
Areas of Crush Nuclear Streaming Should Be Included as Tumor Content in the Era of Molecular Diagnostics


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Eligibility and Sample Preparation
2.2. Assessment of DNA and RNA Quality and Variant Detection Using NGS
2.3. NGS Data Analysis and Validation of Observed Variants
2.4. Statistical Analysis
3. Results
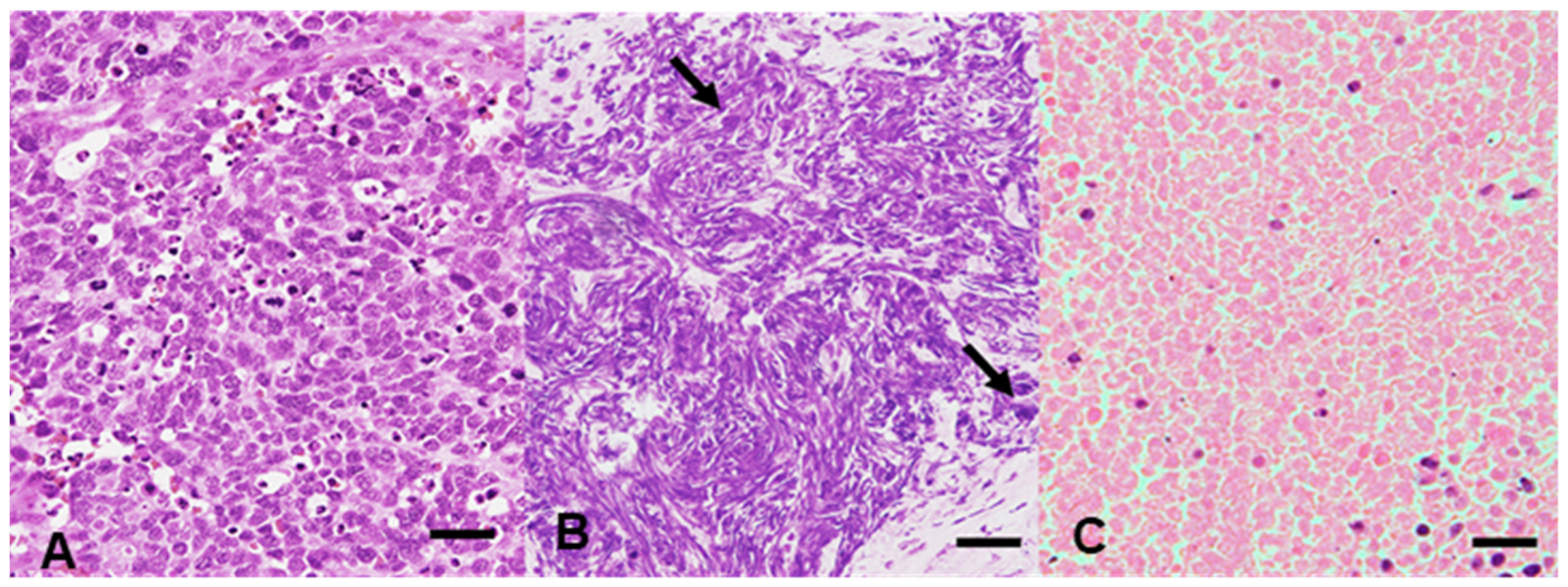
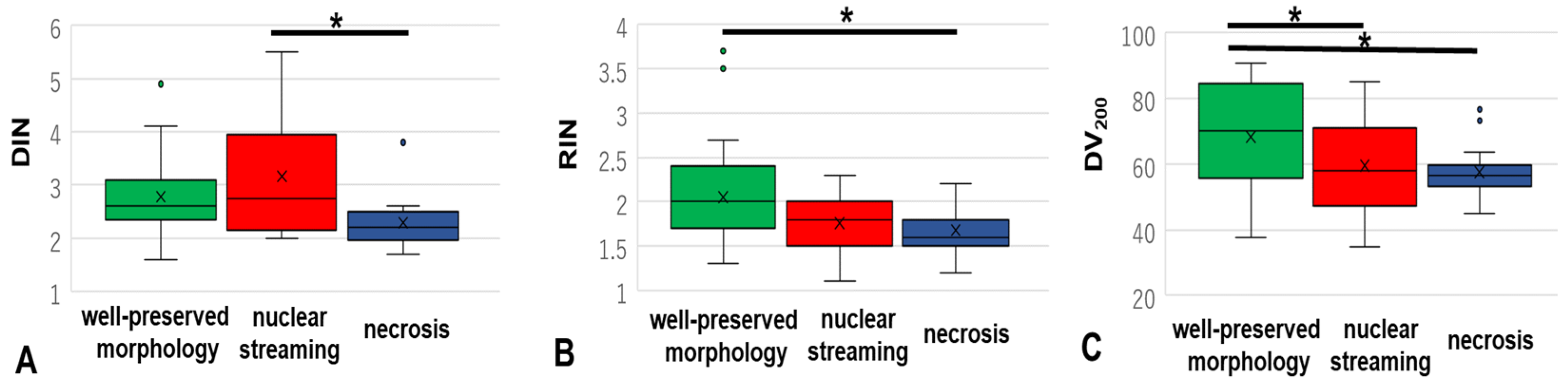
3.1. DIN, RIN, and DV200 of Materials without Degeneration, with Nuclear Streaming, and with Necrosis
3.1.1. DIN
3.1.2. RIN
3.1.3. DV200
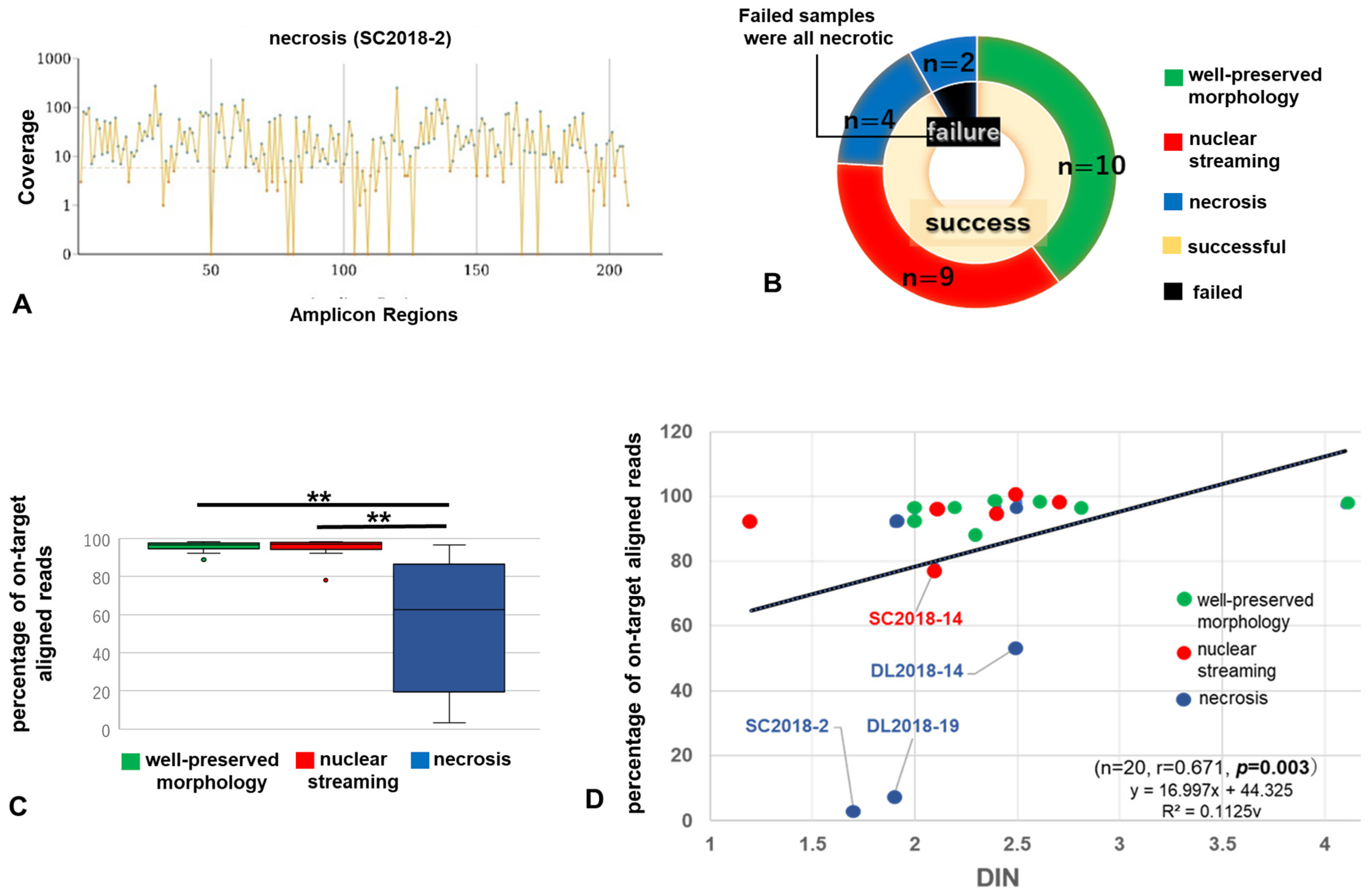
3.2. Results of NGS via AmpliSeq for Illumina Cancer HotSpot Panel v2
3.3. Relationship between Performance in NGS Analysis Using MiSeq and Histological Changes
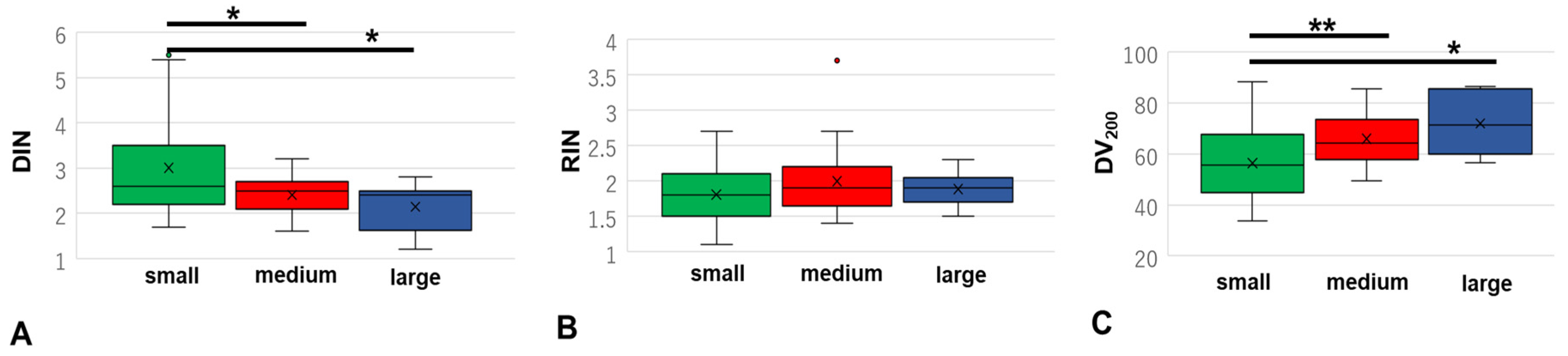
3.4. Differences in DIN, RIN, and DV200 Based on Sample Volume
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bettoni, F.; Koyama, F.C.; Carpinetti, P.D.A.; Galante, P.A.F.; Camargo, A.A.; Asprino, P.F. A straightforward assay to evaluate DNA integrity and optimize next-generation sequencing for clinical diagnosis in oncology. Exp. Mol. Pathol. 2017, 103, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Beigh, M. Next-Generation Sequencing: The Translational Medicine Approach from “Bench to Bedside to Population”. Medicines 2016, 3, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endrullat, C.; Glökler, J.; Franke, P.; Frohme, M. Standardization and quality management in next-generation sequencing. Appl. Transl. Genom. 2016, 10, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Luthra, R.; Goswami, R.; Singh, R.; Roy-Chowdhuri, S. Analysis of Pre-Analytic Factors Affecting the Success of Clinical Next-Generation Sequencing of Solid Organ Malignancies. Cancers 2015, 7, 1699–1715. [Google Scholar] [CrossRef] [PubMed]
- A Cree, I.; Deans, Z.; Ligtenberg, M.J.L.; Normanno, N.; Edsjö, A.; Rouleau, E.; Solé, F.; Thunnissen, E.; Timens, W.; Schuuring, E.; et al. Guidance for laboratories performing molecular pathology for cancer patients. J. Clin. Pathol. 2014, 67, 923–931. [Google Scholar] [CrossRef] [Green Version]
