Bacillus subtilis Lipase A—Lipase or Esterase?

 and
and 
Abstract
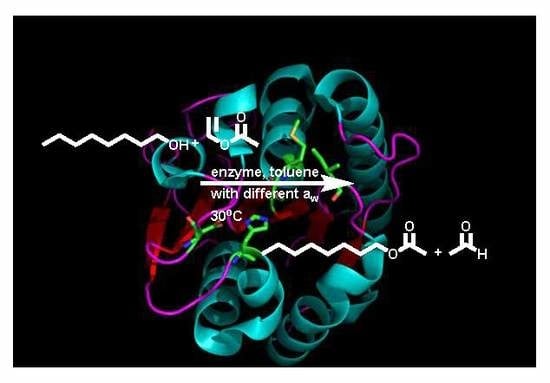
:
1. Introduction
2. Results
2.1. BioGPS
2.2. Amidase Activity
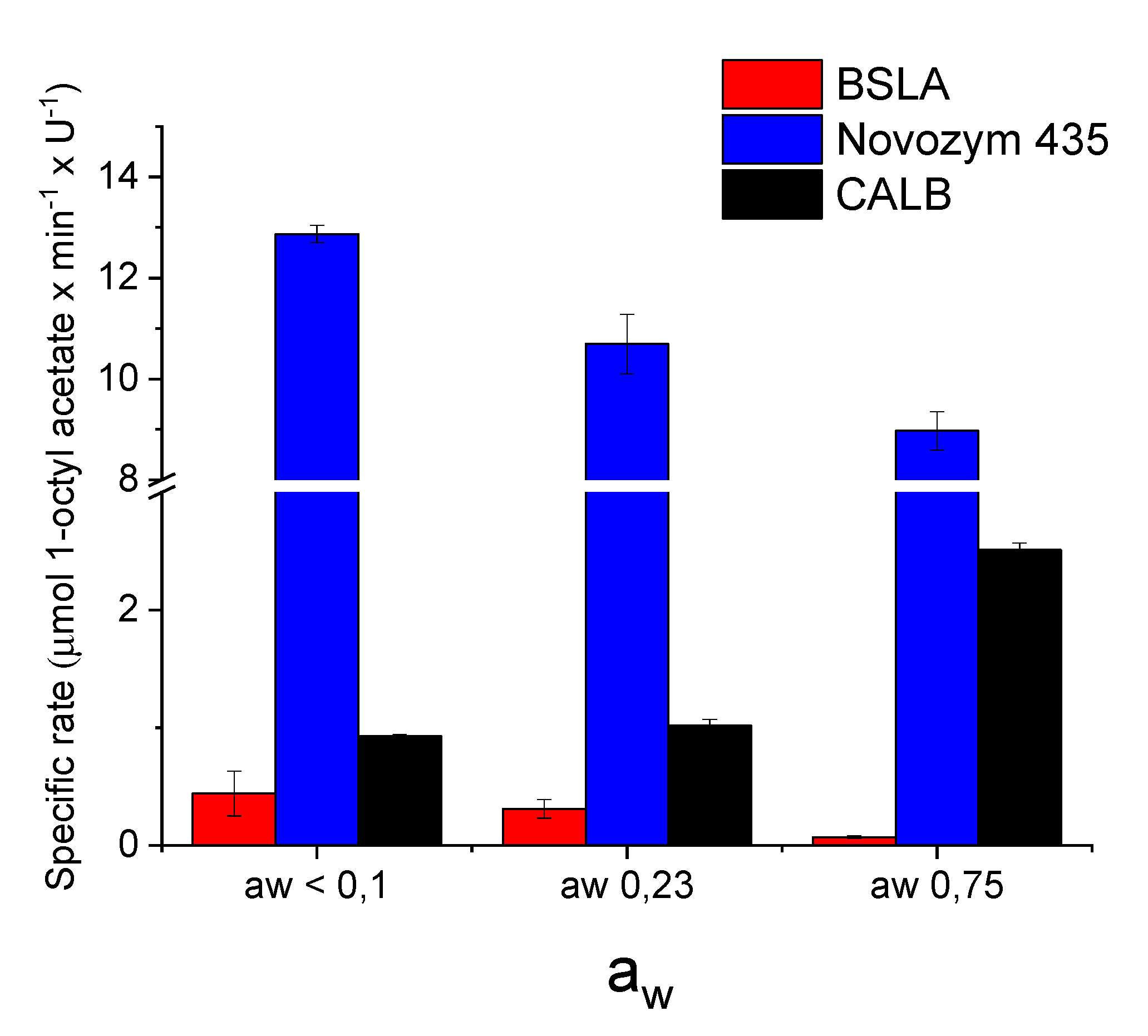
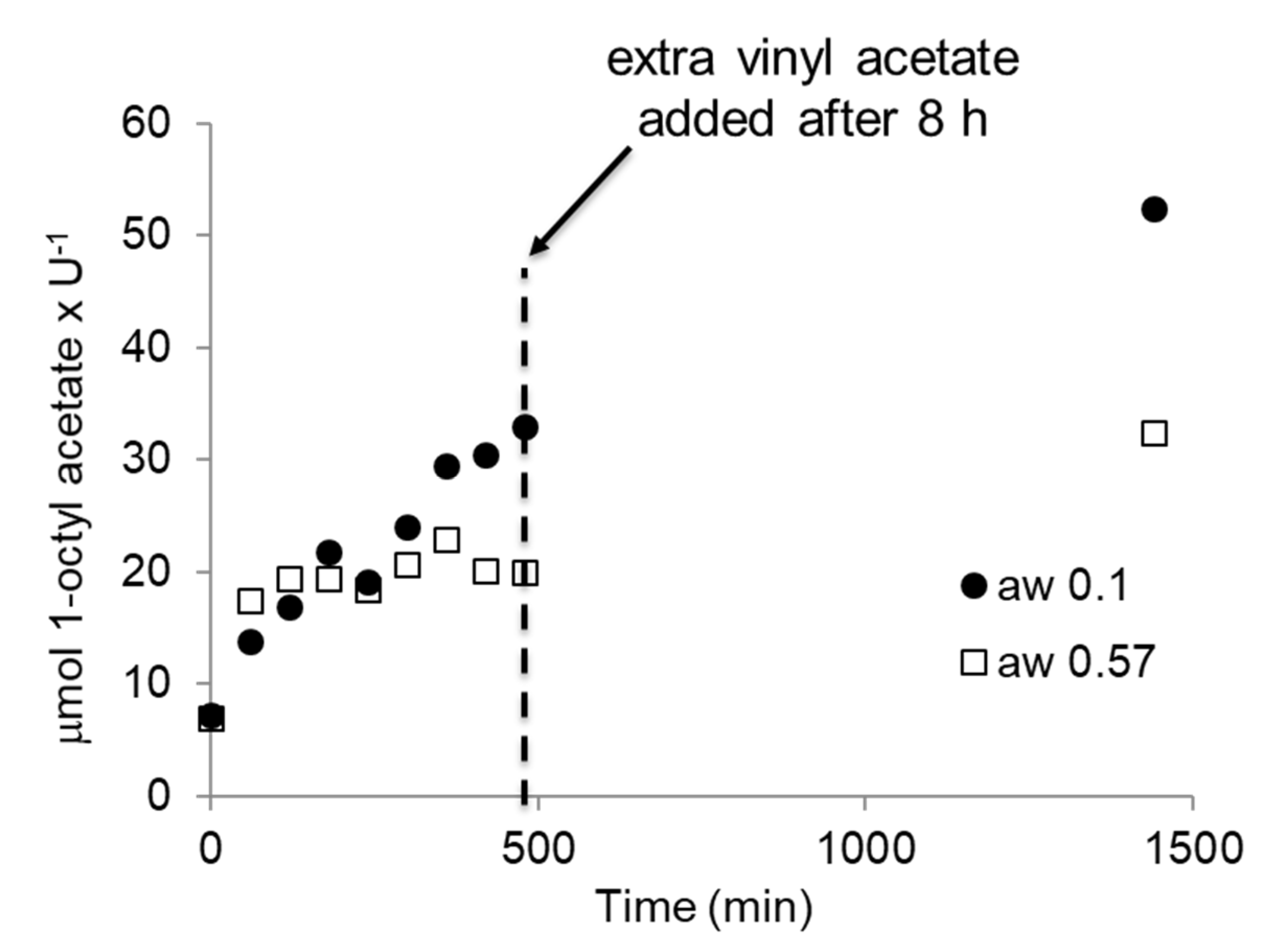
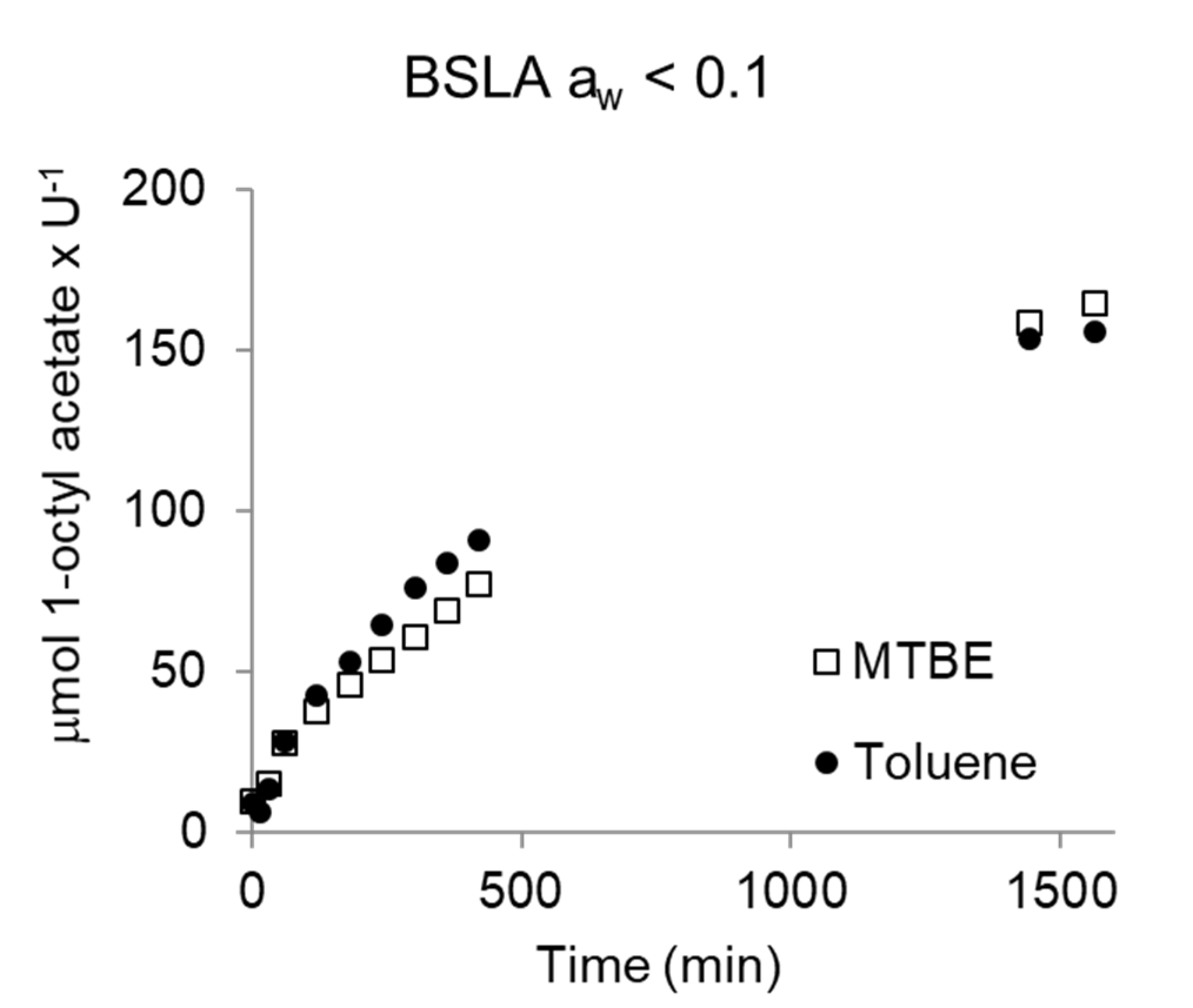
2.3. BSLA Activity in Dry Organic Solvents
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.3. BioGPS Computational Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bornscheuer, U.T.; Kazlauskas, R. Hydrolases in Organic Synthesis; Wiley: Hoboken, NJ, USA, 2005; pp. 61–184. [Google Scholar]
- Ben Ali, Y.; Verger, R.; Abousalham, A. Lipases or Esterases: Does It Really Matter? Toward a New Bio-Physico-Chemical Classification. In Lipases and Phospholipases: Methods and Protocols; Book Series: Methods in Molecular Biology; Sandoval, G., Ed.; Springer Science+Business Media: New York, NY, USA, 2012; Volume 861, pp. 31–51. [Google Scholar]
- Paravidino, M.; Bohm, P.; Gröger, H.; Hanefeld, U. Hydrolysis and Formation of Carboxylic Acid Esters. In Enzyme Catalysis in Organic Synthesis; Wiley: Hoboken, NJ, USA, 2012; pp. 249–362. [Google Scholar]
- Arpigny, J.L.; Jaeger, K.E. Bacterial lipolytic enzymes: Classification and properties. Biochem. J. 1999, 343, 177–183. [Google Scholar] [CrossRef]
- Jaeger, K.-E.; Kovacic, F. Determination of Lipolytic Enzyme Activities. In Pseudomonas Methods and Protocols; Book Series: Methods in Molecular Biology; Filloux, A., Ramos, J.L., Eds.; Springer Science+Business Media: New York, NY, USA, 2014; Volume 1149, pp. 111–134. [Google Scholar]
- Holwerda, K.; Verkade, P.E.; De Willigen, A.H.A. Vergleichende Untersuchungen über die Verseifungsgeschwindigkeit einiger Einsäuriger Triglyceride unter Einfluss von Pankreasextrakt. I. Der Einfluss des Verteilungszustandes der Triglyceride auf die Verseifungsgeschwindigkeit. Rec. Trav. Chim. Pays Bas 1936, 55, 43–57. [Google Scholar] [CrossRef]
- Kirk, O.; Christensen, M.W. Lipases from Candida antarctica: Unique Biocatalysts from a Unique Origin. Org. Proc. Res. Develop. 2002, 6, 446–451. [Google Scholar] [CrossRef]
