Self-Metathesis of Methyl Oleate Using Ru-NHC Complexes: A Kinetic Study

 , , , , and
, , , , and 
Abstract
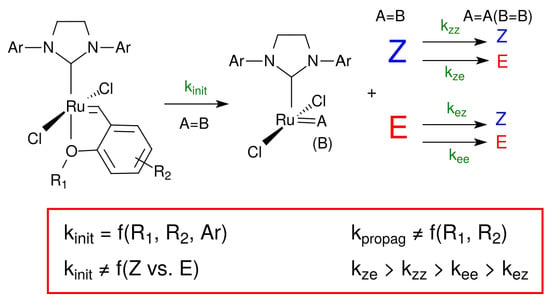
:
1. Introduction
2. Results
- (a)
- Ahr et al. [21] reported a very slight difference in the NMR chemical shifts of the RuCl2(L) = C8H17 (noted RuA, A being the alkyl chain moiety) and RuCl2(L) = C7H14CO2Et (respectively, RuB, with B the ester moiety) alkylidene species (0.02 ppm); during the reaction, both species were also present in the same concentration. This strongly suggests that the Ru coordination sphere is not affected by the presence of an ester group (RuB) and that both species behave similarly. We can now write RuR to indifferently designate the alkylidene species RuA or RuB.
- (b)
- The E olefin configuration is thermodynamically favored. The Z/E selectivity depends on the energy differences between the reaction pathways [22,23,24], and thus the kinetic constants of the reaction steps depend on the configuration of the reacting and the formed olefins. From a reactivity point of view and as fragments A and B are considered equivalent, all Z-olefins, i.e., ABZ, AAZ, and BBZ, are expected to have the same reactivity, thus a single kinetic constant is defined. A similar analysis stands for ABE, AAE, and BBE.
- (c)
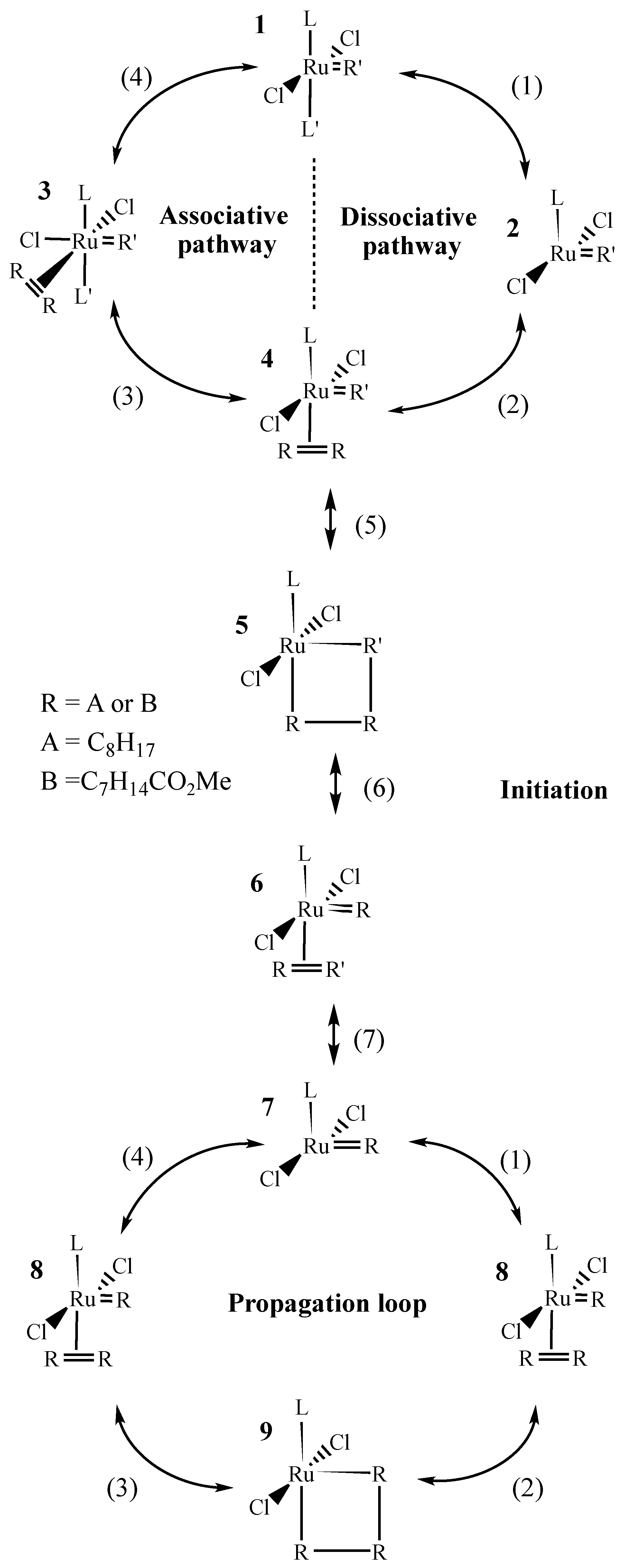
- According to the metathesis mechanism shown in Scheme 2, the active species reacts with an olefin in the propagation loop. it could also react with L’ to regenerate the pre-catalyst. Depending on the ratio between the propagation rate and the regeneration rate, the pre-catalyst concentration after initiation can be either significant or negligible. For Grubbs second-generation catalysts, Sanford et al. [1] reported that the propagation and the regeneration kinetic constants are of the same order of magnitude. Then, at low catalyst loading in the reaction medium, and thus low concentration of initial ligand L’ compared to the substrate, the propagation rate is high compared to the regeneration rate, and thus the regeneration step can be neglected (see Equation (1)).
