A Review of Microwave-Assisted Synthesis-Based Approaches to Reduce Pd-Content in Catalysts


Abstract
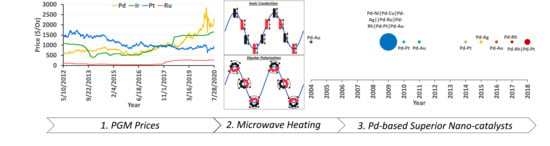
:
1. Introduction
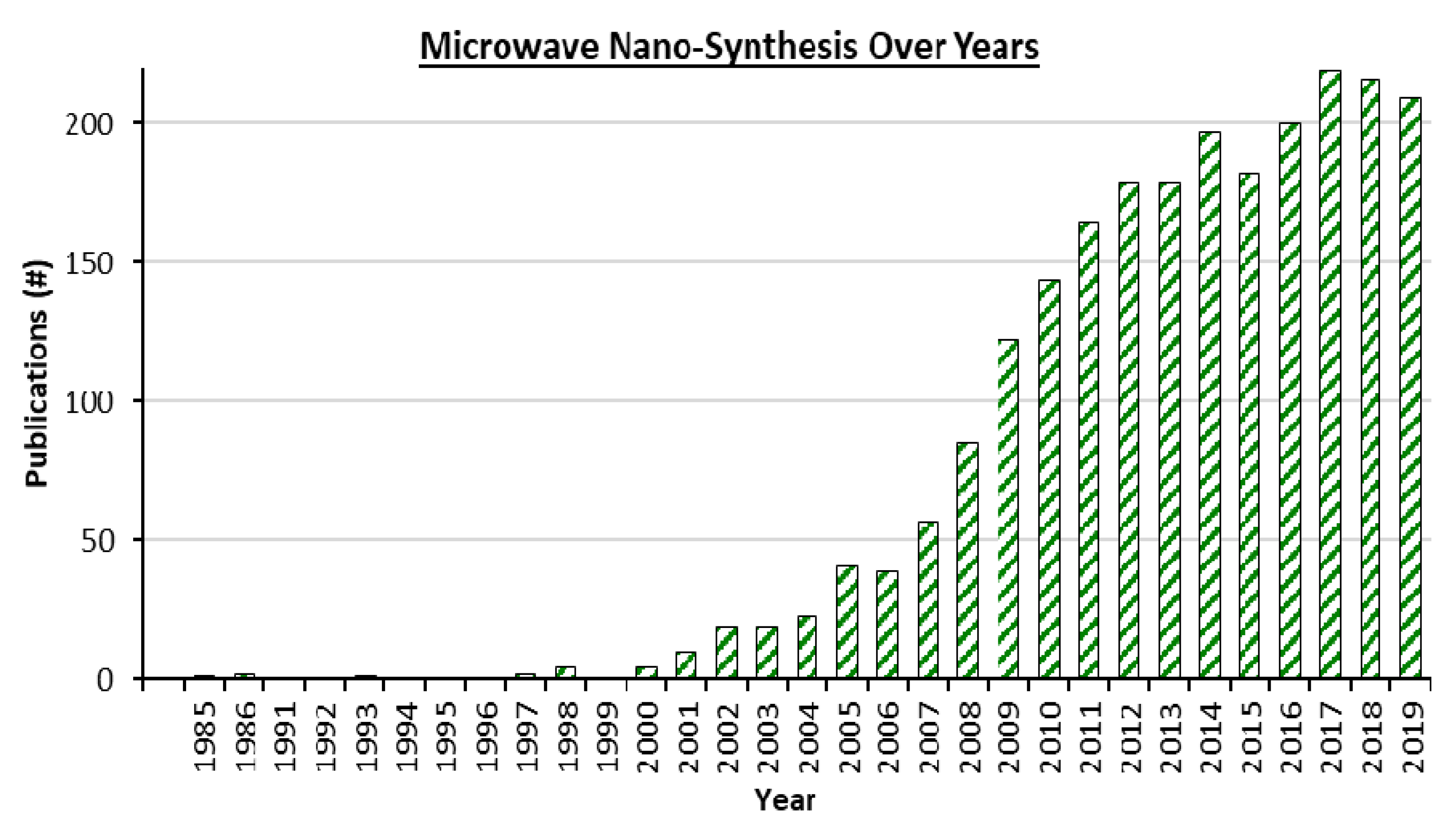
1.1. Basics of Microwave Heating
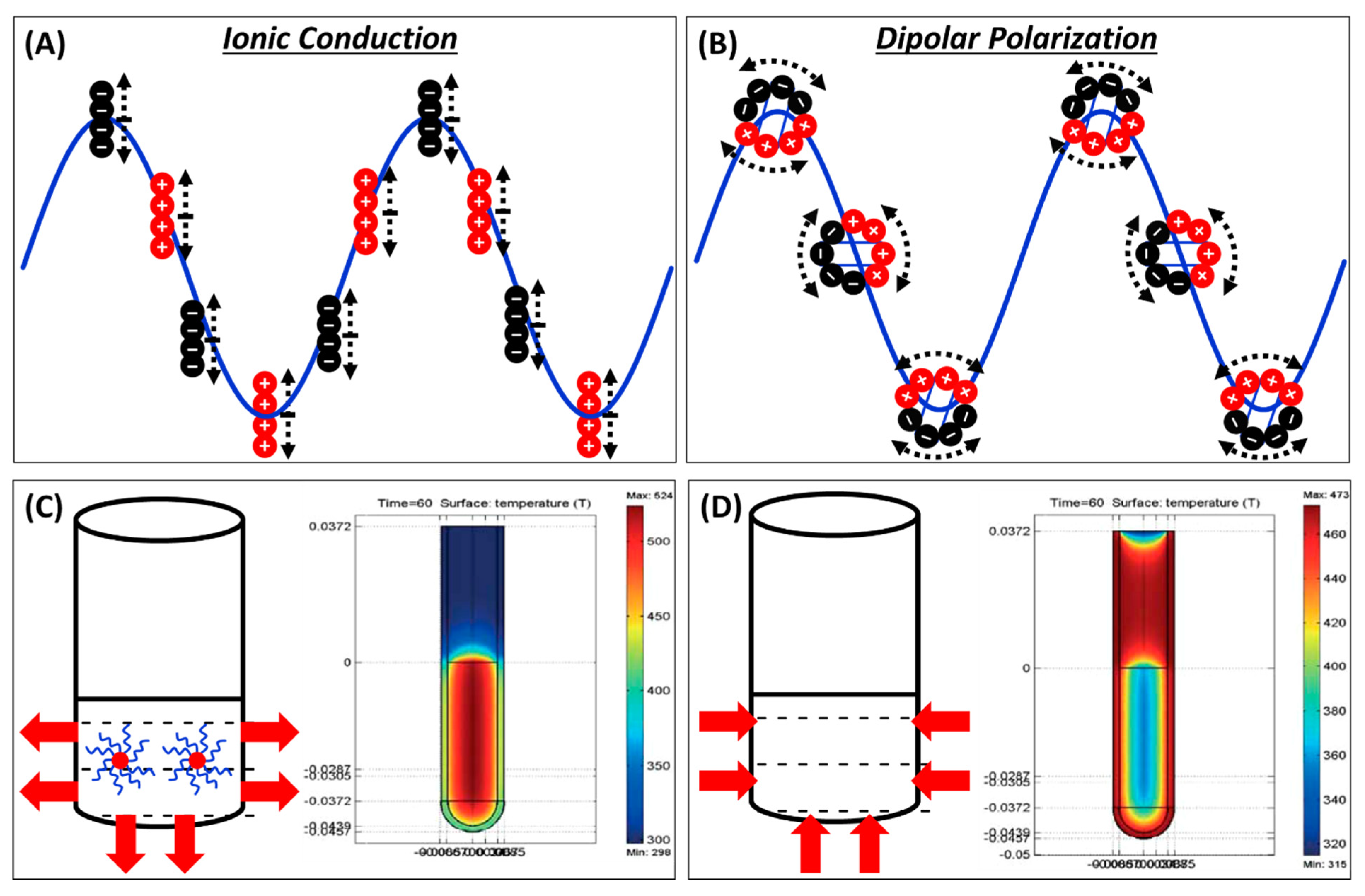
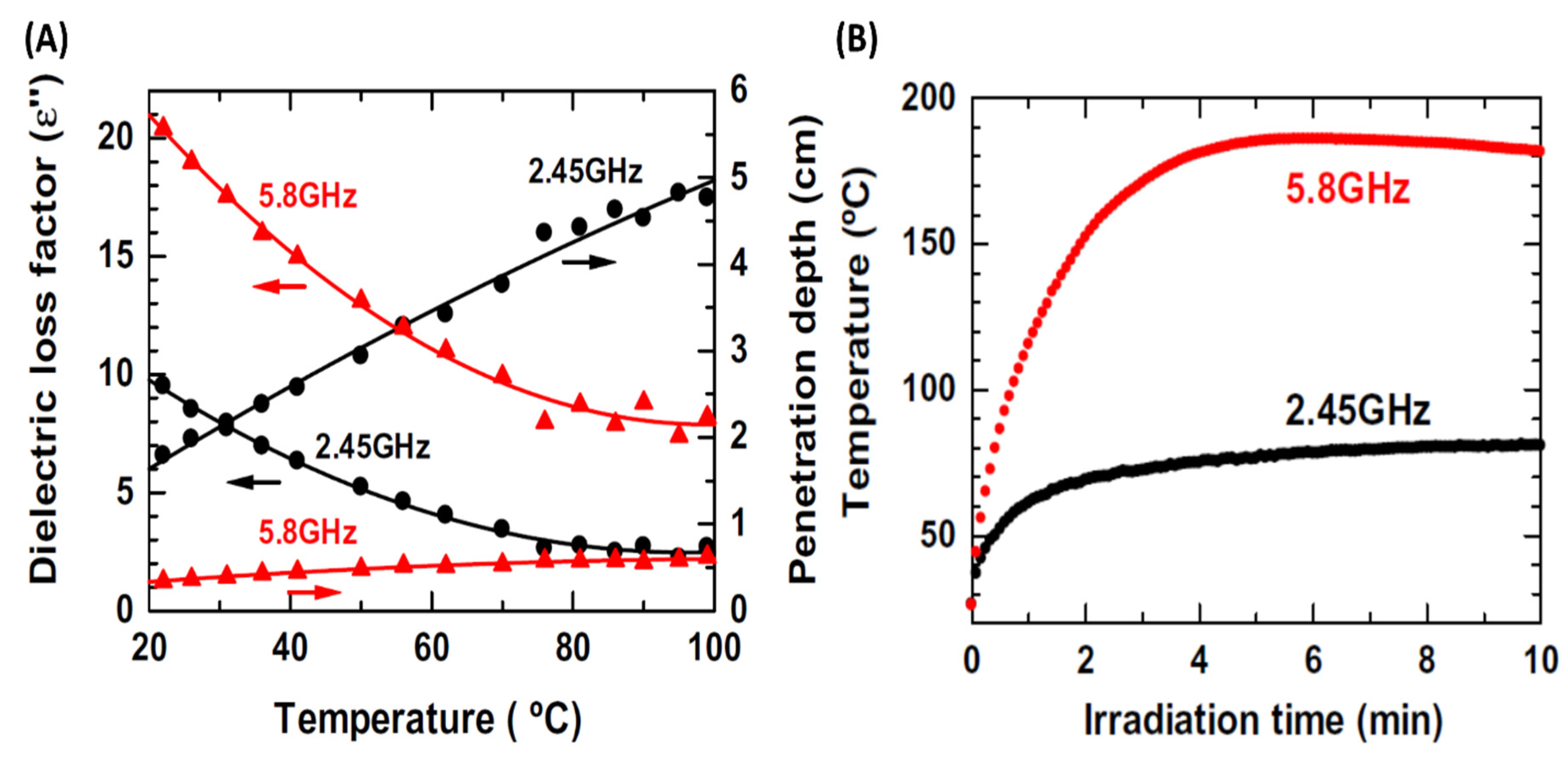
1.2. Mechanism of µw Heating
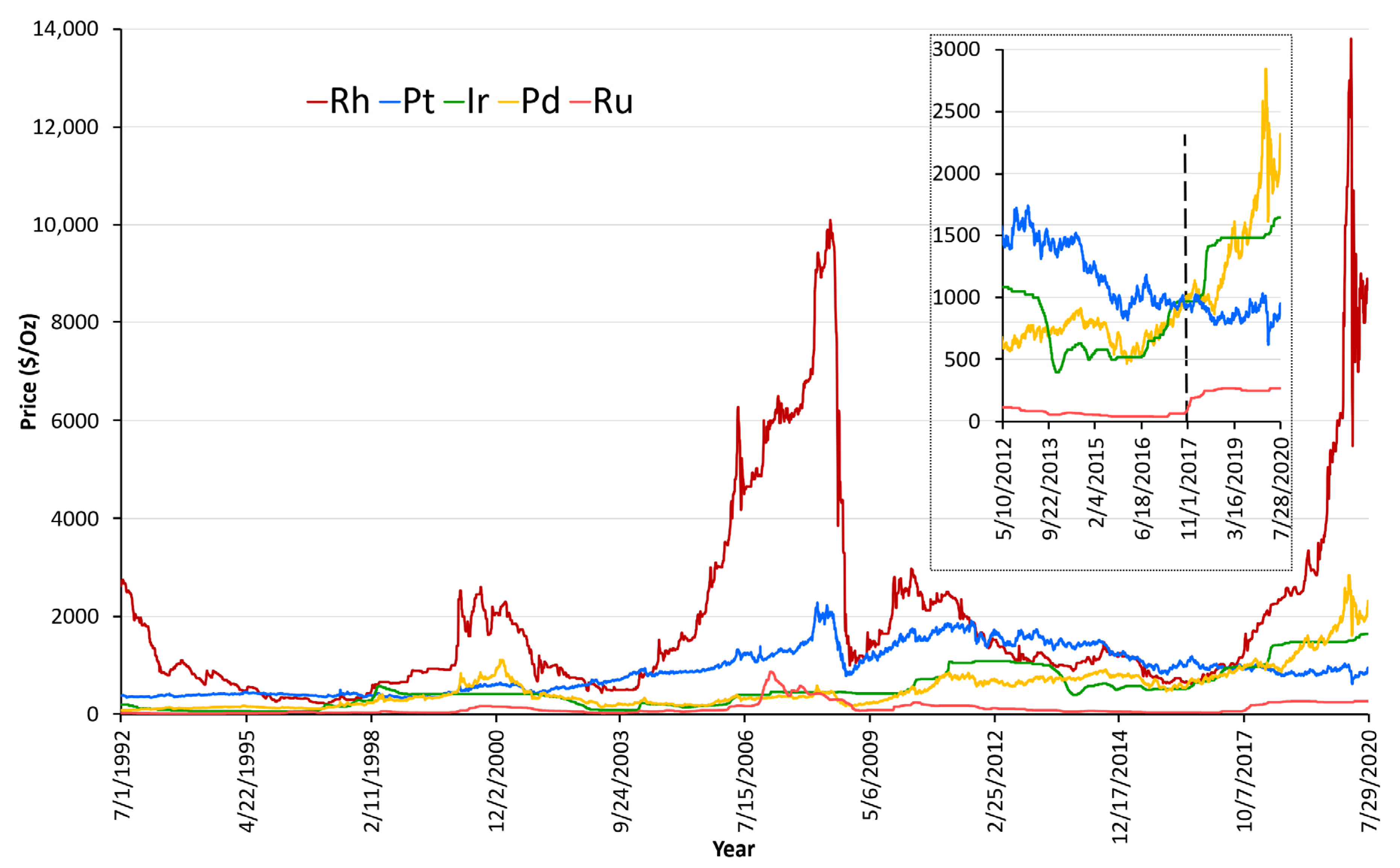
1.3. Importance of Pd in Emission Control
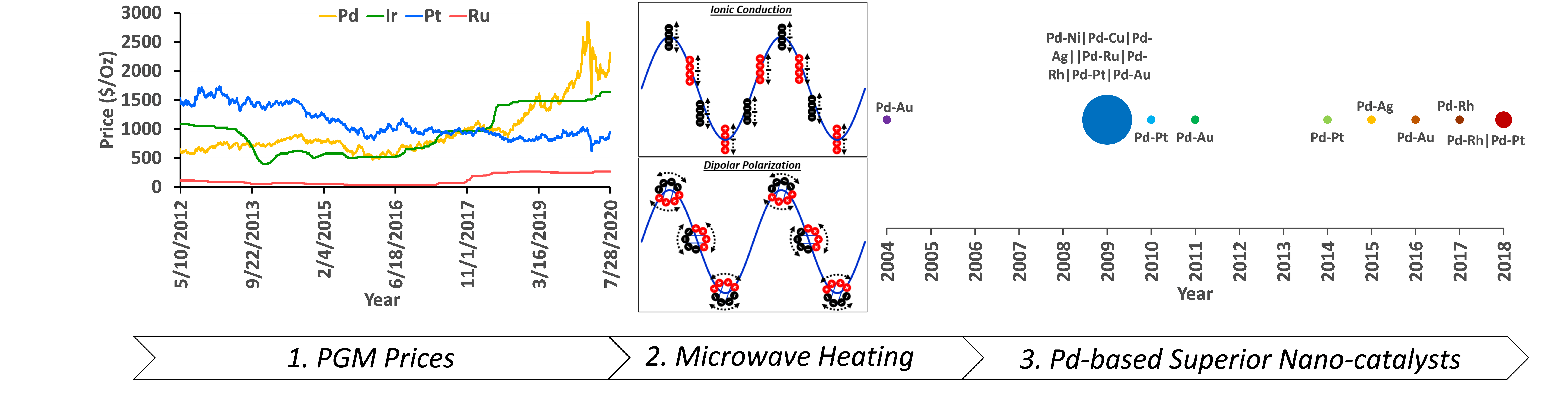
2. Synthesis of Pd-Based Bimetallic NPs using µwH
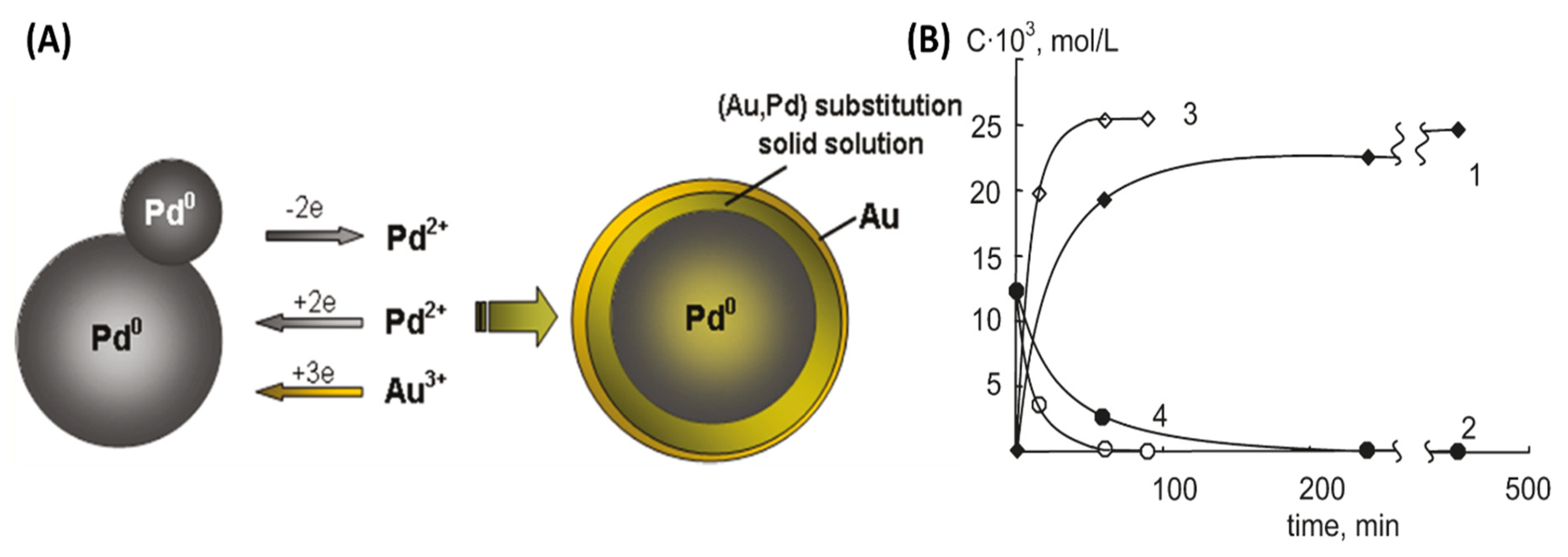
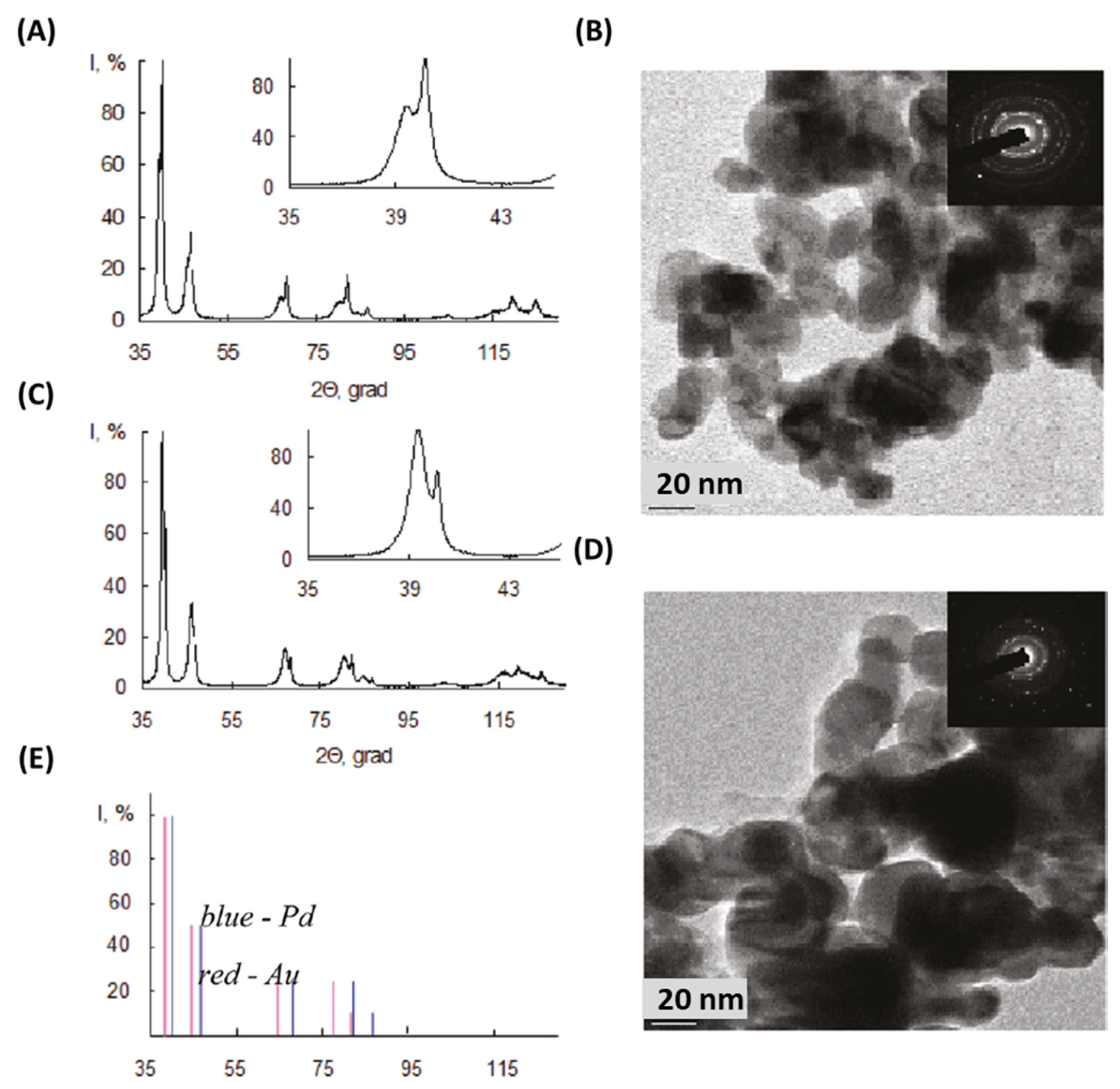
2.1. Microwave Synthesis of Au Core-Pd Shell Nanoparticles
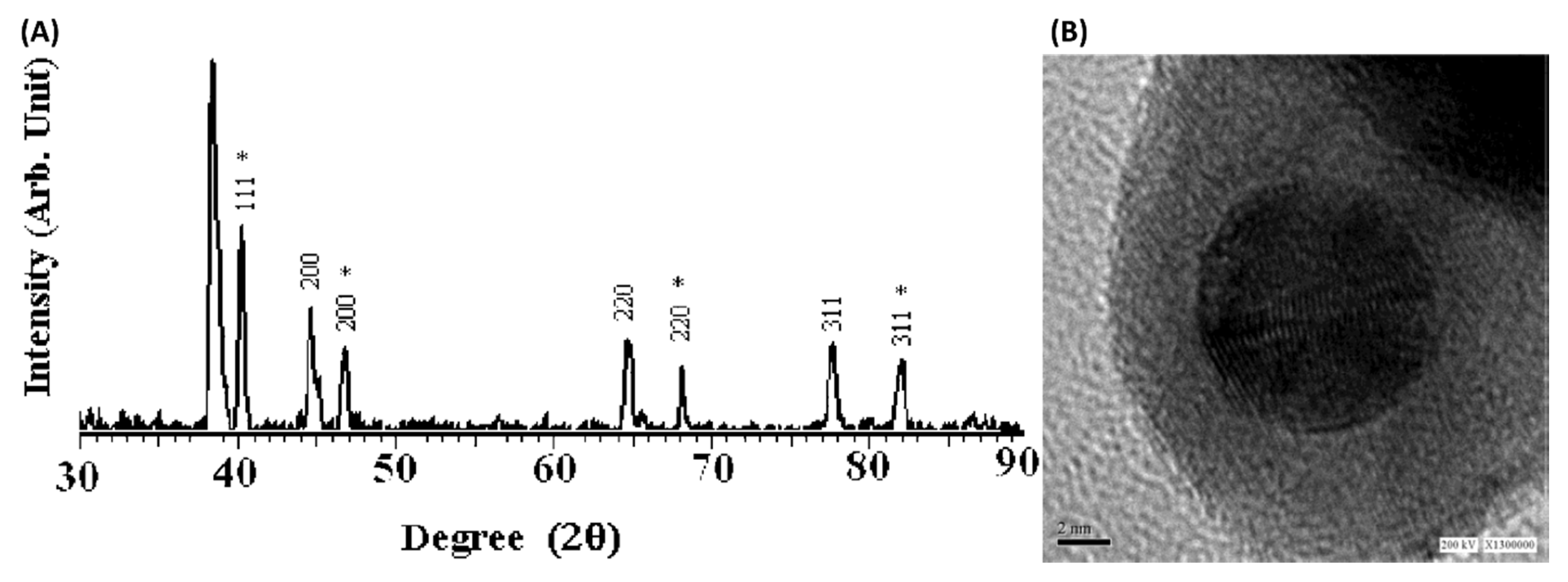
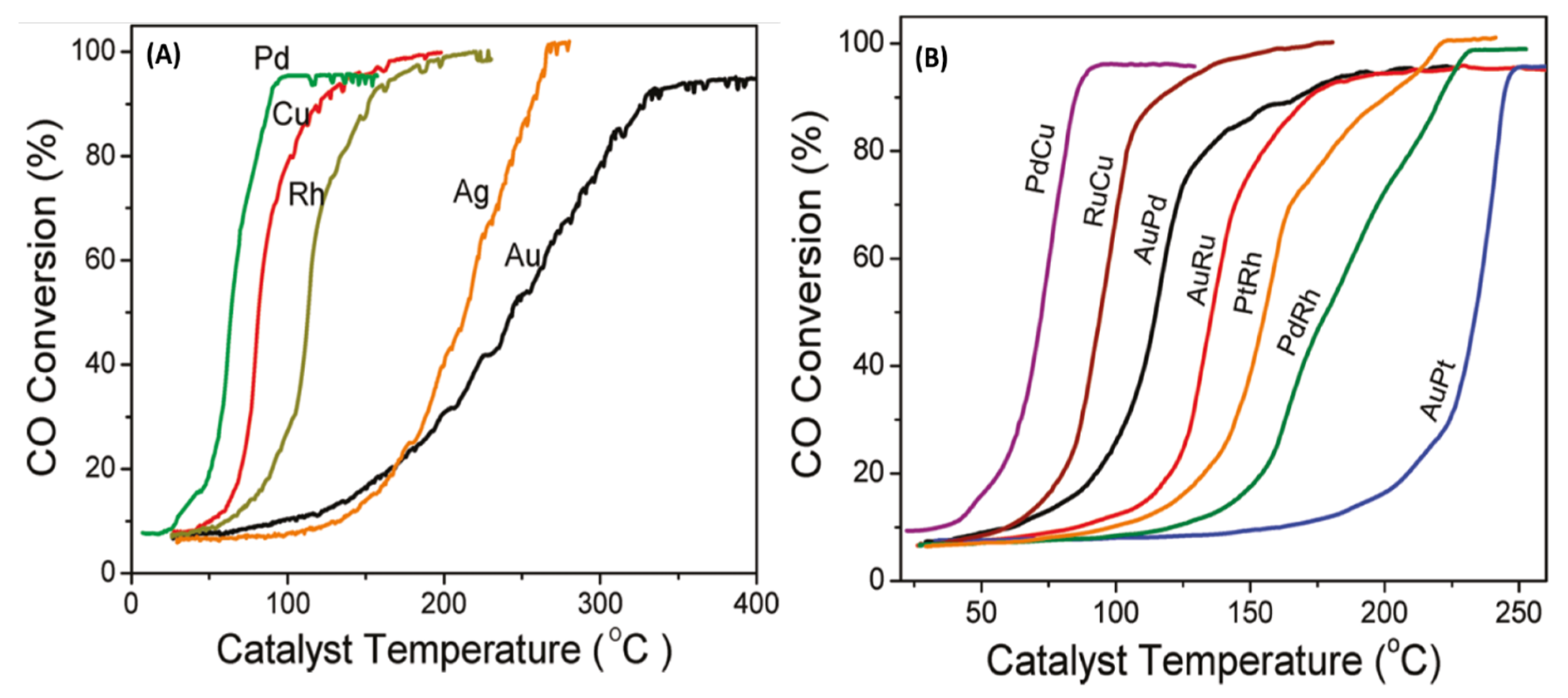
2.2. Microwave Aynthesis of (Ni/Ru/Rh/Pd/Pt)–(Cu/Ag/Au) Alloy NPs for CO Oxidation
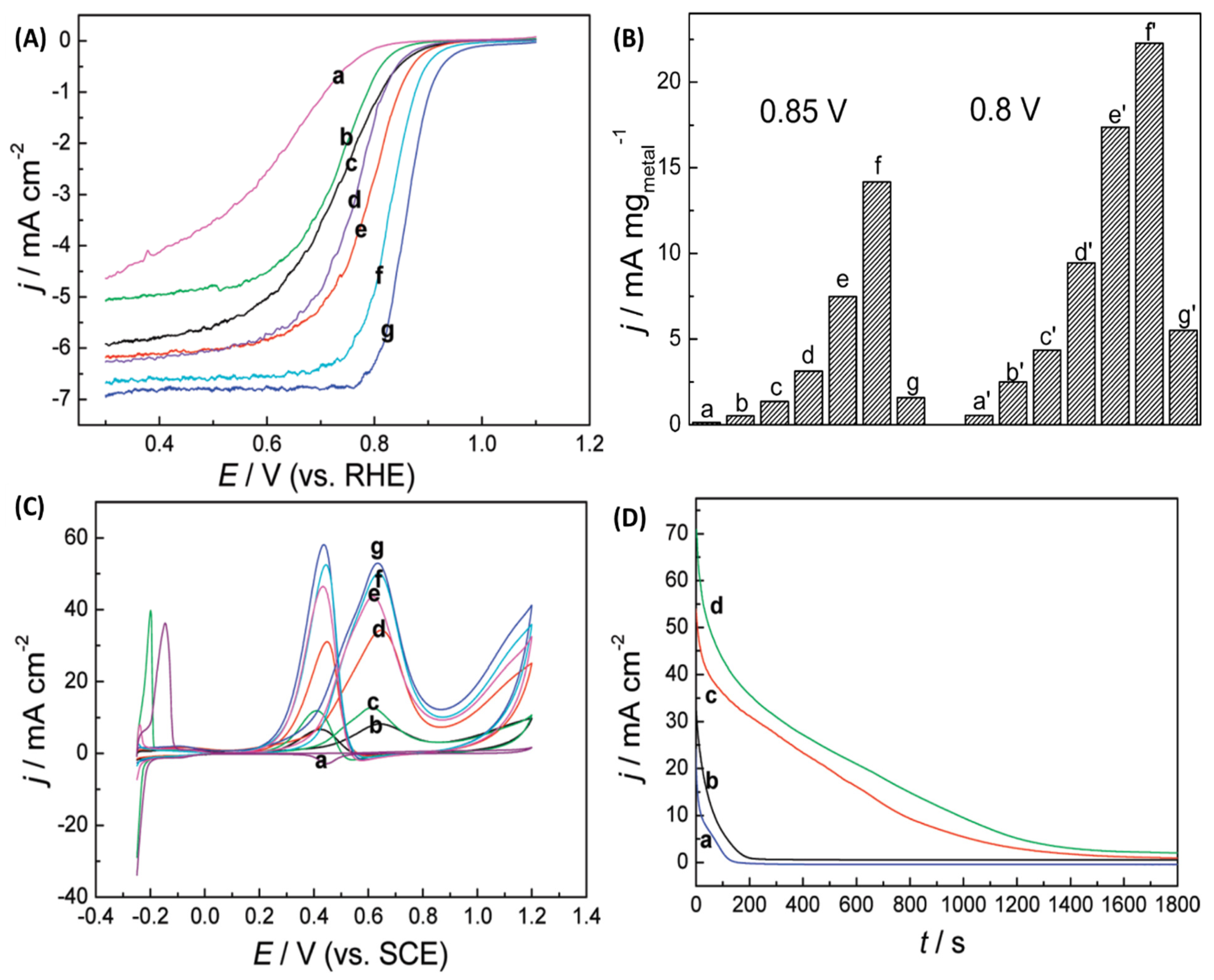
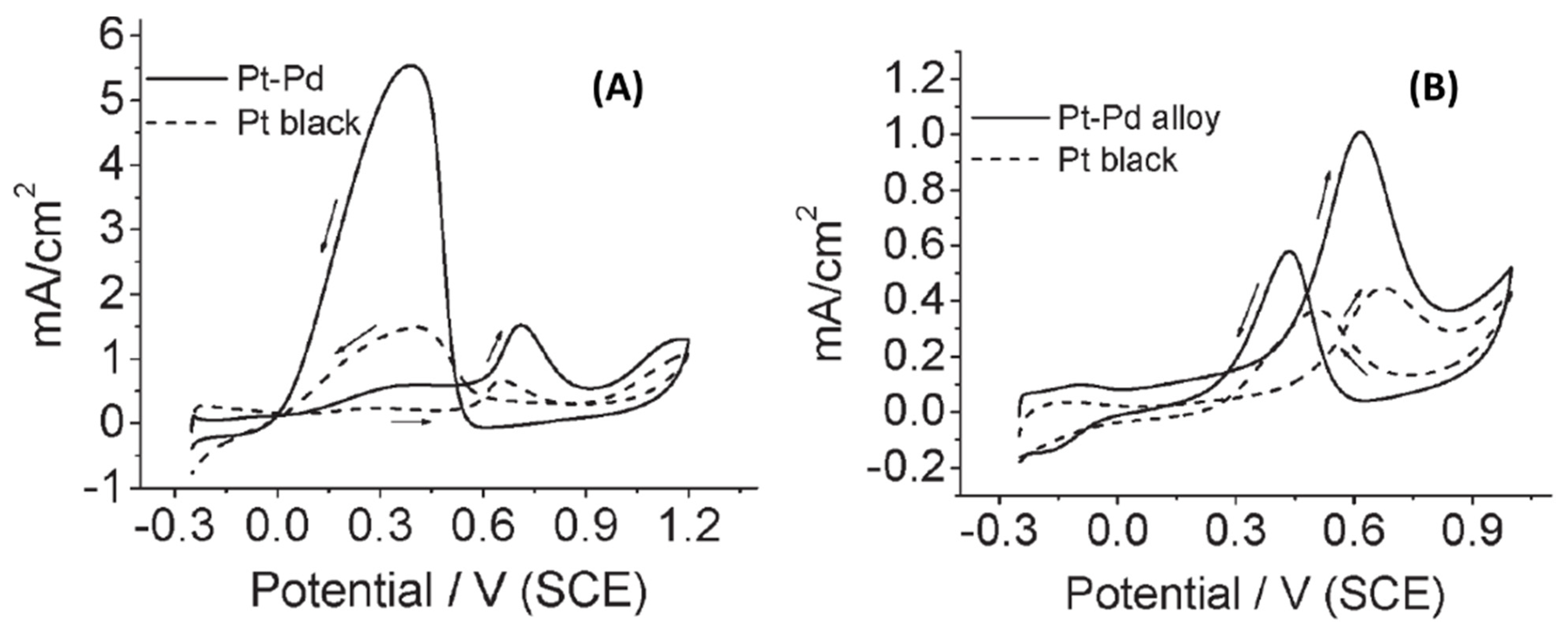
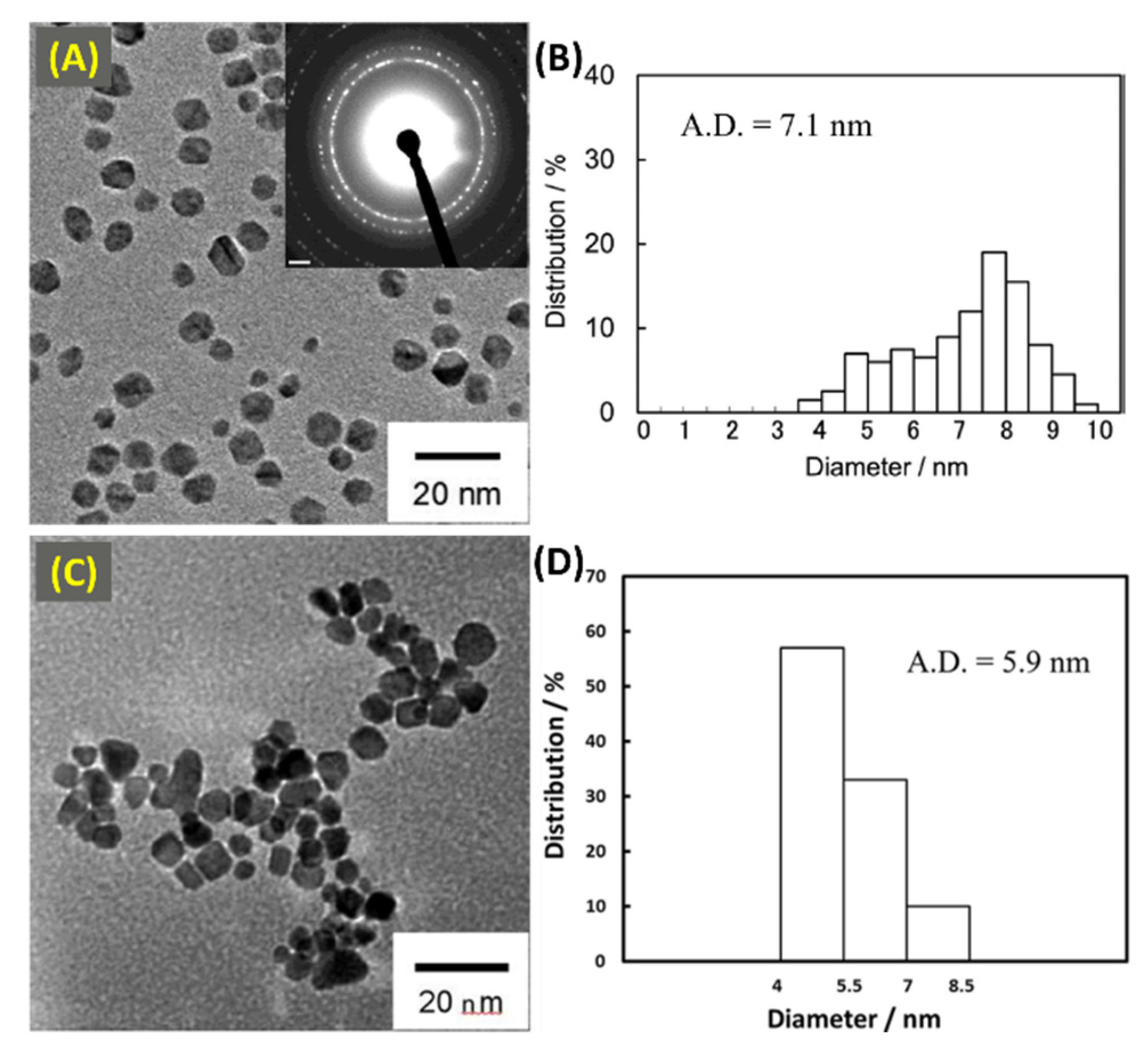
2.3. Synthesis of Composition Tunable Pd-Pt Core-Shell NPs under µwH for Oxygen Reduction and Methanol Oxidation Reactions
2.4. Synthesis of Bimetallic Pd-Au and Pt-Au NPs under µwH and CvH Conditions
2.5. Microwave Synthesis of Pd-Pt Alloy Hypercube Alloy NPs
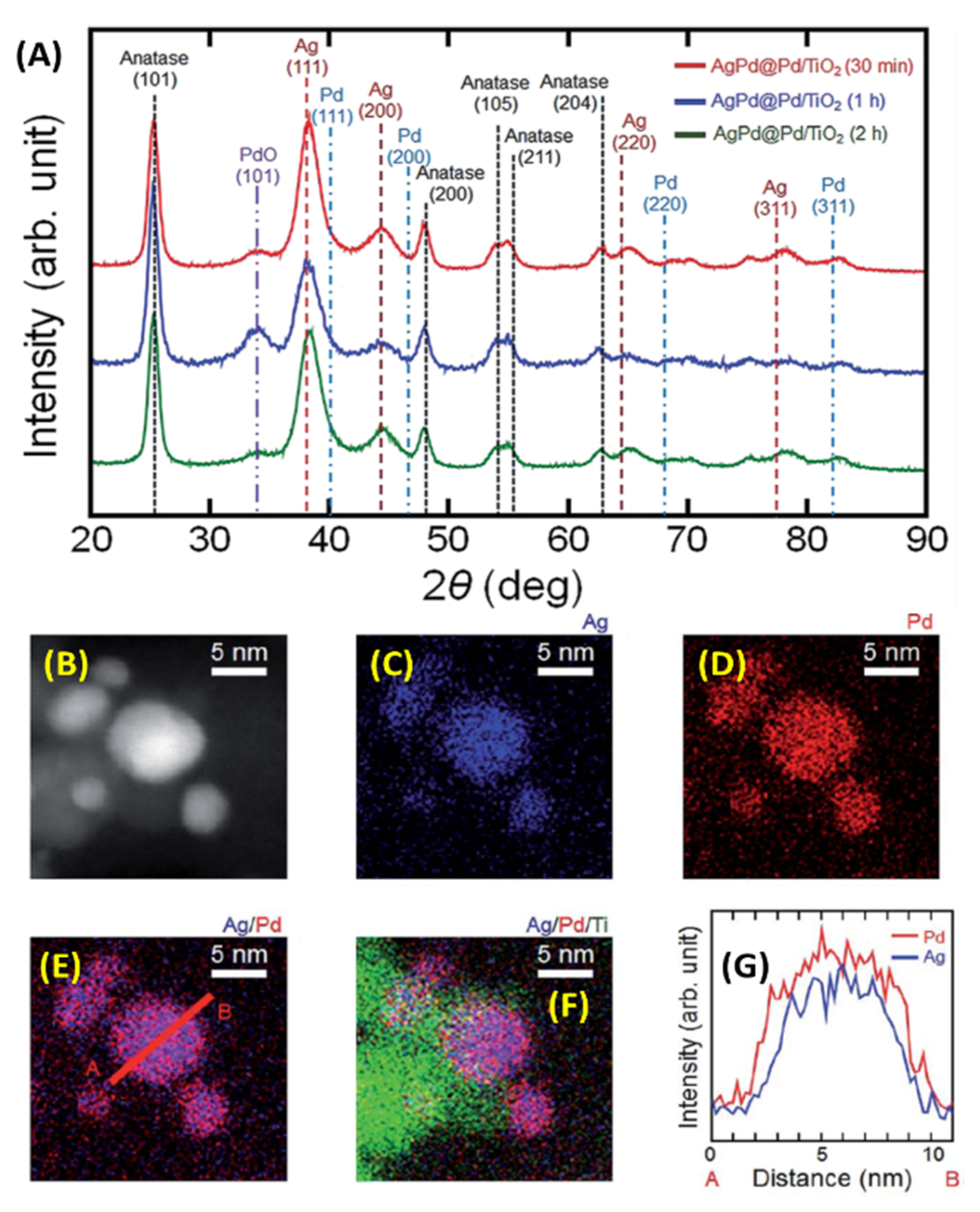
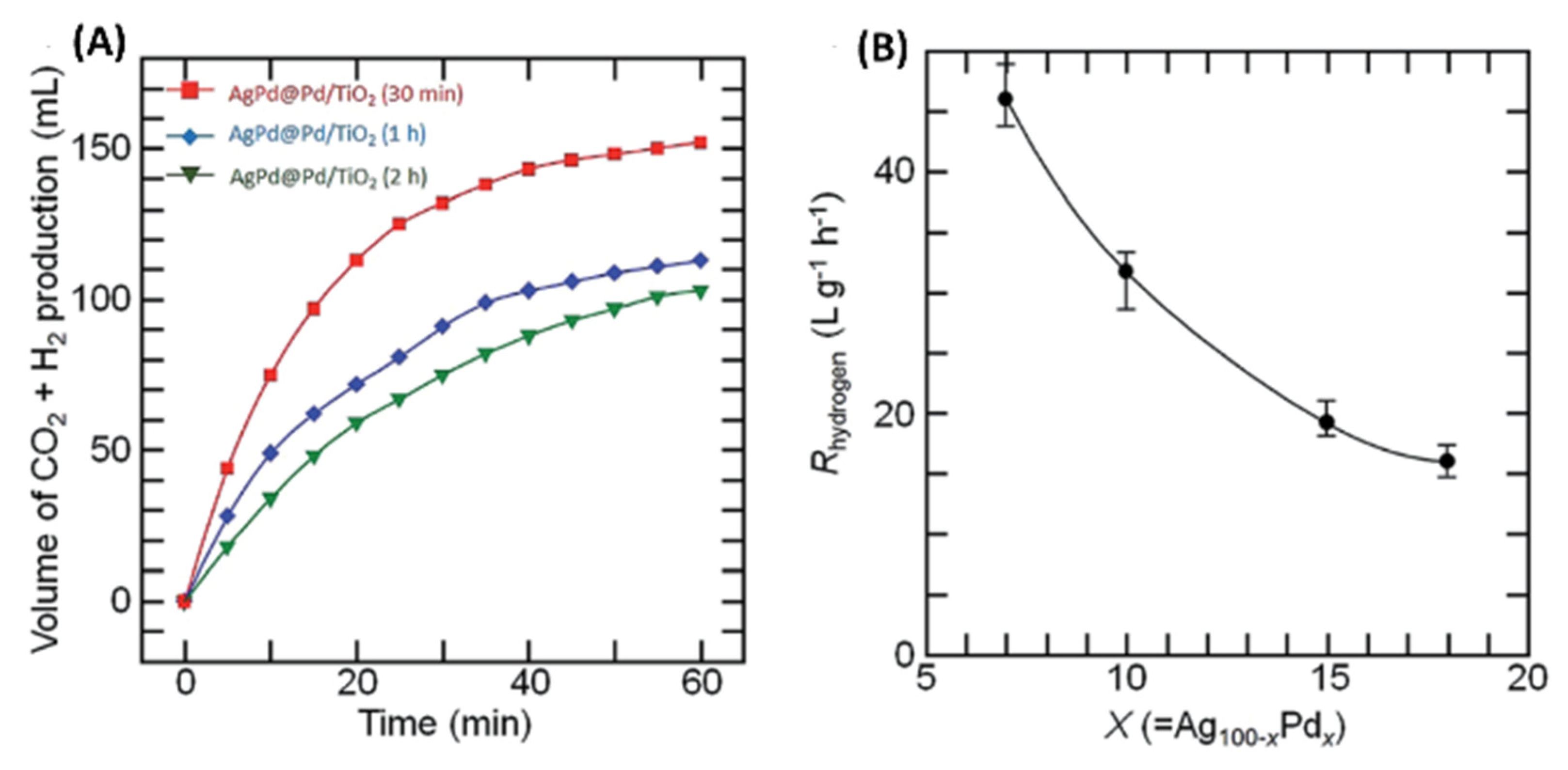
2.6. Microwave Synthesis of AgPd-Pd Core-Shell NPs/TiO2 Nanocatalysts for Hydrogen Production from Formic Acid in Water
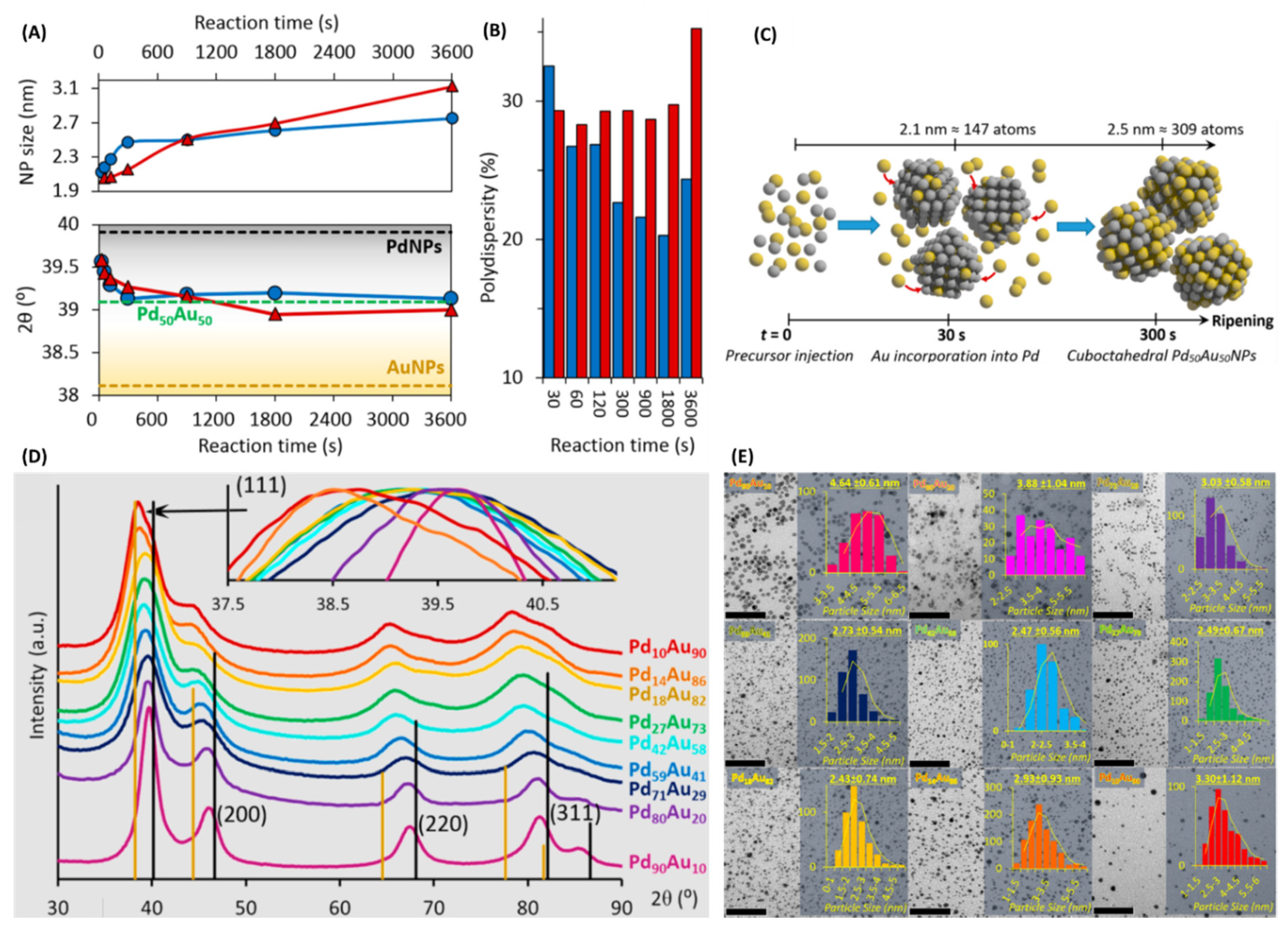
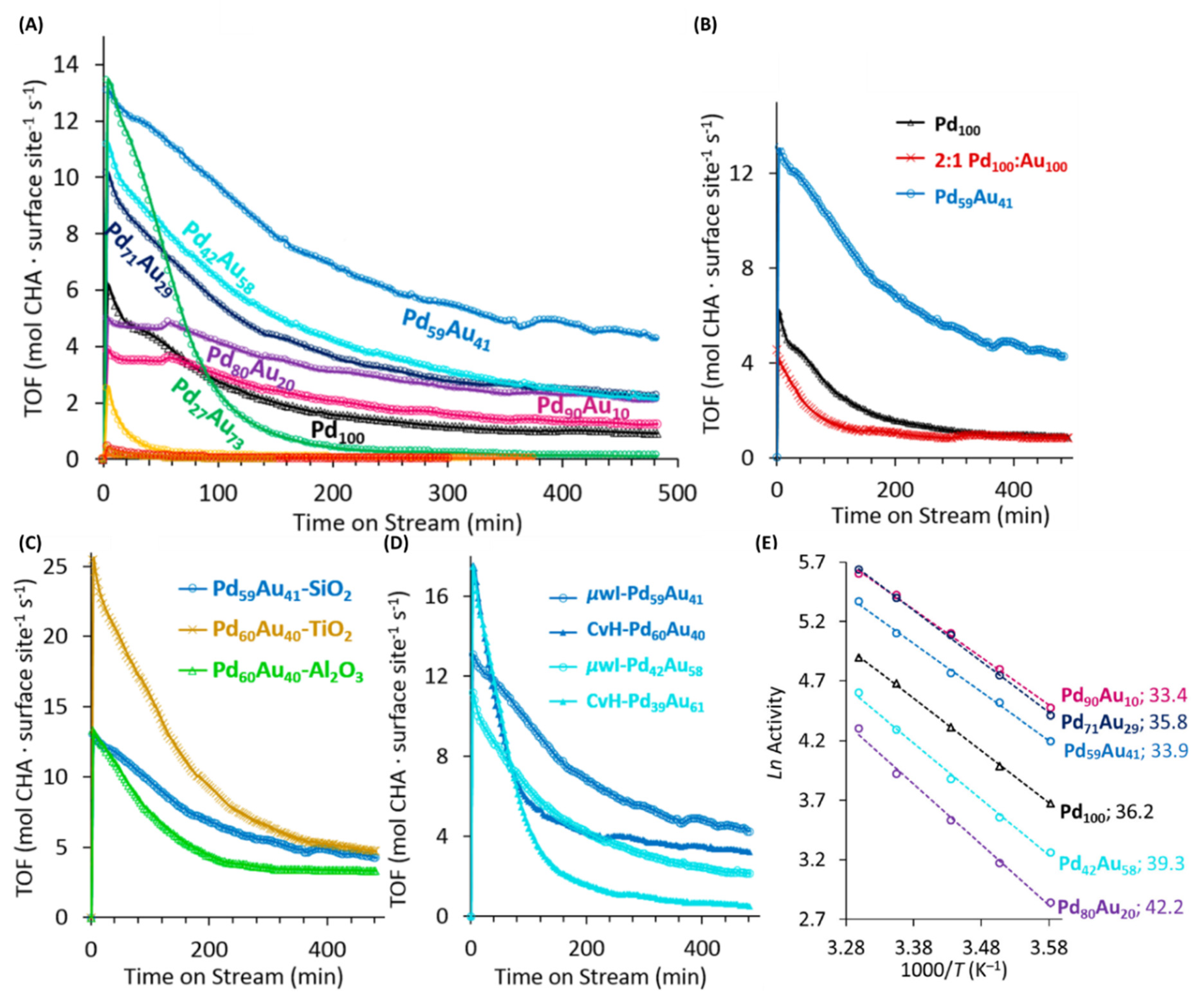
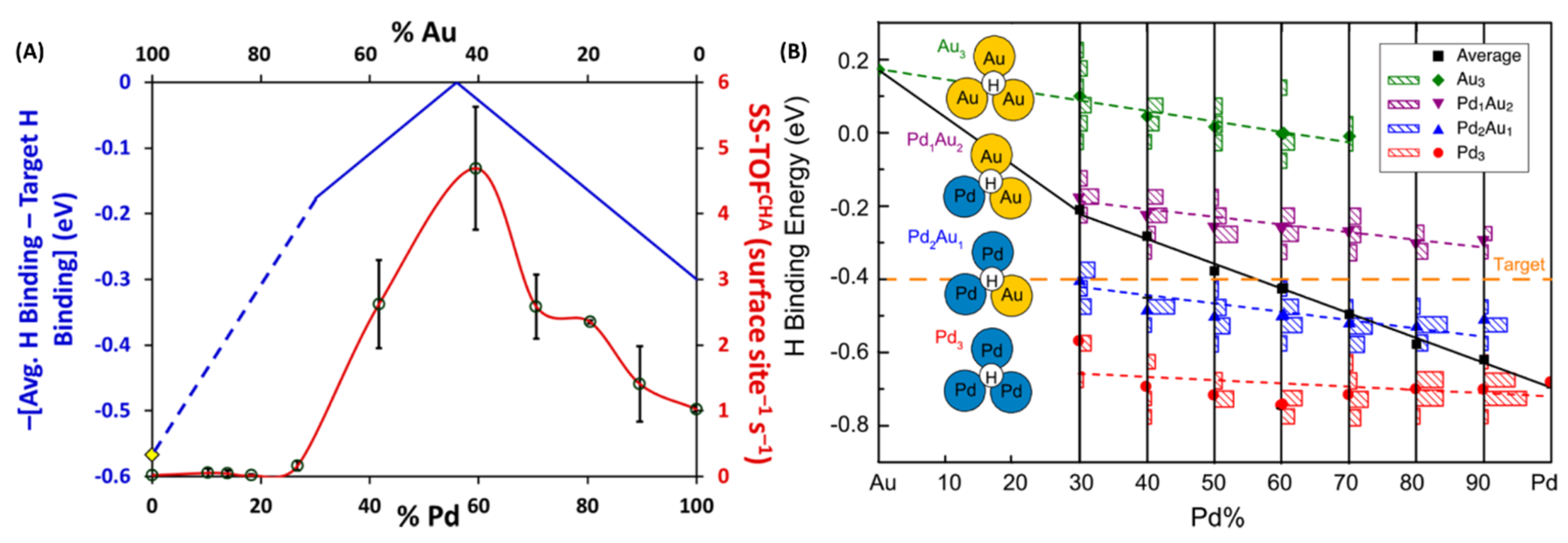
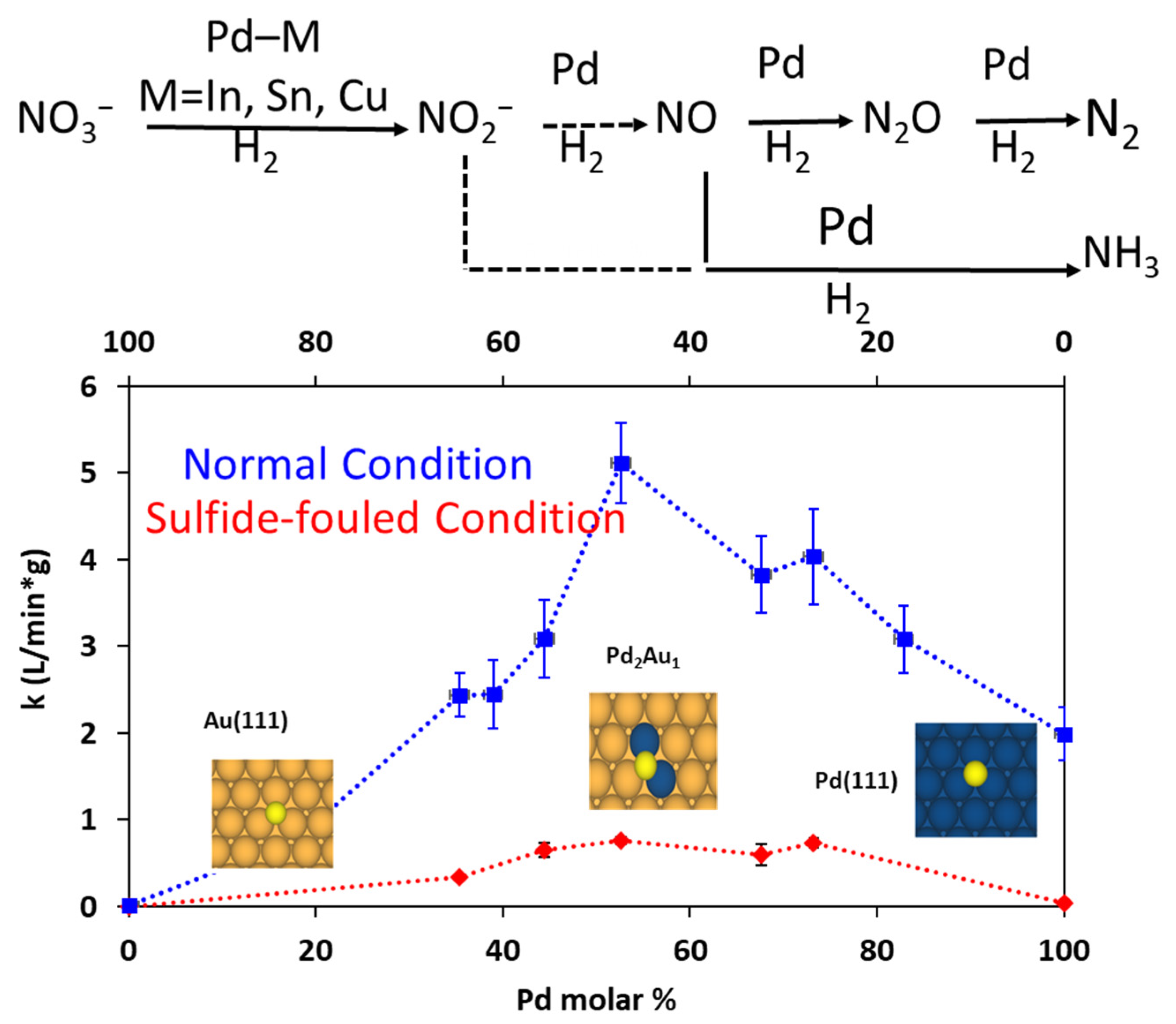
2.7. Synthesis of Various Compositions of PdxAu100-x Alloy NPs under µwH and a Comparative Catalytic Analysis for Probing such Nanostrcutures
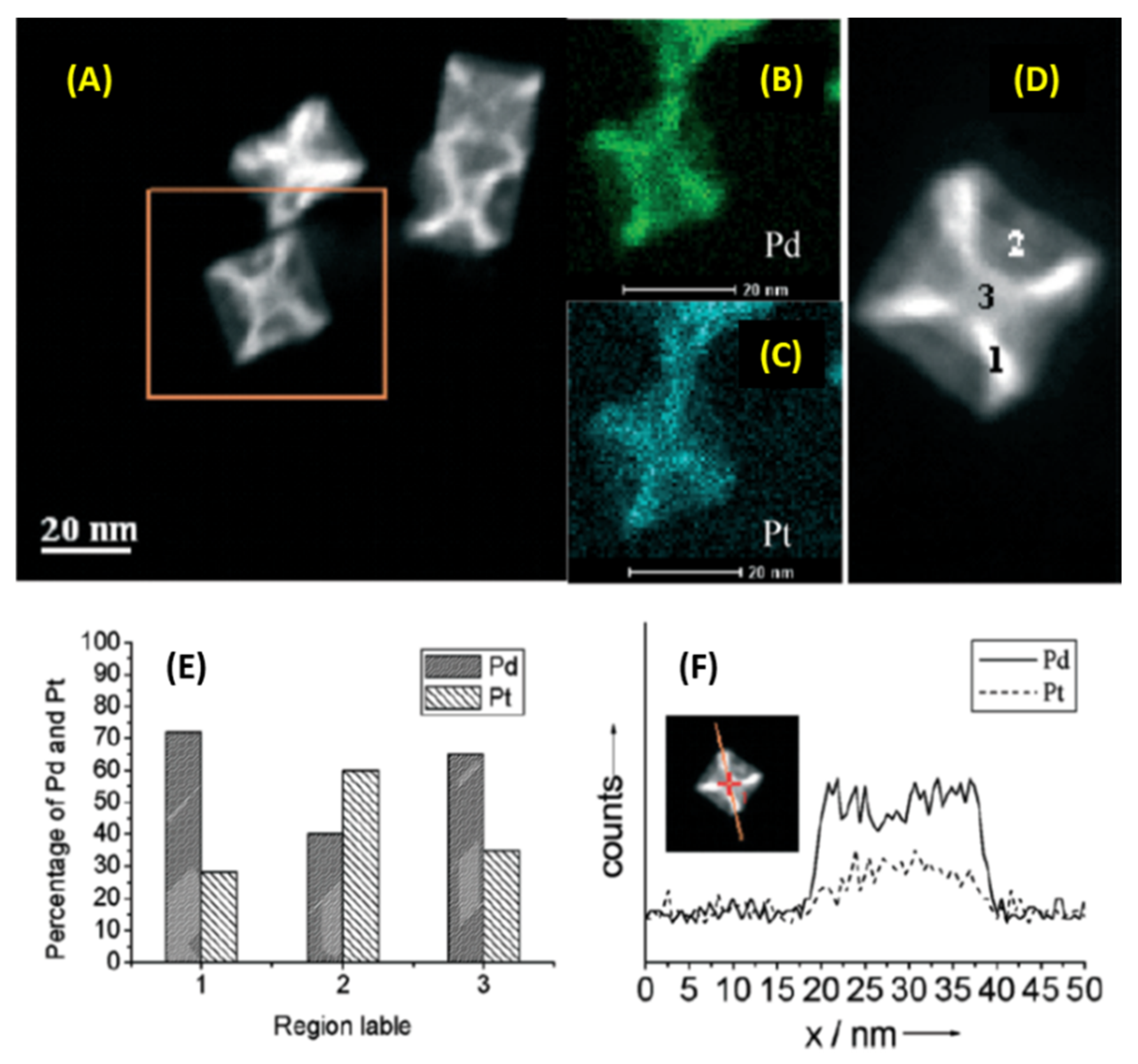
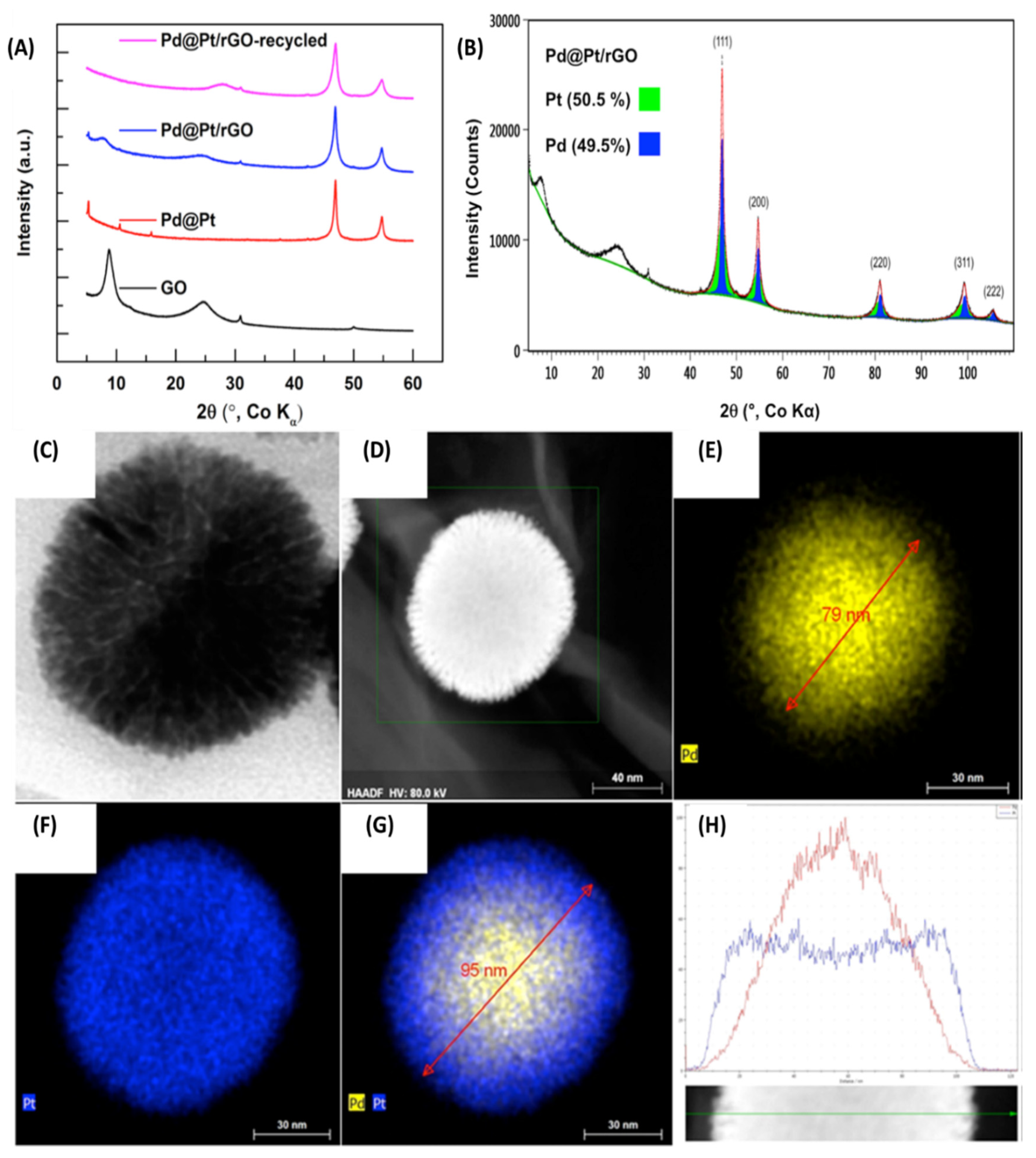
2.8. Microwave Assisted In Situ Generation of Reduced Graphene Oxide Supported Pd-Pt Core-Shell NPs for Dehalogenation Reactions and Olefin Reductions
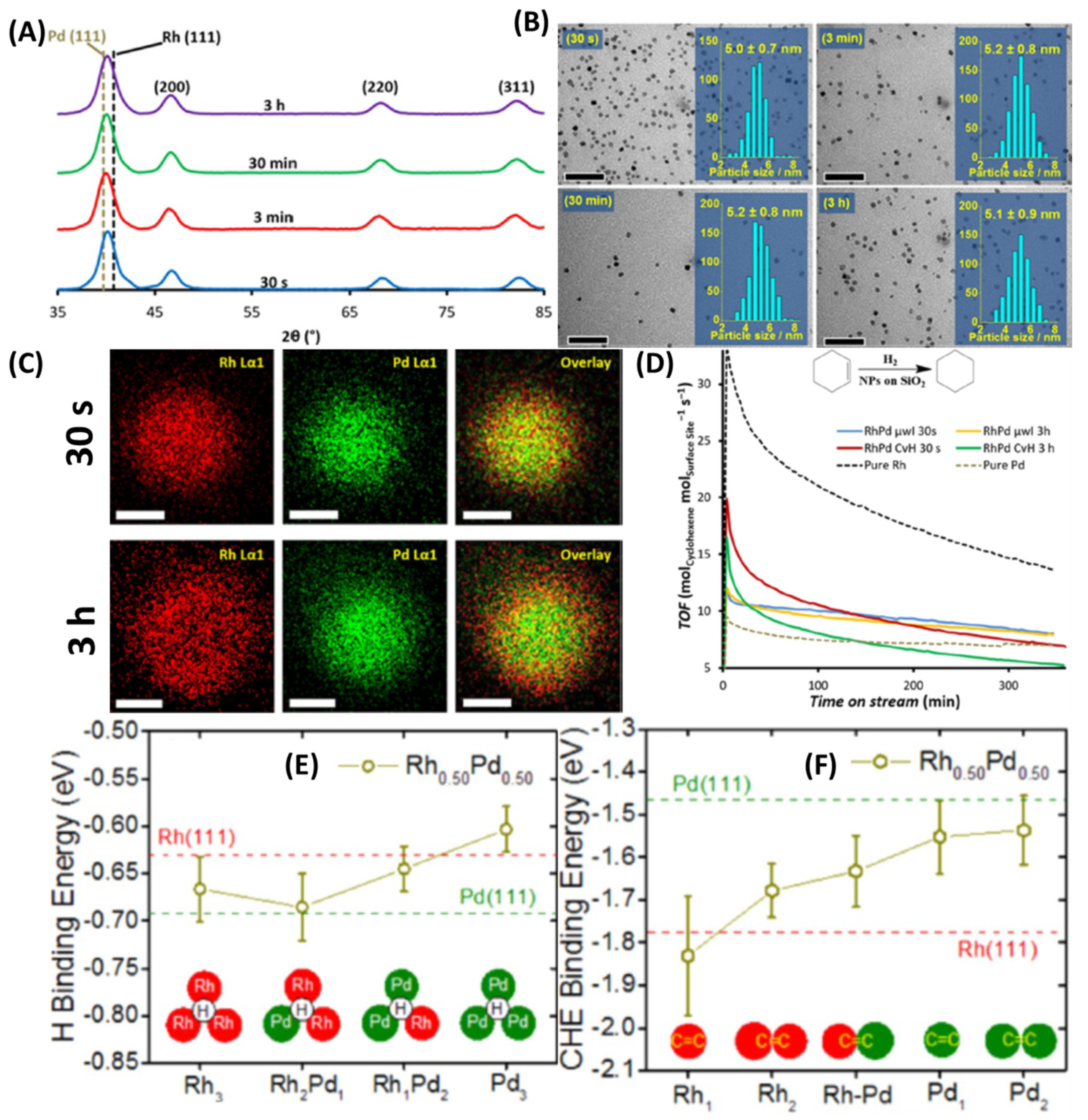
2.9. Rapid Synthesis of Pd-Rh Alloy NPs under µwH