- Nagahashi, M.; Shimada, Y.; Ichikawa, H.; Nakagawa, S.; Sato, N.; Kaneko, K.; Homma, K.; Kawasaki, T.; Kodama, K.; Lyle, S.; et al. Formalin-fixed paraffin-embedded sample conditions for deep next generation sequencing. J. Surg. Res. 2017, 220, 125–132. [Google Scholar] [CrossRef]
- Kanai, Y.; Nishihara, H.; Miyagi, Y.; Tsuruyama, T.; Taguchi, K.; Katoh, H.; Takeuchi, T.; Gotoh, M.; Kuramoto, J.; Arai, E.; et al. The Japanese Society of Pathology Guidelines on the handling of pathological tissue samples for genomic research: Standard operating procedures based on empirical analyses. Pathol. Int. 2018, 68, 63–90. [Google Scholar] [CrossRef]
- Fujii, T.; Uchiyama, T.; Matsuoka, M.; Myojin, T.; Sugimoto, S.; Nitta, Y.; Okabe, F.; Sugimoto, A.; Sekita-Hatakeyama, Y.; Morita, K.; et al. Evaluation of DNA and RNA quality from archival formalin-fixed paraffin-embedded tissue for next-generation sequencing—Retrospective study in Japanese single institution. Pathol. Int. 2020, 70, 602–611. [Google Scholar] [CrossRef]
- Kuwata, T.; Wakabayashi, M.; Hatanaka, Y.; Morii, E.; Oda, Y.; Taguchi, K.; Noguchi, M.; Ishikawa, Y.; Nakajima, T.; Sekine, S.; et al. Impact of DNA integrity on the success rate of tissue-based next-generation sequencing: Lessons from nationwide cancer genome screening project SCRUM-Japan GI-SCREEN. Pathol. Int. 2020, 70, 932–942. [Google Scholar] [CrossRef]
- Hatanaka, Y.; Kuwata, T.; Morii, E.; Kanai, Y.; Ichikawa, H.; Kubo, T.; Hatanaka, K.C.; Sakai, K.; Nishio, K.; Fujii, S.; et al. The Japanese Society of Pathology Practical Guidelines on the handling of pathological tissue samples for cancer genomic medicine. Pathol. Int. 2021, 71, 725–740. [Google Scholar] [CrossRef]
- Goswami, R.; Luthra, R.; Singh, R.R.; Patel, K.P.; Routbort, M.J.; Aldape, K.D.; Yao, H.; Dang, H.D.; Barkoh, B.A.; Manekia, J.; et al. Identification of Factors Affecting the Success of Next-Generation Sequencing Testing in Solid Tumors. Am. J. Clin. Pathol. 2016, 145, 222–237. [Google Scholar] [CrossRef]
- Petrackova, A.; Vasinek, M.; Sedlarikova, L.; Dyskova, T.; Schneiderova, P.; Novosad, T.; Papajik, T.; Kriegova, E. Standardization of Sequencing Coverage Depth in NGS: Recommendation for Detection of Clonal and Subclonal Mutations in Cancer Diagnostics. Front. Oncol. 2019, 9, 851. [Google Scholar] [CrossRef] [PubMed]