- There is only one catalytic species, named RuR.
- The initiation reaction leading to the active species RuR is not reversible.
- No deactivation occurs.
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sanford, M.S.; Love, J.A.; Grubbs, R.H. Mechanism and Activity of Ruthenium Olefin Metathesis Catalysts. J. Am. Chem. Soc. 2001, 123, 6543–6554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Gryp, P.; Marx, S.; Vosloo, H.C.M. Experimental, DFT and Kinetic Study of 1-Octene Metathesis with Hoveyda–Grubbs Second Generation Precatalyst. J. Mol. Catal. Chem. 2012, 355, 85–95. [Google Scholar] [CrossRef]
- du Toit, J.I.; van der Gryp, P.; Loock, M.M.; Tole, T.T.; Marx, S.; Jordaan, J.H.L.; Vosloo, H.C.M. Industrial Viability of Homogeneous Olefin Metathesis: Beneficiation of Linear Alpha Olefins with the Diphenyl-Substituted Pyridinyl Alcoholato Ruthenium Carbene Precatalyst. Catal. Today 2016, 275, 191–200. [Google Scholar] [CrossRef]
- Chikkali, S.; Mecking, S. Refining of Plant Oils to Chemicals by Olefin Metathesis. Angew. Chem. Int. Ed. 2012, 51, 5802–5808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, S. Catalytic transformation of seed oil derivatives via olefin metathesis. HELIA 2014, 30, 133–142. [Google Scholar] [CrossRef]
- Higman, C.S.; Lummiss, J.A.M.; Fogg, D.E. Olefin Metathesis at the Dawn of Implementation in Pharmaceutical and Specialty-Chemicals Manufacturing. Angew. Chem. Int. Ed. 2016, 55, 3552–3565. [Google Scholar] [CrossRef]
- Dinger, M.; Mol, J. High Turnover Numbers with Ruthenium-Based Metathesis Catalysts. Adv. Synth. Catal. 2002, 344, 671–677. [Google Scholar] [CrossRef]
- Marvey, B.B.; Segakweng, C.K.; Vosloo, M.H.C. Ruthenium Carbene Mediated Metathesis of Oleate-Type Fatty Compounds. Int. J. Mol. Sci. 2008, 9, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.A.; Marvey, B.B. Room Temperature Ionic Liquids as Green Solvent Alternatives in the Metathesis of Oleochemical Feedstocks. Molecules 2016, 21, 184. [Google Scholar] [CrossRef] [Green Version]