- Grochulski, P.; Li, Y.; Schrag, J.D.; Cygler, M. Two conformational states of Candida rugosa lipase. Protein Sci. 1994, 3, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Eggert, T.; van Pouderoyen, G.; Pencreac’h, G.; Douchet, I.; Verger, R.; Dijkstra, B.W.; Jaeger, K.-E. Biochemical properties and three-dimensional structures of two extracellular lipolytic enzymes from Bacillus subtilis. Colloids Surf. B Biointerfaces 2002, 26, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Adlercreutz, P. Comparison of lipases and glycoside hydrolases as catalysts in synthesis reactions. Appl. Microbiol. Biotechnol. 2017, 101, 513–519. [Google Scholar] [CrossRef] [Green Version]
- Lopes, D.B.; Fraga, L.P.; Fleuri, L.F.; Macedo, G.A. Lipase and esterase—To what extent can this classification be applied accurately? Ciênc. Tecnol. Aliment. Camp. 2011, 31, 608–613. [Google Scholar] [CrossRef] [Green Version]
- Berlemont, R.; Spee, O.; Delsaute, M.; Lara, Y.; Schuldes, J.; Simon, C.; Power, P.; Daniel, R.; Galleni, M. Novel organic solvent-tolerant esterase isolated by metagenomics: Insights into the lipase/esterase classification. Rev. Argent. Microbiol. 2013, 45, 3–12. [Google Scholar]
- Adlercreutz, P. Fundamentals of Biocatalysis in Neat Organic Solvents. In Organic Synthesis with Enzymes in Non-Aqueous Media; Carrea, G., Riva, S., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 3–24. [Google Scholar]
- Serdakowski, A.L.; Dordick, J.S. Activating Enzymes for Use in Organic Solvents. In Organic Synthesis with Enzymes in Non-Aqueous Media; Carrea, G., Riva, S., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 47–74. [Google Scholar]
- Svensson, I.; Wehtje, E.; Adlercreutz, P.; Mattiasson, B. Effects of water activity on reaction rates and equilibrium positions in enzymatic esterifications. Biotechnol. Bioeng. 1994, 44, 549–556. [Google Scholar] [CrossRef]
- Wehtje, E.; Svensson, I.; Adlercreutz, P.; Mattiasson, B. Continuous control of water activity during biocatalysis in organic media. Biotechnol. Tech. 1993, 7, 873–878. [Google Scholar] [CrossRef]
- Ferrario, V.; Pellis, A.; Cespugli, M.; Guebitz, G.; Gardossi, L. Nature Inspired Solutions for Polymers: Will Cutinase Enzymes Make Polyesters and Polyamides Greener? Catalysts 2016, 6, 205. [Google Scholar] [CrossRef] [Green Version]