- D’Haene, N.; Le Mercier, M.; De Nève, N.; Blanchard, O.; Delaunoy, M.; El Housni, H.; Dessars, B.; Heimann, P.; Remmelink, M.; Demetter, P.; et al. Clinical Validation of Targeted Next Generation Sequencing for Colon and Lung Cancers. PLoS ONE 2015, 10, e0138245. [Google Scholar] [CrossRef]
- Morlote, D.; Janowski, K.M.; Siniard, R.C.; Guo, R.J.; Winokur, T.; DeFrank, G.; Harada, S. Effects of Improved DNA Integrity by Punch From Tissue Blocks as Compared to Pinpoint Extraction From Unstained Slides on Next-Generation Sequencing Quality Metrics. Am. J. Clin. Pathol. 2019, 152, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D. Update on small cell carcinoma and its differentiation from squamous cell carcinoma and other non-small cell carcinomas. Mod. Pathol. 2012, 25 (Suppl. S1), S18–S30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, R.D. Diagnostic value of crush artifact in cytologic specimens. Occurrence in small cell carcinoma of the lung. Acta Cytol. 1990, 34, 502–504. [Google Scholar]
- Wehmas, L.C.; E Wood, C.; Chorley, B.N.; Yauk, C.; Nelson, G.M.; Hester, S.D. Enhanced Quality Metrics for Assessing RNA Derived From Archival Formalin-Fixed Paraffin-Embedded Tissue Samples. Toxicol. Sci. 2019, 170, 357–373. [Google Scholar] [CrossRef]
- Flamminio, F.; Tosi, A.L.; Fellegara, G. Crushing Artifacts Resulting in Small Blue Cellular Clusters That Simulate Small Cell Carcinoma. Int. J. Surg. Pathol. 2011, 19, 487–491. [Google Scholar] [CrossRef]
- Dehghani, M.; Rosenblatt, K.P.; Li, L.; Rakhade, M.; Amato, R.J. Validation and Clinical Applications of a Comprehensive Next Generation Sequencing System for Molecular Characterization of Solid Cancer Tissues. Front. Mol. Biosci. 2019, 6, 82. [Google Scholar] [CrossRef]
- Jabbar, K.; Routbort, M.; Singh, C.; Rashid, A.; Broaddus, R. Impact of tumor necrosis on success of clinical next generation sequencing. In Laboratory Investigation; Nature Publishing Group: New York, NY, USA, 2015; p. 501A. [Google Scholar]
