Palm Oil Conversion to Bio-Jet and Green Diesel Fuels over Cobalt Phosphide on Porous Carbons Derived from Palm Male Flowers

 ,
,
Abstract
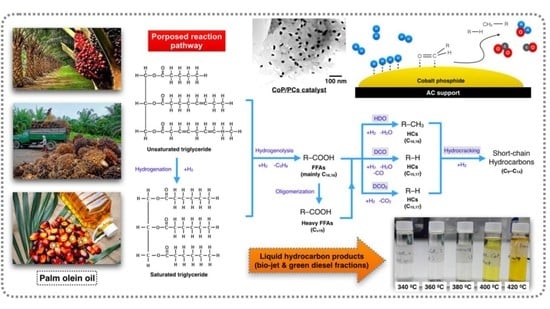
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Cobalt Phosphide@PC Catalysts
2.3. Catalytic Deoxygenation of the Palm Olein Oil
2.4. Characterization of PC and PC-Supported Cobalt Phosphide Catalysts
3. Results and Discussion
3.1. Physicochemical Characteristics of PC
3.2. Characterization of CoP/PC Catalysts
3.3. Palm oil Deoxygenation over CoP/PC Catalysts
3.3.1. Effects of Cobalt Phosphide Species on Palm Oil Deoxygenation Performance
3.3.2. Effects of DO Temperatures
3.3.3. Effects of Liquid Hourly Space Velocity (LHSV)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Bi, P.; Wang, J.; Jiang, P.; Wu, X.; Xue, H.; Liu, J.; Zhou, X.; Li, Q. Production of jet and diesel biofuels from renewable lignocellulosic biomass. Appl. Energy 2015, 150, 128–137. [Google Scholar] [CrossRef]
- Ansari, K.B.; Gaikar, V.G. Investigating production of hydrocarbon rich bio-oil from grassy biomass using vacuum pyrolysis coupled with online deoxygenation of volatile products over metallic iron. Renew. Energy 2019, 130, 305–318. [Google Scholar] [CrossRef]
- Veses, A.; Puértolas, B.; López, J.M.; Callén, M.S.; Solsona, B.; García, T. Promoting deoxygenation of bio-oil by metal-loaded hierarchical ZSM-5 zeolites. ACS Sustain. Chem. Eng. 2016, 4, 1653–1660. [Google Scholar] [CrossRef]
- Ding, R.; Wu, Y.; Chen, Y.; Liang, J.; Liu, J.; Yang, M. Effective hydrodeoxygenation of palmitic acid to diesel-like hydrocarbons over MoO2/CNTs catalyst. Chem. Eng. Sci. 2015, 135, 517–525. [Google Scholar] [CrossRef]
- Pham, L.K.H.; Tran, T.T.V.; Kongparakul, S.; Reubroycharoen, P.; Karnjanakom, S.; Guan, G.; Samart, C. Formation and activity of activated carbon supported Ni2P catalysts for atmospheric deoxygenation of waste cooking oil. Fuel Process. Technol. 2019, 185, 117–125. [Google Scholar] [CrossRef]
- Cao, Y.; Shi, Y.; Liang, J.; Wu, Y.; Huang, S.; Wang, J.; Yang, M.; Hu, H. High iso-alkanes production from palmitic acid over bi-functional Ni/H-ZSM-22 catalysts. Chem. Eng. Sci. 2017, 158, 188–195. [Google Scholar] [CrossRef]
- Cao, Y.; Shi, Y.; Bi, Y.; Wu, K.; Hu, S.; Wu, Y.; Huang, S. Hydrodeoxygenation and hydroisomerization of palmitic acid over bi-functional Co/H-ZSM-22 catalysts. Fuel Process. Technol. 2018, 172, 29–35. [Google Scholar] [CrossRef]
- Kandel, K.; Anderegg, J.W.; Nelson, N.C.; Chaudhary, U.; Slowing, I.I. Supported iron nanoparticles for the hydrodeoxygenation of microalgal oil to green diesel. J. Catal. 2014, 314, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S.; Tan, C.-S. Hydrodeoxygenation of oleic acid in hexane containing pressurized CO2 using Fe/SBA-15 as catalyst. J. Clean. Prod. 2017, 156, 203–213. [Google Scholar] [CrossRef]
- Srifa, A.; Kaewmeesri, R.; Fang, C.; Itthibenchapong, V.; Faungnawakij, K. NiAl2O4 spinel-type catalysts for deoxygenation of palm oil to green diesel. Chem. Eng. J. 2018, 345, 107–113. [Google Scholar] [CrossRef]
- Han-u-domlarpyos, V.; Kuchonthara, P.; Reubroycharoen, P.; Hinchiranan, N. Quality improvement of oil palm shell-derived pyrolysis oil via catalytic deoxygenation over NiMoS/γ-Al2O3. Fuel 2015, 143, 512–518. [Google Scholar] [CrossRef]