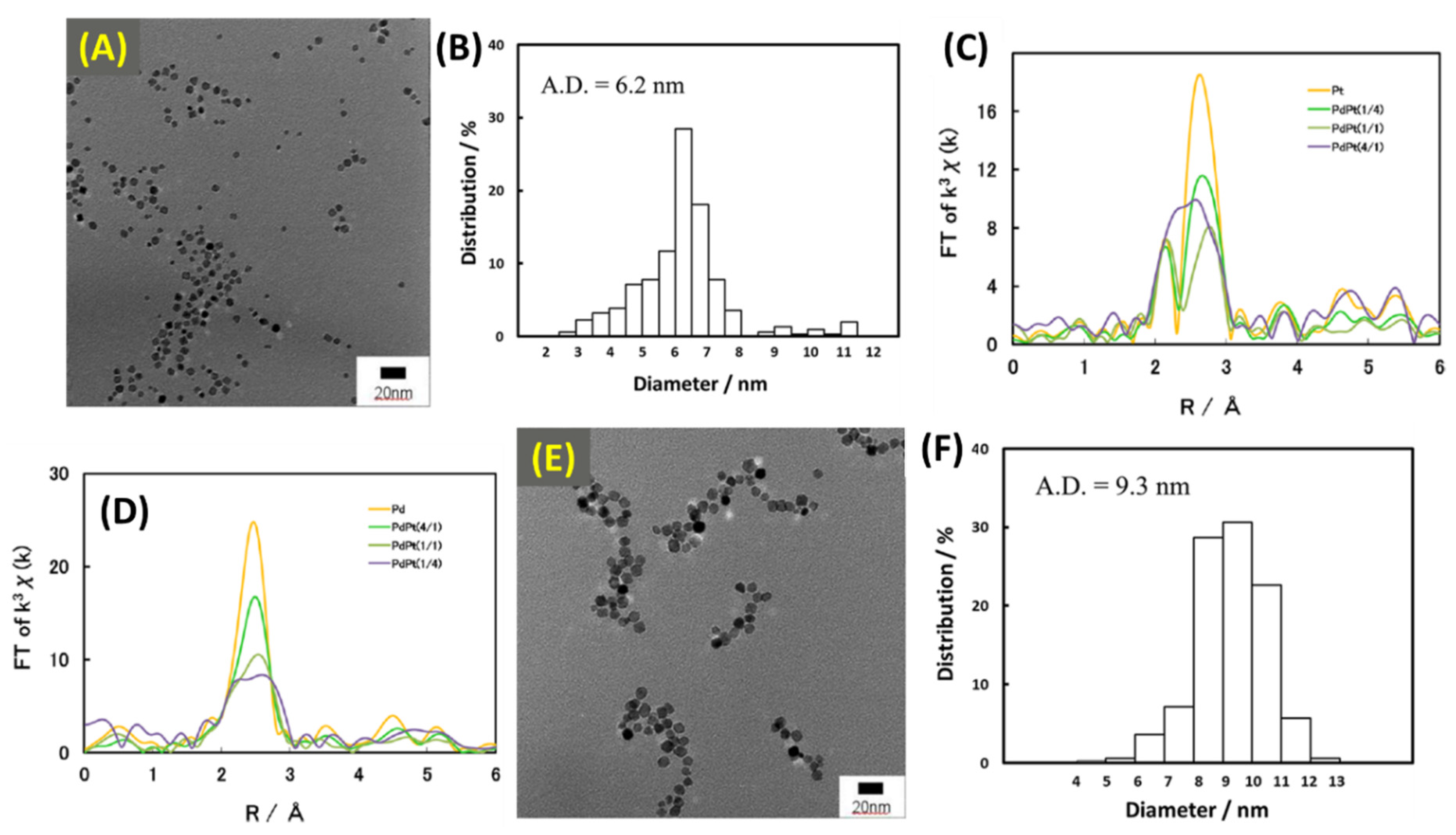
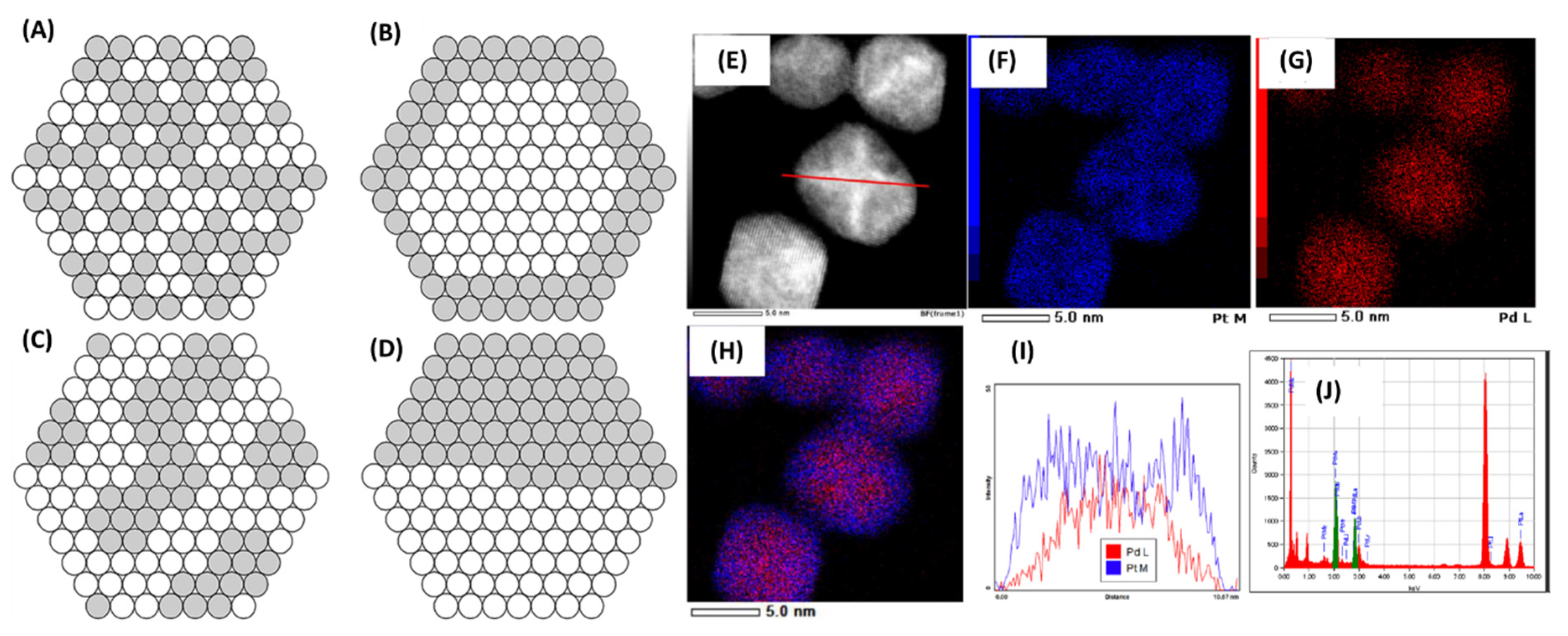
2.10. Synthesis and Structural Evaluations of Pd/Pt NPs under Batch and Continuous Flow Methods
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Komarmeni, S.; Roy, R. Titania gel spheres by a new sol-gel process. Mater. Lett. 1985, 3, 165–167. [Google Scholar] [CrossRef]
- Gedye, R.; Smith, F.; Westway, K.; Ali, H.; Baldisera, L.; Laberge, L.; Rousell, J. The use of microwave ovens for rapid organic synthesis. Tet. Lett. 1986, 27, 279–282. [Google Scholar] [CrossRef]
- Giguerre, R.J.; Bray, T.L.; Duncan, S.M. Application of commercial microwave ovens to organic synthesis. Tet. Lett. 1986, 27, 4945–4948. [Google Scholar] [CrossRef]
- Zhu, Y.-J.; Chen, F. Microwave-assisted preparation of inorganic nanostructures in liquid phase. Chem. Rev. 2014, 114, 6462–6555. [Google Scholar] [CrossRef]
- Schanche, J.-S. Microwave synthesis solutions from personal chemistry. Mol. Divers. 2003, 7, 293–300. [Google Scholar] [CrossRef]
- Horikoshi, S.; Sakai, F.; Kajitani, M.; Abe, M.; Serpone, N. Microwave frequency effects on the photoactivity of TiO2: Dielectric properties and the degradation of 4-chlorophenol, bisphenol A and methylene blue. Chem. Phys. Lett. 2009, 470, 304–307. [Google Scholar] [CrossRef]
- Horikoshi, S.; Iida, S.; Kajitani, M.; Sato, S.; Serpone, N. Chemical reactions with a novel 5.8-GHz microwave apparatus. Characterization of properties of common solvents and application in a Diels—Alder organic synthesis. Org. Process Res. Dev. 2008, 12, 257–263. [Google Scholar] [CrossRef]
- Dudley, G.B.; Richert, R.; Stiegman, A.E. On the existence of and mechanism for microwave-specific reaction rate enhancement. Chem. Sci. 2015, 6, 2144–2152. [Google Scholar] [CrossRef]
- Jacob, J.; Chia, L.H.L.; Boey, F.Y.C. Thermal and non-thermal interaction of microwave radiation with materials. J. Mater. Sci. 1995, 30, 5321–5327. [Google Scholar] [CrossRef]