- Zelin, J.; Trasarti, A.F.; Apesteguía, C.R. Self-metathesis of methyl oleate on silica-supported Hoveyda–Grubbs catalysts. Catal. Commun. 2013, 42, 84–88. [Google Scholar] [CrossRef]
- Pillai, S.K.; Hamoudi, S.; Belkacemi, K. Functionalized Value-Added Products via Metathesis of Methyloleate over Methyltrioxorhenium Supported on ZnCl2-Promoted Mesoporous Alumina. Fuel 2013, 110, 32–39. [Google Scholar] [CrossRef]
- Salameh, A.; Baudouin, A.; Basset, J.M.; Copéret, C. Tuning the Selectivity of Alumina-Supported (CH3)ReO3 by Modifying the Surface Properties of the Support. Angew. Chem. Int. Ed. 2008, 47, 2117–2120. [Google Scholar] [CrossRef] [PubMed]
- Hasib-ur Rahman, M.; Hamoudi, S.; Belkacemi, K. Fatty Acid Methyl Ester Heterogeneous Self-metathesis in Hydrophobic Green Solvent: Mass Transfer Limitations, Catalyst Recyclability, and Stability. Can. J. Chem. Eng. 2018, 96, 223–230. [Google Scholar] [CrossRef]
- Engle, K.M.; Lu, G.; Luo, S.X.; Henling, L.M.; Takase, M.K.; Liu, P.; Houk, K.N.; Grubbs, R.H. Origins of Initiation Rate Differences in Ruthenium Olefin Metathesis Catalysts Containing Chelating Benzylidenes. J. Am. Chem. Soc. 2015, 137, 5782–5792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, V.; Hendann, M.; Wannowius, K.J.; Plenio, H. On the Mechanism of the Initiation Reaction in Grubbs–Hoveyda Complexes. J. Am. Chem. Soc. 2012, 134, 1104–1114. [Google Scholar] [CrossRef]
- Liu, P.; Xu, X.; Dong, X.; Keitz, B.K.; Herbert, M.B.; Grubbs, R.H.; Houk, K.N. Z-Selectivity in Olefin Metathesis with Chelated Ru Catalysts: Computational Studies of Mechanism and Selectivity. J. Am. Chem. Soc. 2012, 134, 1464–1467. [Google Scholar] [CrossRef] [Green Version]
- Trzaskowski, B.; Goddard, W.A.; Grela, K. Faster Initiating Olefin Metathesis Catalysts from Introducing Double Bonds into Cyclopropyl, Cyclobutyl and Cyclopentyl Derivatives of Hoveyda-Grubbs Precatalysts. Mol. Catal. 2017, 433, 313–320. [Google Scholar] [CrossRef]