- Pellis, A.; Ferrario, V.; Zartl, B.; Brandauer, M.; Gamerith, C.; Acero, E.H.; Ebert, C.; Gardossi, L.; Guebitz, G.M. Enlarging the tools for efficient enzymatic polycondensation: Structural and catalytic features of cutinase 1 from Thermobifida cellulosilytica. Catal. Sci. Technol. 2016, 6, 3430–3442. [Google Scholar] [CrossRef] [Green Version]
- Pellis, A.; Ferrario, V.; Cespugli, M.; Corici, L.; Guarneri, A.; Zartl, B.; Acero, E.H.; Ebert, C.; Guebitz, G.; Gardossi, L. Fully renewable polyesters via polycondensation catalyzed by Thermobifida cellulosilytica cutinase 1: An integrated approach. Green Chem. 2017, 19, 490–502. [Google Scholar] [CrossRef]
- Valivety, R.H.; Halling, P.J.; Peilow, A.D.; Macrae, A.R. Relationship between water activity and catalytic activity of lipases in organic media. Effects of supports, loading and enzyme preparation. Eur. J. Biochem. 1994, 222, 461–466. [Google Scholar] [CrossRef]
- Valivety, R.H.; Halling, P.J.; Peilow, A.D.; Macrae, A.R. Lipases from different sources vary widely in dependence of catalytic activity on water activity. Biochim. Biophys. Acta Protein Struct. Mol. Enzym. 1992, 1122, 143–146. [Google Scholar] [CrossRef]
- Gupta, R.; Gupta, N.; Rathi, P. Bacterial lipases: An overview of production, purification and biochemical properties. Appl. Microbiol. Biotechnol. 2004, 64, 763–781. [Google Scholar] [CrossRef]
- Eggert, T.; van Pouderoyen, G.; Dijkstra, B.W.; Jaeger, K.-E. Lipolytic enzymes LipA and LipB from Bacillus subtilis differ in regulation of gene expression, biochemical properties, and three-dimensional structure. FEBS Lett. 2001, 502, 89–92. [Google Scholar] [CrossRef] [Green Version]
- Boersma, Y.L.; Pijning, T.; Bosma, M.S.; van der Sloot, A.; Godinho, L.F.; Dröge, M.J.; Winter, R.T.; van Pouderoyen, G.; Dijkstra, B.W.; Quax, W.J. Loop Grafting of Bacillus subtilis Lipase A: Inversion of Enantioselectivity. Chem. Biol. 2008, 15, 782–789. [Google Scholar] [CrossRef] [Green Version]