- Perreux, L.; Loupy, A. A tentative rationalization of microwave effects in organic synthesis according to the reaction medium, and mechanistic considerations. Tetrahedron 2001, 57, 9199–9223. [Google Scholar] [CrossRef]
- De la Hoz, A.; Díaz-Ortiz, Á.; Moreno, A. Microwaves in organic synthesis. Thermal and non-thermal microwave effects. Chem. Soc. Rev. 2005, 34, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Conner, W.C.; Tompsett, G.A. How could and do microwaves influence chemistry at interfaces? J. Phys. Chem. B 2008, 112, 2110–2118. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.S.; Yang, W.S. Microwave synthesis of zeolite membranes: A review. J. Membr. Sci. 2008, 316, 3–17. [Google Scholar] [CrossRef]
- Gedye, R.N.; Smith, F.E.; Westaway, K.C. The rapid synthesis of organic compounds in microwave ovens. Can. J. Chem. 1988, 66, 17–26. [Google Scholar] [CrossRef]
- Gedye, R.N.; Rank, W.; Westaway, K.C. The rapid synthesis of organic compounds in microwave ovens. II. Can. J. Chem. 1991, 69, 706–711. [Google Scholar] [CrossRef]
- Kappe, C. Unraveling the Mysteries of Microwave Chemistry Using Silicon Carbide Reactor Technology. Acc. Chem. Res. 2013, 46, 1579–1587. [Google Scholar] [CrossRef]
- Dallinger, D.; Kappe, C.O. Microwave-assisted synthesis in water as solvent. Chem. Rev. 2007, 107, 2563–2591. [Google Scholar] [CrossRef]
- Baghbanzadeh, M.; Carbone, L.; Cozzoli, P.D.; Kappe, C.O. Microwave-Assisted Synthesis of Colloidal Inorganic Nanocrystals. Angew. Chem. Int. Ed. 2011, 50, 11312–11359. [Google Scholar] [CrossRef]
- Kappe, C.O. Controlled Microwave Heating in Modern Organic Synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] [CrossRef]
- Robinson, J.; Kingman, S.; Irvine, D.; Licence, P.; Smith, A.; Dimitrakis, G.; Obermayer, D.; Kappe, C.O. Understanding microwave heating effects in single mode type cavities—Theory and experiment. Phys. Chem. Chem. Phys. 2010, 12, 4750–4758. [Google Scholar] [CrossRef]
- Kuhnert, N. Microwave-Assisted Reactions in Organic Synthesis—Are There Any Nonthermal Microwave Effects? Angew. Chem. Int. Ed. 2002, 41, 1863–1866. [Google Scholar] [CrossRef]