- Li, H.; Qiu, Y.F.; Wang, X.L.; Yang, J.; Yu, Y.J.; Chen, Y.Q.; Liu, Y.D. Biochar supported Ni/Fe bimetallic nanoparticles to remove 1,1,1-trichloroethane under various reaction conditions. Chemosphere 2017, 169, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Alsultan, G.A.; Asikin-Mijan, N.; Lee, H.V.; Albazzaz, A.S.; Taufiq-Yap, Y.H. Deoxygenation of waste cooking to renewable diesel over walnut shell-derived nanorode activated carbon supported CaO-La2O3 catalyst. Energy Convers. Manag. 2017, 151, 311–323. [Google Scholar] [CrossRef]
- Duan, Y.; Ding, R.; Shi, Y.; Fang, X.; Hu, H.; Yang, M.; Wu, Y. Synthesis of renewable Diesel range alkanes by hydrodeoxygenation of palmitic acid over 5% Ni/CNTs under mild conditions. Catalysts 2017, 7, 81. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Rao, K.T.V.; Yuan, Z.; He, S.; Rohani, S.; Xu, C. Hydrodeoxygenation of fast pyrolysis oil with novel activated carbon-supported NiP and CoP catalysts. Chem. Eng. Sci. 2018, 178, 248–259. [Google Scholar] [CrossRef]
- Asikin-Mijan, N.; Lee, H.V.; Abdulkareem-Alsultan, G.; Afandi, A.; Taufiq-Yap, Y.H. Production of green Diesel via cleaner catalytic deoxygenation of Jatropha curcas oil. J. Clean. Prod. 2017, 167, 1048–1059. [Google Scholar] [CrossRef]
- Silva, L.N.; Fortes, I.C.P.; de Sousa, F.P.; Pasa, V.M.D. Biokerosene and green Diesel from macauba oils via catalytic deoxygenation over Pd/C. Fuel 2016, 164, 329–338. [Google Scholar] [CrossRef]
- Grilc, M.; Veryasov, G.; Likozar, B.; Jesih, A.; Levec, J. Hydrodeoxygenation of solvolysed lignocellulosic biomass by unsupported MoS2, MoO2, Mo2C and WS2 catalysts. Appl. Catal. B Environ. 2015, 163, 467–477. [Google Scholar] [CrossRef]
- Shim, J.-O.; Jeon, K.-W.; Jang, W.-J.; Na, H.-S.; Cho, J.-W.; Kim, H.-M.; Lee, Y.-L.; Jeong, D.-W.; Roh, H.-S.; Ko, C.H. Facile production of biofuel via solvent-free deoxygenation of oleic acid using a CoMo catalyst. Appl. Catal. B Environ. 2018, 239, 644–653. [Google Scholar] [CrossRef]
- Grilc, M.; Likozar, B.; Levec, J. Hydrodeoxygenation and hydrocracking of solvolysed lignocellulosic biomass by oxide, reduced and sulphide form of NiMo, Ni, Mo and Pd catalysts. Appl. Catal. B Environ. 2014, 150–151, 275–287. [Google Scholar] [CrossRef]
- Wang, W.; Tan, S.; Zhu, G.; Wu, K.; Tan, L.; Li, Y.; Yang, Y. SDBS-assisted hydrothermal synthesis of flower-like Ni–Mo–S catalysts and their enhanced hydrodeoxygenation activity. RSC Adv. 2015, 5, 94040–94045. [Google Scholar] [CrossRef]
- Diaz, E.; Mohedano, A.F.; Casas, J.A.; Rodriguez, J.J. Analysis of the deactivation of Pd, Pt and Rh on activated carbon catalysts in the hydrodechlorination of the MCPA herbicide. Appl. Catal. B Environ. 2016, 181, 429–435. [Google Scholar] [CrossRef]
- Bjelić, A.; Grilc, M.; Huš, M.; Likozar, B. Hydrogenation and hydrodeoxygenation of aromatic lignin monomers over Cu/C, Ni/C, Pd/C, Pt/C, Rh/C and Ru/C catalysts: Mechanisms, reaction micro-kinetic modelling and quantitative structure-activity relationships. Chem. Eng. J. 2019, 359, 305–320. [Google Scholar] [CrossRef]
- Cecilia, J.A.; Infantes-Molina, A.; Rodriguez-Castellon, E. Hydrodechlorination of polychlorinated molecules using transition metal phosphide catalysts. J. Hazard Mater. 2015, 296, 112–119. [Google Scholar] [CrossRef]