- van Pouderoyen, G.; Eggert, T.; Jaeger, K.-E.; Dijkstra, B.W. The crystal structure of Bacillus subtilis lipase: A minimal α/β hydrolase fold enzyme. J. Mol. Biol. 2001, 309, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Augustyniak, W.; Brzezinska, A.A.; Pijning, T.; Wienk, H.; Boelens, R.; Dijkstra, B.W.; Reetz, M.T. Biophysical characterization of mutants of Bacillus subtilis lipase evolved for thermostability: Factors contributing to increased activity retention. Protein Sci. 2012, 21, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Kamal, Z.; Ahmad, S.; Molugu, T.R.; Vijayalakshmi, A.; Deshmukh, M.V.; Sankaranarayanan, R.; Rao, N.M. In Vitro Evolved Non-Aggregating and Thermostable Lipase: Structural and Thermodynamic Investigation. J. Mol. Biol. 2011, 413, 726–741. [Google Scholar] [CrossRef]
- Ahmad, S.; Kamal, Z.; Sankaranarayanan, R.; Rao, N.M. Thermostable Bacillus subtilis Lipases: In Vitro Evolution and Structural Insight. J. Mol. Biol. 2008, 381, 324–340. [Google Scholar] [CrossRef]
- Rajakumara, E.; Acharya, P.; Ahmad, S.; Sankaranaryanan, R.; Rao, N.M. Structural basis for the remarkable stability of Bacillus subtilis lipase (Lip A) at low pH. Biochim. Biophys. Acta Proteins Proteom. 2008, 1784, 302–311. [Google Scholar] [CrossRef]
- Frauenkron-Machedjou, V.J.; Fulton, A.; Zhao, J.; Weber, L.; Jaeger, K.E.; Schwaneberg, U.; Zhu, L. Exploring the full natural diversity of single amino acid exchange reveals that 40–60% of BSLA positions improve organic solvents resistance. Bioresour. Bioprocess. 2018, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Markel, U.; Zhu, L.; Frauenkron-Machedjou, V.J.; Zhao, J.; Bocola, M.; Davari, M.D.; Jaeger, K.-E.; Schwaneberg, U. Are Directed Evolution Approaches Efficient in Exploring Nature’s Potential to Stabilize a Lipase in Organic Cosolvents? Catalysts 2017, 7, 142. [Google Scholar] [CrossRef] [Green Version]
- Laane, C.; Boeren, S.; Vos, K.; Veeger, C. Rules for optimization of biocatalysis in organic solvents. Biotechnol. Bioeng. 1987, 30, 81–87. [Google Scholar] [CrossRef]
- Ferrario, V.; Siragusa, L.; Ebert, C.; Baroni, M.; Foscato, M.; Cruciani, G.; Gardossi, L. BioGPS Descriptors for Rational Engineering of Enzyme Promiscuity and Structure Based Bioinformatic Analysis. PLoS ONE 2014, 9, 109354. [Google Scholar] [CrossRef]
- Cross, S.; Baroni, M.; Goracci, L.; Cruciani, G. GRID-Based Three-Dimensional Pharmacophores I: FLAPpharm, a Novel Approach for Pharmacophore Elucidation. J. Chem. Inf. Model. 2012, 52, 2587–2598. [Google Scholar] [CrossRef]