- Rao, C.R.M.; Reddi, G.S. Platinum group metals (PGM); occurrence, use and recent trends in their determination. TrAC Trends Anal. Chem. 2000, 19, 565–586. [Google Scholar] [CrossRef]
- Yu, W.; Porosoff, M.D.; Chen, J.G. Review of Pt-Based Bimetallic Catalysis: From Model Surfaces to Supported Catalysts. Chem. Rev. 2012, 112, 5780–5817. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, R. Iridium compounds in catalysis. Acc. Chem. Res. 1979, 12, 331–337. [Google Scholar] [CrossRef]
- Hayashi, T.; Yamasaki, K. Rhodium-Catalyzed Asymmetric 1,4-Addition and Its Related Asymmetric Reactions. Chem. Rev. 2003, 103, 2829–2844. [Google Scholar] [CrossRef]
- Phan, N.T.; Van Der Sluys, M.; Jones, C.W. On the Nature of the Active Species in Palladium Catalyzed Mizoroki–Heck and Suzuki–Miyaura Couplings—Homogeneous or Heterogeneous Catalysis, A Critical Review. Advan. Synth. Catal. 2006, 348, 609–679. [Google Scholar] [CrossRef]
- Naota, T.; Takaya, H.; Murahashi, S.I. Ruthenium-Catalyzed Reactions for Organic Synthesis. Chem. Rev. 1998, 98, 2599–2660. [Google Scholar] [CrossRef]
- Schroeder, M. Osmium tetraoxide cis hydroxylation of unsaturated substrates. Chem. Rev. 1980, 80, 187–213. [Google Scholar] [CrossRef]
- Biffis, A.; Centomo, P.; Zotto, A.D.; Zecca, M. Pd metal catalysts for cross-couplings and related reactions in the 21st century: A critical review. Chem. Rev. 2018, 118, 2249–2295. [Google Scholar] [CrossRef]
- Wang, J.; Chen, H.; Hu, Z.; Yao, M.; Li, Y. A Review on the Pd-Based Three-Way Catalyst. Catal. Rev. -Sci. Eng. 2015, 57, 79–144. [Google Scholar] [CrossRef]
- Chaplin, B.P.; Reinhard, M.; Schneider, W.F.; Schüth, C.; Shapley, J.R.; Strathmann, T.J.; Werth, C.J. Critical Review of Pd-Based Catalytic Treatment of Priority Contaminants in Water. Environ. Sci. Technol. 2012, 46, 3655–3670. [Google Scholar] [CrossRef] [PubMed]