- Griffin, M.B.; Baddour, F.G.; Habas, S.E.; Ruddy, D.A.; Schaidle, J.A. Evaluation of Silica-supported metal and metal phosphide nanoparticle catalysts for the hydrodeoxygenation of guaiacol under ex situ catalytic fast pyrolysis conditions. Top. Catal. 2015, 59, 124–137. [Google Scholar] [CrossRef]
- Chen, J.; Shi, H.; Li, L.; Li, K. Deoxygenation of methyl laurate as a model compound to hydrocarbons on transition metal phosphide catalysts. Appl. Catal. B Environ. 2014, 144, 870–884. [Google Scholar] [CrossRef]
- Guan, Q.; Wan, F.; Han, F.; Liu, Z.; Li, W. Hydrodeoxygenation of methyl palmitate over MCM-41 supported nickel phosphide catalysts. Catal. Today 2016, 259, 467–473. [Google Scholar] [CrossRef]
- Yang, Y.; Ochoa-Hernández, C.; de la Peña O’Shea, V.A.; Coronado, J.M.; Serrano, D.P. Ni2P/SBA-15 as a hydrodeoxygenation catalyst with enhanced selectivity for the conversion of methyl oleate into n-octadecane. ACS Catal. 2012, 2, 592–598. [Google Scholar] [CrossRef]
- Zarchin, R.; Rabaev, M.; Vidruk-Nehemya, R.; Landau, M.V.; Herskowitz, M. Hydroprocessing of soybean oil on nickel-phosphide supported catalysts. Fuel 2015, 139, 684–691. [Google Scholar] [CrossRef]
- Alvarez-Galvan, M.C.; Blanco-Brieva, G.; Capel-Sanchez, M.; Morales-delaRosa, S.; Campos-Martin, J.M.; Fierro, J.L.G. Metal phosphide catalysts for the hydrotreatment of non-edible vegetable oils. Catal. Today 2018, 302, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.; Guo, K.; Li, D.; Yang, H.; Hu, C. Production of high-grade diesel from palmitic acid over activated carbon-supported nickel phosphide catalysts. Appl. Catal. B Environ. 2016, 187, 375–385. [Google Scholar] [CrossRef]
- Foo, K.Y.; Hameed, B.H. Microwave-assisted preparation and adsorption performance of activated carbon from biodiesel industry solid reside: Influence of operational parameters. Bioresour. Technol. 2012, 103, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Moshood Abioye, A.; Ani, F.N. Advancement in the production of activated carbon from biomass using microwave heating. J. Teknol. 2017, 79, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Dizbay-Onat, M.; Vaidya, U.K.; Lungu, C.T. Preparation of industrial sisal fiber waste derived activated carbon by chemical activation and effects of carbonization parameters on surface characteristics. Ind. Crops Prod. 2017, 95, 583–590. [Google Scholar] [CrossRef]
- Sayğılı, H.; Güzel, F. High surface area mesoporous activated carbon from tomato processing solid waste by zinc chloride activation: Process optimization, characterization and dyes adsorption. J. Clean. Prod. 2016, 113, 995–1004. [Google Scholar] [CrossRef]
- Borchard, N.; Wolf, A.; Laabs, V.; Aeckersberg, R.; Scherer, H.W.; Moeller, A.; Amelung, W. Physical activation of biochar and its meaning for soil fertility and nutrient leaching—A greenhouse experiment. Soil Use Manag. 2012, 28, 177–184. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Xing, Z.-J.; Duan, Z.-K.; Meng, L.; Wang, Y. Effects of steam activation on the pore structure and surface chemistry of activated carbon derived from bamboo waste. Appl. Surf. Sci. 2014, 315, 279–286. [Google Scholar] [CrossRef]
- Marrakchi, F.; Ahmed, M.J.; Khanday, W.A.; Asif, M.; Hameed, B.H. Mesoporous-activated carbon prepared from chitosan flakes via single-step sodium hydroxide activation for the adsorption of methylene blue. Int. J. Biol. Macromol. 2017, 98, 233–239. [Google Scholar] [CrossRef]