- Veum, L.; Kanerva, L.T.; Halling, P.J.; Maschmeyer, T.; Hanefeld, U. Optimisation of the Enantioselective Synthesis of Cyanohydrin Esters. Adv. Synth. Catal. 2005, 347, 1015–1021. [Google Scholar] [CrossRef]
- Henke, E.; Bornscheuer, U.T. Fluorophoric Assay for the High-Throughput Determination of Amidase Activity. Anal. Chem. 2003, 75, 255–260. [Google Scholar] [CrossRef]
- Mathews, I.; Soltis, M.; Saldajeno, M.; Ganshaw, G.; Sala, R.; Weyler, W.; Cervin, M.A.; Whited, G.; Bott, R. Structure of a Novel Enzyme That Catalyzes Acyl Transfer to Alcohols in Aqueous Conditions. Biochemistry 2007, 46, 8969–8979. [Google Scholar] [CrossRef] [PubMed]
- Mestrom, L.; Claessen, J.G.R.; Hanefeld, U. Enzyme-Catalyzed Synthesis of Esters in Water. ChemCatChem 2019, 11, 2004–2010. [Google Scholar] [CrossRef] [Green Version]
- Land, H.; Hendil-Forssell, P.; Martinelle, M.; Berglund, P. One-pot biocatalytic amine transaminase/acyl transferase cascade for aqueous formation of amides from aldehydes or ketones. Catal. Sci. Technol. 2016, 6, 2897–2900. [Google Scholar] [CrossRef] [Green Version]
- Contente, M.L.; Farris, S.; Tamborini, L.; Molinari, F.; Paradisi, F. Flow-based enzymatic synthesis of melatonin and other high value tryptamine derivatives: A five-minute intensified process. Green Chem. 2019, 21, 3263–3266. [Google Scholar] [CrossRef] [Green Version]
- Hanefeld, U. Reagents for (ir)reversible enzymatic acylations. Org. Biomol. Chem. 2003, 1, 2405–2415. [Google Scholar] [CrossRef]
- Paravidino, M.; Hanefeld, U. Enzymatic acylation: Assessing the greenness of different acyl donors. Green Chem. 2011, 13, 2651–2657. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, C.; Ferreira, M.L.; Barbosa, O.; dos Santos, J.C.S.; Rodrigues, R.C.; Berenguer-Murcia, Á.; Briand, E.L.; Fernandez-Lafuente, R. Novozym 435: The “perfect” lipase immobilized biocatalyst? Catal. Sci. Technol. 2019, 9, 2380–2420. [Google Scholar] [CrossRef] [Green Version]
- Basso, A.; Braiuca, P.; Cantone, S.; Ebert, C.; Linda, P.; Spizzo, P.; Caimi, P.; Hanefeld, U.; Degrassi, G.; Gardossi, L. In Silico Analysis of Enzyme Surface and Glycosylation Effect as a Tool for Efficient Covalent Immobilisation of CalB and PGA on Sepabeads®. Adv. Synth. Catal. 2007, 349, 877–886. [Google Scholar] [CrossRef]