- Li, Y.; Xie, X. Deconvolving tumor purity and ploidy by integrating copy number alterations and loss of heterozygosity. Bioinformatics 2014, 30, 2121–2129. [Google Scholar] [CrossRef] [Green Version]
- Strom, S.P. Current practices and guidelines for clinical next-generation sequencing oncology testing. Cancer Biol. Med. 2016, 13, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiväli, Ü.; Paier, A.; Tenson, T. When stable RNA becomes unstable: The degradation of ribosomes in bacteria and beyond. Biol. Chem. 2013, 394, 845–855. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | Mean | SD | SE | 95% CI | vs. Without Degeneration p-Value | vs. Nuclear Streaming p-Value | vs. Necrosis p-Value | |
|---|---|---|---|---|---|---|---|---|
| DIN (n = 71) | ||||||||
| Well-preserved morphology | 30 | 2.78 | 0.68702 | 0.12543 | 2.523–3.037 | - | 0.222 | 0.212 |
| Nuclear streaming | 28 | 3.16071 | 1.11963 | 0.21159 | 2.727–3.593 | 0.222 | - | 0.011 |
| Necrosis | 13 | 2.29230 | 0.52830 | 0.14652 | 1.973–2.612 | 0.213 | 0.011 | |
| RIN (n = 72) | ||||||||
| Well-preserved morphology | 31 | 2.05161 | 0.59602 | 0.10704 | 1.833–2.270 | - | 0.05 | 0.026 |
| Nuclear streaming | 30 | 1.75666 | 0.31697 | 0.05787 | 1.638–1.875 | 0.05 | - | 0.747 |
| Necrosis | 11 | 1.68181 | 0.27863 | 0.08401 | 1.495–1.869 | 0.026 | 0.747 | - |
| DV200 (n = 79) | ||||||||
| Well-preserved morphology | 32 | 68.34093 | 16.40420 | 2.89988 | 62.4266–74.2553 | - | 0.041 | 0.031 |
| Nuclear streaming | 30 | 59.56033 | 13.70592 | 2.39534 | 54.4425–64.6872 | 0.041 | - | 0.878 |
| Necrosis | 17 | 57.49411 | 8.15700 | 1.97836 | 53.3002–61.6881 | 0.031 | 0.878 | - |
| Year-No. | Status | Organ | Status | Percent Q30 Bases | Total PF Reads | Percentage On-target Aligned Reads | Uniformity of Coverage [Pct > 0.2 × Mean] | Amplicon Mean Coverage | DIN (≥2.3) |
|---|---|---|---|---|---|---|---|---|---|
| SC 2015-1 | R | uterus | well-preserved morphology | 90.3 | 690,942 | 95.81 | 93.24 | 2803.9 | 2.0 |
| nuclear streaming | 90.37 | 396,176 | 92.33 | 90.82 | 1547 | 1.2 | |||
| necrosis | 89.64 | 543,100 | 71.48 | 60.87 | 1494.5 | 1.4 | |||
| SC 2015-2 | B | lung | well-preserved morphology | 79.69 | 695,098 | 88.79 | 67.15 | 1253.5 | 2.3 |