- Li, S.; Han, K.; Li, J.; Li, M.; Lu, C. Preparation and characterization of super activated carbon produced from gulfweed by KOH activation. Micropor. Mesopor. Mat. 2017, 243, 291–300. [Google Scholar] [CrossRef]
- Choi, G.G.; Oh, S.J.; Lee, S.J.; Kim, J.S. Production of bio-based phenolic resin and activated carbon from bio-oil and biochar derived from fast pyrolysis of palm kernel shells. Bioresour. Technol. 2015, 178, 99–107. [Google Scholar] [CrossRef]
- dos Reis, G.S.; Wilhelm, M.; Silva, T.C.d.A.; Rezwan, K.; Sampaio, C.H.; Lima, E.C.; de Souza, S.M.A.G.U. The use of design of experiments for the evaluation of the production of surface rich activated carbon from sewage sludge via microwave and conventional pyrolysis. Appl. Therm. Eng. 2016, 93, 590–597. [Google Scholar] [CrossRef]
- Yang, X.; Ni, L. Synthesis of hybrid hydrogel of poly(AM co DADMAC)/silica sol and removal of methyl orange from aqueous solutions. Chem. Eng. J. 2012, 209, 194–200. [Google Scholar] [CrossRef]
- Deng, H.; Li, G.; Yang, H.; Tang, J.; Tang, J. Preparation of activated carbons from cotton stalk by microwave assisted KOH and K2CO3 activation. Chem. Eng. J. 2010, 163, 373–381. [Google Scholar] [CrossRef]
- Yao, Z.; Wang, G.; Shi, Y.; Zhao, Y.; Jiang, J.; Zhang, Y.; Wang, H. One-step synthesis of nickel and cobalt phosphide nanomaterials via decomposition of hexamethylenetetramine-containing precursors. Dalton Trans. 2015, 44, 14122–14129. [Google Scholar] [CrossRef] [PubMed]
- Maneeprakorn, W.; Malik, M.A.; O’Brien, P. The preparation of cobalt phosphide and cobalt chalcogenide (CoX, X = S, Se) nanoparticles from single source precursors. J. Mater. Chem. 2010, 20. [Google Scholar] [CrossRef]
- Tan, M.; Wang, X.; Wang, X.; Zou, X.; Ding, W.; Lu, X. Influence of calcination temperature on textural and structural properties, reducibility, and catalytic behavior of mesoporous γ-alumina-supported Ni–Mg oxides by one-pot template-free route. J. Catal. 2015, 329, 151–166. [Google Scholar] [CrossRef]
- Wang, H.; Li, X.; Lan, X.; Wang, T. Supported ultrafine NiCo bimetallic alloy nanoparticles derived from bimetal–organic frameworks: A highly active catalyst for furfuryl alcohol hydrogenation. ACS Catal. 2018, 8, 2121–2128. [Google Scholar] [CrossRef]
- Liu, Y.; Sotelo-Boyás, R.; Murata, K.; Minowa, T.; Sakanishi, K. Hydrotreatment of vegetable oils to produce bio-hydrogenated diesel and liquefied petroleum gas fuel over catalysts containing sulfided Ni–Mo and solid acids. Energy Fuels 2011, 25, 4675–4685. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Tan, S.; Wu, K.; Zhu, G.; Liu, Y.; Xu, Y.; Yang, Y. Preparation of NiS2//MoS2 catalysts by two-step hydrothermal method and their enhanced activity for hydrodeoxygenation of p-cresol. Fuel 2016, 179, 1–9. [Google Scholar] [CrossRef]
- Patil, S.J.; Vaidya, P.D. On the production of bio-hydrogenated diesel over hydrotalcite-like supported palladium and ruthenium catalysts. Fuel Process. Technol. 2018, 169, 142–149. [Google Scholar] [CrossRef]
- Onyestyák, G.; Harnos, S.; Szegedi, Á.; Kalló, D. Sunflower oil to green diesel over Raney-type Ni-catalyst. Fuel 2012, 102, 282–288. [Google Scholar] [CrossRef]