- Ma, L.; Persson, M.; Adlercreutz, P. Water activity dependence of lipase catalysis in organic media explains successful transesterification reactions. Enzym. Microb. Technol. 2002, 31, 1024–1029. [Google Scholar] [CrossRef]
- Paravidino, M.; Sorgedrager, M.J.; Orru, R.V.A.; Hanefeld, U. Activity and Enantioselectivity of the Hydroxynitrile Lyase MeHNL in Dry Organic Solvents. Chem. Eur. J. 2010, 16, 7596–7604. [Google Scholar] [CrossRef] [Green Version]
- Secundo, F.; Carrea, G.; Soregaroli, C.; Varinelli, D.; Morrone, R. Activity of different Candida antarctica lipase B formulations in organic solvents. Biotechnol. Bioeng. 2001, 73, 157–163. [Google Scholar] [CrossRef]
- Valivety, R.H.; Halling, P.J.; Macrae, A.R. Reaction rate with suspended lipase catalyst shows similar dependence on water activity in different organic solvents. Biochim. Biophys. Acta Protein Struct. Mol. Enzym. 1992, 1118, 218–222. [Google Scholar] [CrossRef]
- Fontes, N.; Partridge, J.; Halling, P.J.; Barreiros, S. Zeolite molecular sieves have dramatic acid-base effects on enzymes in nonaqueous media. Biotechnol. Bioeng. 2002, 77, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Halling, P.J. Salt hydrates for water activity control with biocatalysts in organic media. Biotechnol. Tech. 1992, 6, 271–276. [Google Scholar] [CrossRef]
- Greenspan, L. Humidity fixed points of binary saturated aqueous solutions. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1977, 81, 89–96. [Google Scholar] [CrossRef]
- Johnson, J.R.; Affsprung, H.E.; Christian, S.D. The molecular complexity of water in organic solvents. Part II. J. Chem. Soc. A 1966, 77–78. [Google Scholar] [CrossRef]
- Gupta, R.; Rathi, P.; Gupta, N.; Bradoo, S. Lipase assays for conventional and molecular screening: An overview. Biotechnol. Appl. Biochem. 2003, 37, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Mateos-Díaz, E.; Rodríguez, J.A.; de los Ángeles Camacho-Ruiz, M.; Mateos-Díaz, J.C. High-Throughput Screening Method for Lipases/Esterases. In Lipases and Phospholipases: Methods and Protocols; Book Series: Methods in Molecular Biology; Sandoval, G., Ed.; Springer Science + Business Media: New York, NY, USA, 2012; Volume 861, pp. 89–100. [Google Scholar]