| nuclear streaming | 80.92 | 654,338 | 97.35 | 82.61 | 1358.2 | 2.5 | |||
| SC 2018-2 | R | lung | well-preserved morphology | 80.63 | 553,474 | 97.15 | 88.89 | 1173.5 | - |
| nuclear streaming | 79.97 | 482,590 | 96.97 | 85.99 | 1009.7 | 2.4 | |||
| necrosis | 86.36 | 270,754 | 3.19 | 78.74 | 29.2 | 1.7 | |||
| SC 2018-3 | R | lung | well-preserved morphology | 90.33 | 677,400 | 96.45 | 75.36 | 2737.3 | 2.8 |
| necrosis | 90.84 | 668,770 | 96.52 | 71.01 | 2673.8 | 2.5 | |||
| SC 2018-4 | R | brain | well-preserved morphology | 79.74 | 665,558 | 95.3 | 85.99 | 1347.4 | - |
| nuclear streaming | 79.88 | 501,748 | 78.14 | 82.61 | 780.7 | 2.1 | |||
| SC 2019-8 | R | brain | well-preserved morphology | 80.94 | 662,300 | 97.09 | 82.13 | 1402.6 | 2.2 |
| nuclear streaming | 92.26 | 396,976 | 96.18 | 66.67 | 1626.7 | 2.1 | |||
| SC 2019-9 | B | uterus | well-preserved morphology | 91.89 | 949,214 | 97.39 | 88.41 | 3901.8 | 4.1 |
| nuclear streaming | 81.02 | 945,082 | 97.87 | 93.24 | 1947.3 | - | |||
| DL 2018-14 | R | intestine | well-preserved morphology | 80.36 | 641,642 | 98.47 | 83.57 | 1375.6 | 2.6 |
| nuclear streaming | 80.84 | 648,072 | 98.14 | 79.23 | 1387.9 | 2.7 | |||
| necrosis | 76.66 | 737,332 | 53.64 | 82.13 | 756.4 | 2.5 | |||
| DL 2018-19 | R | brain | well-preserved morphology | 80.46 | 421,506 | 92.23 | 79.23 | 845.9 | 2 |
| nuclear streaming | 80.13 | 593,102 | 95.93 | 82.13 | 1236.5 | - | |||
| necrosis | 75.26 | 517,606 | 8.05 | 76.33 | 82.6 | 1.9 | |||
| DL 2020-2 | R | colon | well-preserved morphology | 80.83 | 464,446 | 98.34 | 80.68 | 1003.7 | 2.4 |
| nuclear streaming | 80.25 | 645,404 | 98.46 | 87.44 | 1393.1 | 2.5 | |||
| necrosis | 79.49 | 555,934 | 91.64 | 82.61 | 1079.7 | 1.9 |
| Year-No. | Status | Detected Mutation (n) | Mutations Passed Filter with Quality 100 * (n) | Mutations * Having PP (n) | Gene (Variant) | PP | VAF | Total RD | Recommended RD † |
|---|---|---|---|---|---|---|---|---|---|
| -SC 2015-1 | preserved morphology | 31 | 23 | 3 | KDR(T > T/A) | B | 66.81 | 473 | 9 |
| TP53(A > A/G) | D | 40.4 | 1222 | 9 | |||||
| TP53(G > G/C) | B | 24.4 | 573 | 42 | |||||