- Luo, S.X.; Engle, K.M.; Dong, X.; Hejl, A.; Takase, M.K.; Henling, L.M.; Liu, P.; Houk, K.N.; Grubbs, R.H. An Initiation Kinetics Prediction Model Enables Rational Design of Ruthenium Olefin Metathesis Catalysts Bearing Modified Chelating Benzylidenes. ACS Catal. 2018, 8, 4600–4611. [Google Scholar] [CrossRef] [Green Version]
- Benitez, D.; Tkatchouk, E.; Goddard, W.A., III. Relevance of cis- and trans-dichloride Ru intermediates in Grubbs-II olefin metathesis catalysis (H2IMesCl2RuCHR). Chem. Commun. 2008, 6194–6196. [Google Scholar] [CrossRef] [Green Version]
- Kajetanowicz, A.; Sytniczuk, A.; Grela, K. Metathesis of renewable raw materials—Influence of ligands in the indenylidene type catalysts on self-metathesis of methyl oleate and cross-metathesis of methyl oleate with (Z)-2-butene-1,4-diol diacetate. Green Chem. 2014, 16, 1579–1585. [Google Scholar] [CrossRef]
- Ahr, M.; Thieuleux, C.; Copéret, C.; Fenet, B.; Basset, J.M. Noels’ vs. Grubbs’ Catalysts: Evidence for One Unique Active Species for Two Different Systems! Adv. Synth. Catal. 2007, 349, 1587–1591. [Google Scholar] [CrossRef]
- Dang, Y.; Wang, Z.X.; Wang, X. A Thorough DFT Study of the Mechanism of Homodimerization of Terminal Olefins through Metathesis with a Chelated Ruthenium Catalyst: From Initiation to Z Selectivity to Regeneration. Organometallics 2012, 31, 7222–7234. [Google Scholar] [CrossRef]
- Keitz, B.K.; Endo, K.; Patel, P.R.; Herbert, M.B.; Grubbs, R.H. Improved Ruthenium Catalysts for Z-Selective Olefin Metathesis. J. Am. Chem. Soc. 2012, 134, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luján, C.; Nolan, S.P. E/Z selectivity in ruthenium-mediated cross metathesis. Catal. Sci. Technol. 2012, 2, 1027–1032. [Google Scholar] [CrossRef]
- Bates, J.M.; Lummiss, J.A.M.; Bailey, G.A.; Fogg, D.E. Operation of the Boomerang Mechanism in Olefin Metathesis Reactions Promoted by the Second-Generation Hoveyda Catalyst. ACS Catal. 2014, 4, 2387–2394. [Google Scholar] [CrossRef]