- Híreš, M.; Rapavá, N.; Šimkovič, M.; Varečka, Ľ.; Berkeš, D.; Kryštofová, S. Development and Optimization of a High-Throughput Screening Assay for Rapid Evaluation of Lipstatin Production by Streptomyces Strains. Curr. Microbiol. 2018, 75, 580–587. [Google Scholar] [CrossRef]
- Nalder, T.D.; Ashton, T.D.; Pfeffer, F.M.; Marshall, S.N.; Barrow, C.J. 4-Hydroxy-N-propyl-1,8-naphthalimide esters: New fluorescence-based assay for analysing lipase and esterase activity. Biochimie 2016, 128–129, 127–132. [Google Scholar] [CrossRef]
- Corici, L.; Ferrario, V.; Pellis, A.; Ebert, C.; Lotteria, S.; Cantone, S.; Voinovich, D.; Gardossi, L. Large scale applications of immobilized enzymes call for sustainable and inexpensive solutions: Rice husk as renewable alternative to fossil-based organic resins. RSC Adv. 2016, 6, 63256–63270. [Google Scholar] [CrossRef]
- Secundo, F.; Carrea, G. Lipase activity and conformation in neat organic solvents. J. Mol. Catal. B Enzym. 2002, 19–20, 93–102. [Google Scholar] [CrossRef]
- Hanefeld, U.; Gardossi, L.; Magner, E. Understanding enzyme immobilisation. Chem. Soc. Rev. 2009, 38, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Stauch, B.; Fisher, S.J.; Cianci, M. Open and closed states of Candida antarctica lipase B: Protonation and the mechanism of interfacial activation. J. Lipid Res. 2015, 56, 2348–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banik, S.D.; Nordblad, M.; Woodley, J.M.; Peters, G.H. Effect of Water Clustering on the Activity of Candida antarctica Lipase B in Organic Medium. Catalysts 2017, 7, 227. [Google Scholar] [CrossRef] [Green Version]
- Ebert, C.; Gardossi, L.; Linda, P. Control of enzyme hydration in penicillin amidase catalysed synthesis of amide bond. Tetrahedron Lett. 1996, 37, 9377–9380. [Google Scholar] [CrossRef]
- Ru, M.T.; Dordick, J.S.; Reimer, J.A.; Clark, D.S. Optimizing the salt-induced activation of enzymes in organic solvents: Effects of lyophilization time and water content. Biotechnol. Bioeng. 1999, 63, 233–241. [Google Scholar] [CrossRef]