- Epling, W.S.; Campbell, L.E.; Yezerets, A.; Currier, N.W.; Parks, J.E. Overview of the Fundamental Reactions and Degradation Mechanisms of NOx Storage/Reduction Catalysts. Catal. Rev. -Sci. Eng. 2004, 46, 163–245. [Google Scholar] [CrossRef]
- Toops, T.J.; Smith, D.B.; Epling, W.S.; Parks, J.E.; Partridge, W.P. Quantified NOx adsorption on Pt/K/gamma-Al2O3 and the effects of CO2 and H2O. Appl. Catal. B 2005, 58, 255–264. [Google Scholar] [CrossRef]
- Littman, F.E.; Magill, P.L. Some unique aspects of air pollution in Los Angeles. Air Repair 1953, 3, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Voltz, S.E.; Morgan, C.R.; Liederman, D.; Jacob, S.M. Kinetic study of carbon monoxide and propylene oxidation on platinum catalysts. Ind. Eng. Chem. Prod. Res. Develop. 1973, 12, 294–301. [Google Scholar] [CrossRef]
- Dwyer, F.G. Catalysis for Control of Automotive Emissions. Catal. Rev.-Sci. Eng. 1972, 6, 261–291. [Google Scholar] [CrossRef]
- Meguerian, G.H.; Hirschberg, E.H.; Rakowsky, F.W. Catalyst for Treating Exhaust Gas from Internal Combustion Engine. U.S. Patent 4006103, 1 February 1977. [Google Scholar]
- Hegedus, L.; Summers, J.C. Platinum-Rhodium Catalyst for Automotive Emission Control. U.S. Patent 4128506, 5 December 1978. [Google Scholar]
- Acres, G.J.K.; Cooper, B.J. Platinum Catalysts for Exhaust Emission Control: The Mechanism of Catalyst Poisoning by Lead and Phosphorus Compounds. Platinum. Metals. Rev. 2010, 54, 61–62. [Google Scholar] [CrossRef]
- Hu, Z.; Allen, F.M.; Wan, C.Z.; Heck, R.M.; Steger, J.J.; Lakis, R.E.; Lyman, C.E. Performance and Structure of Pt–Rh Three-Way Catalysts: Mechanism for Pt/Rh Synergism. J. Catal. 1998, 174, 13. [Google Scholar] [CrossRef]
- Hu, Z. A Pt–Rh synergism in Pt/Rh three-way catalysts. Chem. Commun. 1996, 879–880. [Google Scholar] [CrossRef]
- Matsuura, S.; Hirai, A.; Arimura, K.; Shinjoh, H. Development of Three-Way Catalyst with Using Only Pd as Activator. SAE Trans. 1995, 302–309. [Google Scholar] [CrossRef]
- Available online: http://www.platinum.matthey.com/prices/price-charts (accessed on 29 July 2020).
