structure | Administration | Safety | CRS/ICANS | Responses | Ongoing Studies |
|---|---|---|---|---|---|---|---|
| Teclistamab | MajesTEC-1 NCT03145181 | humanized, IgG Fc | Teclistamab 1.5 mg/kg weekly S/C with a 2-step-up priming dose regimen (0.06 mg/kg and 0.3 mg/kg) | Anaemia 52%, neutropenia 71%, thrombocytopenia 40%, infections 76% (grade 3–4 45%), neurotoxicity 15% | CRS 72% (all but one case grade 1–2), ICANS 3% (all grade 1–2) | ORR 63%, 39% CR or better, median DOR 18.4 months | Several MagesTEC studies ongoing using teclistamab in RRMM and NDMM in combination therapies |
| Elranatamab | MagnetisMM-3 NCT04649359 Cohort A | full length, humanized, IgG2a | Elranatamab 76 mg weekly S/C on a 28 day cycles with a 2-step-up priming dose regimen (12 mg and 32 mg) | Anaemia 56%, neutropenia 53%, thrombocytopenia 27%, infection 62% (grade 3–4 32%), %, peripheral neuropathy 17%, nausea 30%, diarrhoea 45% | CRS 56% (all grade 1–2), ICANS 3% (all grade 1–2) | ORR 61%, median DOR not reached | Several MagnetisMM studies ongoing using elranatamab in RRMM and NDMM in combination therapies |
| AMG 420 | NCT02514239 | BiTE | Continuous 28 day IV infusion followed by 2 week break. Dose-escalation from 0.2–800 μg/day | Infection 33%, polyneuropathy 5%, 12% deranged liver enzymes | CRS 38% (94% Grade 1–2) | ORR 31% across all doses, 70% for the 400 ug/day cohort | Development discontinued by Amgen |
| AMG 701 | NCT03287908 | extended half-life, scFvs plus Fc region | Weekly IV. Dose-escalation from 5 μg–12 mg | Anaemia 43%, neutropenia 23%, thrombocytopenia 20%, diarrhoea 31%, fatigue 25%, infection 17%, elevated pancreatic enzymes 3%. | CRS 61% (90% Grade 1–2) | ORR 36% for 3–12 mg doses | Development discontinued by Amgen |
| Linvoseltamab (REGN5458) | NCT03761108 | Fc Fab arms | IV weekly, then every 2 weeks. Dose escalation over 9 dose levels. | Anaemia 37%, neutropenia 29%, thrombocytopenia 21%, fatigue 34% | CRS 48% (all but one case Grade 1–2) | ORR 41% for doses <200 mg and 75% ≥200 mg, median DOR not reached | Phase 2 study of 200 mg REGN5458 is recruiting |
| Alnuctamab (CC-93269) | NCT03486067 | 2 arm humanized IgG1 Fc | Dose escalation of IV alnuctamab from 0.15–10 mg. S/C alnuctamab given on D1, 4, 8, 15 and 22 of C1, weekly in C2–3, every other week in C4–6 and every 28 days thereafter. Dose escalation from 10–60 mg | Anaemia 34%, neutropenia 34% | CRS 53% (all grade 1–2), 1 grade 1 ICANS | IV alnuctamab ORR 39%, median PFS 13 weeks, median DOR in responding patients 146 weeks. S/C alnuctamab ORR 51% across all doses, 77% for doses ≥30 mg | Ongoing recruitment to the phase 1 study |
| Abbv-383 | NCT03933735 | IgG4 Fc. 2 heavy chain only anti-BCMA moieties | Dose escalation and expansion cohorts (n = 6 in 40 mg cohort, n = 60 in 60 mg cohort) | Infections in 50% of 40 mg cohort and 43% of 60 mg cohort, neutropenia in 67%/40%, anaemia in 33%/32%, thrombocytopenia 33%/25% | CRS 83% (all grade 1–2) in 40 mg cohort and 72% (2% grade 3–4) in 60 mg cohort | ORR 57% across all groups, 83% at 40 mg and 60% at 60 mg. ≥CR 67% at 40 mg and 29% at 60 mg | Phase 1b study planned NCT05650632 |
| Study | BsAb | Target | IMiD-Based Combination Therapy |
|---|---|---|---|
| MajesTEC-2 NCT04722146 | Teclistamab | BCMA | Tec/dara/pom, tec/dara/bort/len, tec/dara/len |
| MajesTEC-7 NCT05552222 | Teclistamab | BCMA | Tec/dara/len vs. Dara/len/dex |
| MagnetisMM-4 NCT05090566 | Elranatamab | BCMA | Elran/len/dex |
| LINKER-MM2 NCT05137054 | Linvoseltamab | BCMA | Linvo/len, linvo/pom |
| NCT04910568 | Cevostamab | FcRH5 | Cevo/Pom/dex |
| NCT05050097 | Talquetamab | GPRC5D | TalqLen, talqdara/len, talq/pom |
| Study | Design | Treatment | Eligibility |
|---|---|---|---|
| MajesTEC-2 NCT04722146 | Multi-arm phase 1b study | Teclistamab with other MM therapies (daratumumab, pomalidomide, lenalidomide, bortezomib, nirogacestat, in various combinations, arms A-F) | Elligibility differs according to treatment arm. Arm B) tec/dara/len/bort (Q21): NDMM or RRMM naïve to lenalidomide. Arm E) tec/dara/len: NDMM or RRMM with 1–3 prior lines including PI/ImID. Arm F) tec/dara/len/bor (Q28): NDMM only. |
| MajesTEC-4 NCT05243797 | Randomised, open-label, multicentre phase 3 study | Tec/len vs. lenalidomide maintenance post ASCT | NDMM patients who have undergone induction and ASCT |
| MajesTEC-7 NCT05552222 | Phase 3 randomised study | Tec/dara/len vs. Dara/len/dex | NDMM patients either ineligible or not suitable for ASCT |
| MagnetisMM-6 NCT05623020 | Open-label, 2 arm, multicentre, randomised study | Elran/dara/len vs. Dara/len/dex | NDMM ineligible for ASCT |
| MagnetisMM-7 NCT05317416 | Randomised, 2-arm, phase 3 study | Elranatamab vs. lenalidomide monotherapy | NDMM patients who are MRD positive post ASCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swan, D.; Murphy, P.; Glavey, S.; Quinn, J. Bispecific Antibodies in Multiple Myeloma: Opportunities to Enhance Efficacy and Improve Safety. Cancers 2023, 15, 1819. https://doi.org/10.3390/cancers15061819
Swan D, Murphy P, Glavey S, Quinn J. Bispecific Antibodies in Multiple Myeloma: Opportunities to Enhance Efficacy and Improve Safety. Cancers. 2023; 15(6):1819. https://doi.org/10.3390/cancers15061819
Chicago/Turabian StyleSwan, Dawn, Philip Murphy, Siobhan Glavey, and John Quinn. 2023. "Bispecific Antibodies in Multiple Myeloma: Opportunities to Enhance Efficacy and Improve Safety" Cancers 15, no. 6: 1819. https://doi.org/10.3390/cancers15061819
APA StyleSwan, D., Murphy, P., Glavey, S., & Quinn, J. (2023). Bispecific Antibodies in Multiple Myeloma: Opportunities to Enhance Efficacy and Improve Safety. Cancers, 15(6), 1819. https://doi.org/10.3390/cancers15061819