| nuclear streaming | 44 | 15 | 2 | KDR(T > A/A) | B | 98.1 | 160 | 2 | |
| TP53(A > A/G) | D | 26.9 | 309 | 37 | |||||
| necrosis | 28 | 13 | 2 | KDR(T > T/A) | B | 84.8 | 33 | 4 | |
| TP53(A > A/G) | D | 43 | 467 | 17 | |||||
| SC 2015-2 | preserved morphology | 13 | 12 | 1 | TP53(G > C/C) | B | 100 | 48 | 1 |
| nuclear streaming | 14 | 13 | 2 | TP53(A > C/C) | D | 92.7 | 355 | 3 | |
| TP53(G > C/C) | B | 100 | 138 | 1 | |||||
| SC 2018-2 | preserved morphology | 15 | 15 | 4 | KIT(G > G/C) | D | 47.8 | 1468 | 15 |
| PTEN(A > A/G) | D | 52.5 | 240 | 13 | |||||
| KRAS(G >G/T) | D | 27.4 | 880 | 37 | |||||
| TP53(G > C/C) | D | 90.1 | 333 | 3 | |||||
| nuclear streaming | 16 | 14 | 4 | KIT(G > G/C) | D | 47.8 | 1895 | 15 | |
| PTEN(A > A/G) | D | 50.2 | 317 | 14 | |||||
| PIK3CA(A >A/G) | D | 23.1 | 1617 | 44 | |||||
| KRAS(G > G/T) | D | 30.5 | 1189 | 32 | |||||
| necrosis | 15 | 2 | 1 | PTEN(A > A/G) | D | 75 | 12 | 5 | |
| SC 2018-3 | preserved morphology | 30 | 22 | 5 | ERBB4(C > C/A) | D | 35.5 | 346 | 27 |
| KIT(A > A/C) | B | 34.5 | 2041 | 28 | |||||
| KDR(T > T/A) | B | 63.6 | 294 | 10 | |||||
| MET(A > A/G) | B | 41.4 | 336 | 18 | |||||
| TP53(G > C/C) | D | 90.1 | 154 | 2 | |||||
| necrosis | 26 | 20 | 5 | ERBB4(C > C/A) | D | 76.6 | 184 | 5 | |
| KIT(A > A/C) | B | 9 | 1699 | 195 | |||||
| KDR(T > T/A) | B | 90.1 | 203 | 3 | |||||
| MET(A > A/G) | B | 16.4 | 644 | 75 | |||||
| TP53(G > C/C) | D | 100 | 114 | 1 | |||||
| SC 2018-4 | preserved morphology | 15 | 14 | 2 | TP53(T > T/C) | D | 68 | 747 | 9 |
| TP53(G > C/C) | D | 100 | 217 | 1 | |||||
| nuclear streaming | 15 | 14 | 2 | TP53(T > T/C) | D | 91.5 | 177 | 3 | |
| TP53(G > C/C) | B | 96.9 | 64 | 2 | |||||
| SC 2019-8 | preserved morphology | 10 | 5 | 0 | - | - | - | - | - |
| nuclear streaming | 21 | 17 | 3 | KIT(G > G/C) | B | 48.5 | 1333 | 15 | |
| TP53(T > T/C) | D | 62.7 | 59 | 10 | |||||
| TP53(G > C/C) | B | 100 | 12 | 1 | |||||
| SC 2019-9 | preserved morphology | 27 | 18 | 2 | TP53(G > C/C) | B | 99.6 | 233 | 2 |
| STK11(C > C/G) | B | 47.4 | 352 | 15 | |||||
| nuclear streaming | 16 | 14 | 2 | TP53(G > C/C) | B | 99.5 | 360 | 2 | |
| STK11(C > C/G) | B | 100 | 506 | 1 | |||||