- Gandhi, H.S.; Williamson, W.B.; Logothetis, E.M.; Tabock, J.; Peters, C.; Hurley, M.D.; Shelef, M. Affinity of lead for noble metals on different supports. Surf. Interface Anal. 1984, 6, 149–161. [Google Scholar] [CrossRef]
- Gandhi, H.S.; Shelef, M. Effects of sulphur on noble metal automotive catalysts. Appl. Catal. 1991, 77, 175–186. [Google Scholar] [CrossRef]
- Chattha, M.S.; Watkins, W.L.H.; Gandhi, H.S. Three-Way Catalyst for Automotive Emission Control and Method of Making the Catalyst. U.S. Patent 4992405, 12 February 1991. [Google Scholar]
- Shelef, M.; McCabe, R.W. Twenty-five years after introduction of automotive catalysts: What next? Catal. Today 2000, 62, 35–50. [Google Scholar] [CrossRef]
- Gandhi, H.S.; Graham, G.W.; McCabe, R.W. Automotive exhaust catalysis. J. Catal. 2003, 216, 433–442. [Google Scholar] [CrossRef]
- Harpness, R.; Gedanken, A. Microwave Synthesis of Core−Shell Gold/Palladium Bimetallic Nanoparticles. Langmuir 2004, 20, 3431–3434. [Google Scholar] [CrossRef]
- Abdelsayed, V.; Alijarash, A.; El-Shall, M.S.; Al Othmon, Z.A.; Alghamdi, A.H. Microwave Synthesis of Bimetallic Nanoalloys and CO Oxidation on Ceria-Supported Nanoalloys. Chem. Mater. 2009, 21, 2825–2834. [Google Scholar] [CrossRef]
- Abdelsayed, V.; Glaspell, G.; Nguyen, M.; Howe, J.M.; El-Shall, M.S. Laser synthesis of bimetallic nanoalloys in the vapor and liquid phases and the magnetic properties of PdM and PtM nanoparticles (M = Fe, Co and Ni). Faraday Discuss. 2008, 138, 163–180. [Google Scholar] [CrossRef]
- Fox, E.B.; Velu, S.; Engelhard, M.H.; Chin, Y.H.; Miller, J.T.; Kropf, J.; Song, C. Characterization of CeO2-supported Cu–Pd bimetallic catalyst for the oxygen-assisted water–gas shift reaction. J. Catal. 2008, 260, 358–370. [Google Scholar] [CrossRef]
- Wang, F.; Zhao, K.; Zhang, H.; Dong, Y.; Wang, T.; He, D. Low temperature CO catalytic oxidation over supported Pd–Cu catalysts calcined at different temperatures. Chem. Eng. J. 2014, 242, 10–18. [Google Scholar] [CrossRef]