- Partridge, J.; Dennison, P.R.; Moore, B.D.; Halling, P.J. Activity and mobility of subtilisin in low water organic media: Hydration is more important than solvent dielectric. Biochim. Biophys. Acta Protein Struct. Mol. Enzym. 1998, 1386, 79–89. [Google Scholar] [CrossRef]
- Valivety, R.H.; Halling, P.J.; Macrae, A.R. Rhizomucor miehei lipase remains highly active at water activity below 0.0001. FEBS Lett. 1992, 301, 258–260. [Google Scholar] [CrossRef] [Green Version]
- Syrén, P.-O.; Hult, K. Amidases Have a Hydrogen Bond that Facilitates Nitrogen Inversion, but Esterases Have Not. ChemCatChem 2011, 3, 853–860. [Google Scholar] [CrossRef]
- Larsen, M.W.; Zielinska, D.F.; Martinelle, M.; Hidalgo, A.; Jensen, L.J.; Bornscheuer, U.T.; Hult, K. Suppression of Water as a Nucleophile in Candida antarctica Lipase B Catalysis. ChemBioChem 2010, 11, 796–801. [Google Scholar] [CrossRef]
- Studier, F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
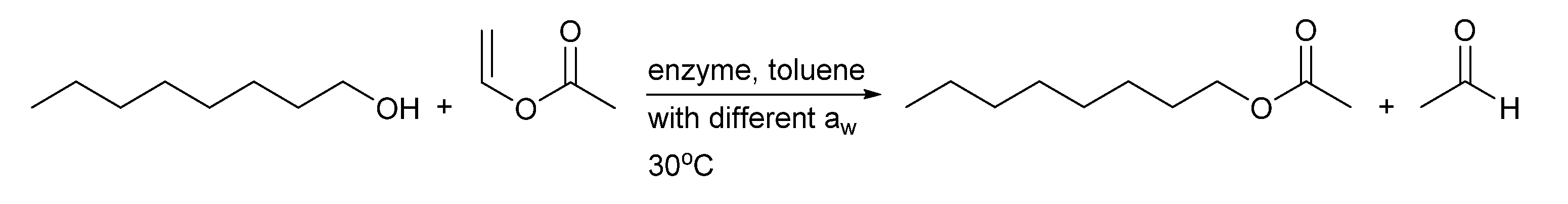{kind=link}
| aw | Agent (Vapor Phase or Salt Pair) | Moles of H2O/mol of Salt | Water Content (ppm) |
|---|---|---|---|
| <0.1 | Mol. sieves | 0 | ~20 |
| 0.25 | NaAc anhydr. (salt pair) | 1.5 | ~180 |
| 0.57 | Na2HPO4 anhydr. (salt pair) | 5.0 | ~360 |
| 0.23 | KAc (vapor phase) | NA a) | ~120 |
| 0.75 | NaCl (vapor phase) | NA a) | ~400 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bracco, P.; van Midden, N.; Arango, E.; Torrelo, G.; Ferrario, V.; Gardossi, L.; Hanefeld, U. Bacillus subtilis Lipase A—Lipase or Esterase? Catalysts 2020, 10, 308. https://doi.org/10.3390/catal10030308
Bracco P, van Midden N, Arango E, Torrelo G, Ferrario V, Gardossi L, Hanefeld U. Bacillus subtilis Lipase A—Lipase or Esterase? Catalysts. 2020; 10(3):308. https://doi.org/10.3390/catal10030308
Chicago/Turabian StyleBracco, Paula, Nelleke van Midden, Epifanía Arango, Guzman Torrelo, Valerio Ferrario, Lucia Gardossi, and Ulf Hanefeld. 2020. "Bacillus subtilis Lipase A—Lipase or Esterase?" Catalysts 10, no. 3: 308. https://doi.org/10.3390/catal10030308
APA StyleBracco, P., van Midden, N., Arango, E., Torrelo, G., Ferrario, V., Gardossi, L., & Hanefeld, U. (2020). Bacillus subtilis Lipase A—Lipase or Esterase? Catalysts, 10(3), 308. https://doi.org/10.3390/catal10030308