| DL 2018-14 | preserved morphology | 16 | 15 | 2 | PDGFR(C > C/G) | B | 38.1 | 1203 | 25 |
| TP53(G > C/C) | B | 99.8 | 832 | 2 | |||||
| nuclear streaming | 17 | 14 | 2 | PDGFR(C > C/G) | B | 34.1 | 947 | 28 | |
| TP53(G > C/C) | B | 98.8 | 257 | 2 | |||||
| necrosis | 16 | 16 | 2 | PDGFR(C > C/G) | B | 32.4 | 345 | 30 | |
| TP53(G > C/C) | B | 100 | 55 | 1 | |||||
| DL 2018-19 | preserved morphology | 11 | 9 | 1 | TP53(G > G/C) | B | 58.2 | 79 | 12 |
| nuclear streaming | 11 | 9 | 1 | TP53(G > G/C) | B | 40.8 | 172 | 19 | |
| necrosis | 10 | 5 | 0 | - | - | - | - | - | |
| DL 2020-2 | preserved morphology | 12 | 11 | 2 | KIT(A > A/C) | B | 45.1 | 1367 | 16 |
| TP53(G > C/C) | B | 99.6 | 634 | 2 | |||||
| nuclear streaming | 14 | 11 | 2 | KIT(A > A/C) | B | 45.1 | 2209 | 16 | |
| TP53(G > C/C) | B | 99.8 | 604 | 2 | |||||
| necrosis | 14 | 11 | 2 | KIT(A > A/C) | B | 46.7 | 1537 | 16 | |
| TP53(G > C/C) | B | 99.8 | 566 | 2 |
| Metric | n | Mean | SD | SE | 95% CI | vs. With Well-Preserved Morphology p-Value | vs. Nuclear Streaming p-Value | vs. Necrosis p-Value |
|---|---|---|---|---|---|---|---|---|
| Percentage of Q30 bases | ||||||||
| Well-preserved morphology | 10 | 83.517 | 5.087 | 1.608 | 79.8776–87.1564 | - | 0.954 | 0.988 |
| Nuclear streaming | 9 | 82.848 | 4.841 | 1.613 | 79.1277–86.5700 | 0.954 | - | 0.998 |
| Necrosis | 6 | 83.041 | 6.771 | 2.764 | 75.9355–90.1478 | 0.988 | 0.998 | - |
| Total PF reads | ||||||||
| Well-preserved morphology | 10 | 642.158 | 145.218.283 | 45.922.053 | 53,8275.10–7406,040.9 | - | 0.716 | 0.498 |
| Nuclear streaming | 9 | 584.832 | 169.773.8 | 56,591.263 | 454,332.31–715,331.69 | 0.716 | - | 0.910 |
| Necrosis | 6 | 548.916 | 160.189.302 | 65,397.009 | 380,807.64–717,024.36 | 0.498 | 0.910 | - |
| Percentage of on-target aligned reads | ||||||||
| Well-preserved morphology | 10 | 95.702 | 3.019 | 0.954 | 93.5420–97.8620 | - | 0.992 | 0.001 |
| Nuclear streaming | 9 | 94.596 | 6.438 | 2.146 | 89.6475–99.5459 | 0.992 | - | 0.02 |
| Necrosis | 6 | 54.086 | 40.554 | 16.556 | 11.5271–96.6462 | 0.001 | 0.02 | - |
| Uniformity of coverage | ||||||||