- Zhou, F.; Du, X.; Yu, J.; Mao, D.; Lu, G. Highly water-resistant carbon nanotube supported PdCl2–CuCl2 catalysts for low temperature CO oxidation. RSC Adv. 2016, 6, 66553–66563. [Google Scholar] [CrossRef]
- Rappé, K.G.; DiMaggio, C.; Pihl, J.A.; Theis, J.R.; Oh, S.H.; Fisher, G.B.; Parks, J.; Easterling, V.G.; Yang, M.; Stewart, M.L.; et al. Aftertreatment Protocols for Catalyst Characterization and Performance Evaluation: Low-Temperature Oxidation, Storage, Three-Way, and NH3-SCR Catalyst Test Protocols. Emiss. Control Sci. Technol. 2019, 5, 183–214. [Google Scholar] [CrossRef]
- Zhang, J.; Mo, Y.; Vukmirovic, M.B.; Klie, R.; Sasaki, K.; Adzic, R.R. Platinum Monolayer Electrocatalysts for O2 Reduction: Pt Monolayer on Pd (111) and on Carbon-Supported Pd Nanoparticles. J. Phys. Chem. B 2004, 108, 10955–10964. [Google Scholar] [CrossRef]
- Golikand, A.N.; Lohrasbi, E.; Maragheh, M.G.; Asgari, M.J. Carbon nano-tube supported Pt–Pd as methanol-resistant oxygen reduction electrocatalyts for enhancing catalytic activity in DMFCs. Appl. Electrochem. 2009, 39, 2421–2431. [Google Scholar] [CrossRef]
- Zhang, H.; Yin, Y.; Hu, Y.; Li, C.; Wu, P.; Wei, S.; Cai, C.J. Pd@ Pt core—Shell nanostructures with controllable composition synthesized by a microwave method and their enhanced electrocatalytic activity toward oxygen reduction and methanol oxidation. Phys. Chem. B 2010, 114, 11861–11867. [Google Scholar] [CrossRef]
- Zhang, J.L.; Vukmirovic, M.B.; Xu, Y.; Mavrikakis, M.; Adzic, R.R. Controlling the catalytic activity of platinum-monolayer electrocatalysts for oxygen reduction with different substrates. Angew. Chem. Int. Ed. 2005, 44, 2132. [Google Scholar] [CrossRef]
- Hammer, B.; Norskov, J.K. Theoretical surface science and catalysis—Calculations and concepts. Adv. Catal. 2000, 45, 71–129. [Google Scholar] [CrossRef]
- Belousov, O.V.; Belousova, N.T.; Sirotina, A.V.; Solovyov, L.A.; Zhyzhaev, A.M.; Zharkov, S.M.; Mikhlin, Y.L. Formation of Bimetallic Au–Pd and Au–Pt Nanoparticles under Hydrothermal Conditions and Microwave Irradiation. Langmuir 2011, 27, 11697–11703. [Google Scholar] [CrossRef]