- Kim, S.K.; Han, J.Y.; Lee, H.-S.; Yum, T.; Kim, Y.; Kim, J. Production of renewable diesel via catalytic deoxygenation of natural triglycerides: Comprehensive understanding of reaction intermediates and hydrocarbons. Appl. Energy 2014, 116, 199–205. [Google Scholar] [CrossRef]
- Pinto, F.; Varela, F.T.; Gonçalves, M.; Neto André, R.; Costa, P.; Mendes, B. Production of bio-hydrocarbons by hydrotreating of pomace oil. Fuel 2014, 116, 84–93. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conditions | Proximate Analysis (As-Received Basis, w/w) | Ultimate Analysis (As-Received Basis, w/w) | ||||||
|---|---|---|---|---|---|---|---|---|
| M | VM | FC | A | C | H | N | O | |
| PMF | 7.6 | 61.1 | 24.0 | 7.3 | 43.7 | 2.4 | 1.2 | 52.6 |
| Carbonized PMF | 2.1 | 26.7 | 64.4 | 6.6 | 68.7 | 1.3 | 0.9 | 29.1 |
| PC 700W (s) | 2.9 | 13.7 | 74.6 | 8.9 | 79.1 | 0.9 | 0.9 | 19.2 |
| Conditions | Pore Characteristics | DXRD (nm) | NH3 Uptake (μmol/g) | |||
|---|---|---|---|---|---|---|
| SBET (m2/g) | VT (cm3/g) | Vmic (%) | Vmes (%) | |||
| PC 700W (s) | 964.0 | 0.57 | 77.92 | 22.08 | - | - |
| CoP/PC-600 | 822.9 | 0.43 | 68.79 | 31.21 | 3.4 | 52.5 |
| CoP/PC-700 | 750.5 | 0.35 | 66.85 | 33.15 | 11.7 | 85.2 |
| CoP/PC-800 | 629.2 | 0.31 | 62.06 | 37.94 | 26.8 | 113.2 |
| CoP/PC-900 | 578.2 | 0.29 | 59.72 | 40.28 | 21.7 | 118.1 |
| Catalyst | Reagent | Condition | Conversion (%) | Product | Reference |
|---|---|---|---|---|---|
| MoO2/CNTs | Palmitic acid | 190–260 °C 40 bar, 300 rpm (batch reactor) | 53.8–100 | Diesel-like hydrocarbons | [4] |
| Ni/H-ZSM-22 | Palmitic acid | 150–260 °C 40 bar, 300 rpm (batch reactor) | 72.2–100 | Green diesel | [6] |
| Ni–Co/MWCNTs | Jatropha curcas oil | 350 °C 10 bar, 1 h, 400 rpm (batch reactor) | 100 | Green diesel | [16] |
| NiP/SiO2 | Soybean oil | 340–420 °C, 30 bar, LHSV = 1 h−1 (continuous reactor) | 100 | Green diesel | [29] |
| NiP/AC | Palmitic acid | 350 °C 1 bar, (continuous reactor) | 86.2–100 | High grade diesel | [31] |
| CoP/Porous carbon | Palm olein oil | 340–420 °C, 50 bar, LHSV = 1 h−1 (continuous reactor) | 100 | Bio-jet fuel, Green diesel | This study |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaewtrakulchai, N.; Kaewmeesri, R.; Itthibenchapong, V.; Eiad-Ua, A.; Faungnawakij, K. Palm Oil Conversion to Bio-Jet and Green Diesel Fuels over Cobalt Phosphide on Porous Carbons Derived from Palm Male Flowers. Catalysts 2020, 10, 694. https://doi.org/10.3390/catal10060694
Kaewtrakulchai N, Kaewmeesri R, Itthibenchapong V, Eiad-Ua A, Faungnawakij K. Palm Oil Conversion to Bio-Jet and Green Diesel Fuels over Cobalt Phosphide on Porous Carbons Derived from Palm Male Flowers. Catalysts. 2020; 10(6):694. https://doi.org/10.3390/catal10060694
Chicago/Turabian StyleKaewtrakulchai, Napat, Rungnapa Kaewmeesri, Vorranutch Itthibenchapong, Apiluck Eiad-Ua, and Kajornsak Faungnawakij. 2020. "Palm Oil Conversion to Bio-Jet and Green Diesel Fuels over Cobalt Phosphide on Porous Carbons Derived from Palm Male Flowers" Catalysts 10, no. 6: 694. https://doi.org/10.3390/catal10060694
APA StyleKaewtrakulchai, N., Kaewmeesri, R., Itthibenchapong, V., Eiad-Ua, A., & Faungnawakij, K. (2020). Palm Oil Conversion to Bio-Jet and Green Diesel Fuels over Cobalt Phosphide on Porous Carbons Derived from Palm Male Flowers. Catalysts, 10(6), 694. https://doi.org/10.3390/catal10060694