- Johns, A.M.; Ahmed, T.S.; Jackson, B.W.; Grubbs, R.H.; Pederson, R.L. High Trans Kinetic Selectivity in Ruthenium-Based Olefin Cross-Metathesis through Stereoretention. Org. Lett. 2016, 18, 772–775. [Google Scholar] [CrossRef] [Green Version]
- Forcina, V.; García-Domínguez, A.; Lloyd-Jones, G.C. Kinetics of Initiation of the Third Generation Grubbs Metathesis Catalyst: Convergent Associative and Dissociative Pathways. Faraday Discuss. 2019, 220, 179–195. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
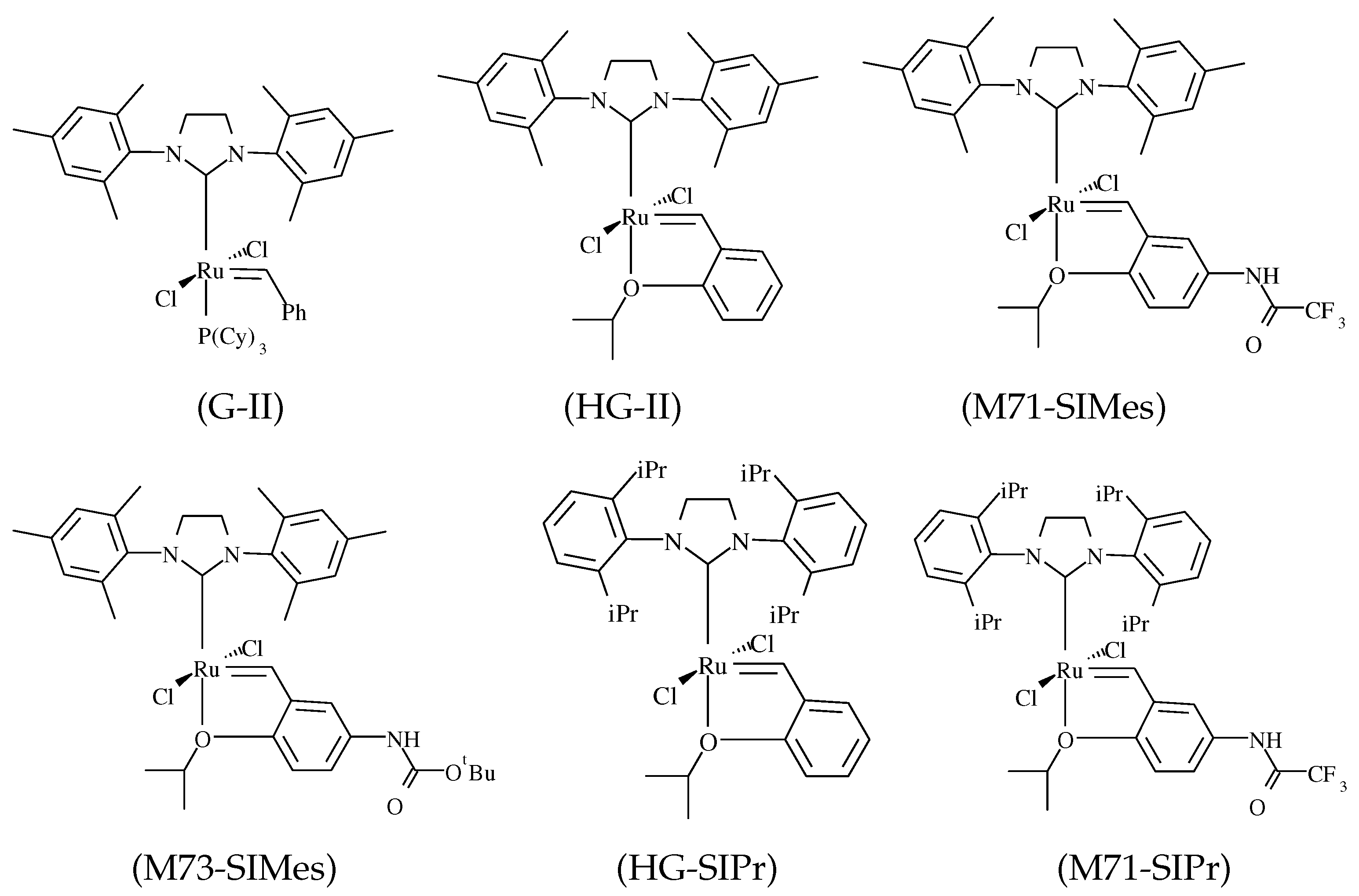
| Pre-Catalyst | |||||
|---|---|---|---|---|---|
| (s) | (L·mol·s) | (L·mol·s) | (L·mol·s) | (L·mol·s) | |
| G-II | 1.8 ± 0.1 | 120 ± 9 | 260 ± 15 | 58 ± 4 | 79 ± 5 |
| HG-II | 3.2 ± 0.2 | ||||
| M71-SIMes | 1.2 ± 0.1 | ||||
| M73-SIMes | 1.9 ± 0.3 |
| Pre-Catalyst | |||||
|---|---|---|---|---|---|
| (s) | (L·mol·s) | (L·mol·s) | (L·mol·s) | (L·mol·s) | |
| HG-SIPr | 1.8 ± 0.9 | 325 ± 5 | 650 ± 10 | 190 ± 5 | 410 ± 15 |
| M71-SIPr | 7.3 ± 0.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renom Carrasco, M.; Nikitine, C.; Hamou, M.; de Bellefon, C.; Thieuleux, C.; Meille, V. Self-Metathesis of Methyl Oleate Using Ru-NHC Complexes: A Kinetic Study. Catalysts 2020, 10, 435. https://doi.org/10.3390/catal10040435
Renom Carrasco M, Nikitine C, Hamou M, de Bellefon C, Thieuleux C, Meille V. Self-Metathesis of Methyl Oleate Using Ru-NHC Complexes: A Kinetic Study. Catalysts. 2020; 10(4):435. https://doi.org/10.3390/catal10040435
Chicago/Turabian StyleRenom Carrasco, Marc, Clémence Nikitine, Mohamed Hamou, Claude de Bellefon, Chloé Thieuleux, and Valérie Meille. 2020. "Self-Metathesis of Methyl Oleate Using Ru-NHC Complexes: A Kinetic Study" Catalysts 10, no. 4: 435. https://doi.org/10.3390/catal10040435
APA StyleRenom Carrasco, M., Nikitine, C., Hamou, M., de Bellefon, C., Thieuleux, C., & Meille, V. (2020). Self-Metathesis of Methyl Oleate Using Ru-NHC Complexes: A Kinetic Study. Catalysts, 10(4), 435. https://doi.org/10.3390/catal10040435