| Well-preserved morphology | 10 | 82.465 | 7.491 | 2.369 | 77.1056–87.8244 | - | 0.960 | 0.237 |
| Nuclear streaming | 9 | 83.415 | 7.705 | 2.568 | 77.4922–89.3389 | 0.960 | - | 0.181 |
| Necrosis | 6 | 75.281 | 8.241 | 3.364 | 66.6328–83.9305 | 0.237 | 0.181 | - |
| Variants with filter pass and quality 100 (%) | ||||||||
| Well-preserved morphology | 10 | 81.70 | 15.571 | 4.924 | 70.56–92.84 | - | 0.974 | 0.332 |
| Nuclear streaming | 9 | 80.0 | 18.000 | 6.000 | 66.16–93.84 | 0.974 | - | 0.401 |
| Necrosis | 6 | 60.83 | 30.825 | 12.684 | 28.48–93.18 | 0.332 | 0.401 | - |
| Variants without filter pass or with quality < 100 (%) | ||||||||
| Well-preserved morphology | 10 | 18.30 | 15.571 | 4.924 | 7.16–29.44 | - | 0.967 | 0.332 |
| Nuclear streaming | 9 | 20.22 | 17.894 | 5.965 | 6.47–33.98 | 0.967 | - | 0.408 |
| Necrosis | 6 | 39.17 | 30.825 | 12.584 | 6.82–71.52 | 0.332 | 0.408 | - |
| Variant allele frequency (TP53) | ||||||||
| Well-preserved morphology | 9 | 84.01 | 22.695 | 7.5653 | 66.566–101.457 | - | 0.650 | 0.960 |
| Nuclear streaming | 9 | 83.80 | 28.657 | 9.5524 | 61.772–105.828 | 0.650 | - | 0.552. |
| Necrosis | 4 | 85.77 | 28.466 | 14.2334 | 40.403–130.997 | 0.960 | 0.552 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noda, Y.; Yamaka, R.; Atsumi, N.; Higasa, K.; Tsuta, K. Areas of Crush Nuclear Streaming Should Be Included as Tumor Content in the Era of Molecular Diagnostics. Cancers 2023, 15, 1910. https://doi.org/10.3390/cancers15061910
Noda Y, Yamaka R, Atsumi N, Higasa K, Tsuta K. Areas of Crush Nuclear Streaming Should Be Included as Tumor Content in the Era of Molecular Diagnostics. Cancers. 2023; 15(6):1910. https://doi.org/10.3390/cancers15061910
Chicago/Turabian StyleNoda, Yuri, Ryosuke Yamaka, Naho Atsumi, Koichiro Higasa, and Koji Tsuta. 2023. "Areas of Crush Nuclear Streaming Should Be Included as Tumor Content in the Era of Molecular Diagnostics" Cancers 15, no. 6: 1910. https://doi.org/10.3390/cancers15061910
APA StyleNoda, Y., Yamaka, R., Atsumi, N., Higasa, K., & Tsuta, K. (2023). Areas of Crush Nuclear Streaming Should Be Included as Tumor Content in the Era of Molecular Diagnostics. Cancers, 15(6), 1910. https://doi.org/10.3390/cancers15061910