- Buffat, P.H.; Borel, J.-P. Size effect on the melting temperature of gold particles. Phys. Rev. A 1976, 13, 2287–2298. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; Zhao, Y.; Chi, Q.; Huang, T.; Liu, H. Controlled synthesis of Pd–Pt alloy nanohypercubes under microwave irradiation. Cryst. Eng. Comm. 2014, 16, 5206–5211. [Google Scholar] [CrossRef]
- Hattori, M.; Shimamoto, D.; Ago, H.; Tsuji, M. AgPd@Pd/TiO2 nanocatalyst synthesis by microwave heating in aqueous solution for efficient hydrogen production from formic acid. J. Mater. Chem. A 2015, 3, 10666–10670. [Google Scholar] [CrossRef]
- Tedsree, K.; Li, T.; Jones, S.; Chan, C.W.A.; Yu, K.M.K.; Bagot, P.A.J.; Marquis, E.A.; Smith, G.D.W.; Tsnag, S.C.E. Hydrogen production from formic acid decomposition at room temperature using a Ag–Pd core–shell nanocatalyst. Nat. Nanotechnol. 2011, 6, 302–307. [Google Scholar] [CrossRef]
- Zhou, X.; Huang, Y.; Xing, W.; Liu, C.; Liao, J.; Lu, T. High-quality hydrogen from the catalyzed decomposition of formic acid by Pd–Au/C and Pd–Ag/C. Chem. Commun. 2008, 3540–3542. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhou, X.; Yin, M.; Liu, C.; Xing, W. Novel PdAu@Au/C Core−Shell Catalyst: Superior Activity and Selectivity in Formic Acid Decomposition for Hydrogen Generation. Chem. Mater. 2010, 22, 5122–5128. [Google Scholar] [CrossRef]
- Hattori, M.; Einaga, H.; Daio, T.; Tsuji, M. Efficient hydrogen production from formic acid using TiO2-supported AgPd@Pd nanocatalysts. J. Mater. Chem. A 2015, 3, 4453–4461. [Google Scholar] [CrossRef]
- Denton, A.R.; Ashcroft, N.W. Vegard’s Law. Phys. Rev. A 1991, 43, 3161–3164. [Google Scholar] [CrossRef]
- Steiner, P.; Hufner, S. Thermochemical analysis of PdxAg1-x alloys from XPS core-level binding energy shifts. Solid State Commun. 1981, 37, 79–81. [Google Scholar] [CrossRef]
- Wang, Z.-L.; Yan, J.-M.; Ping, Y.; Wang, H.-L.; Zheng, W.-T.; Jiang, Q. An Efficient CoAuPd/C Catalyst for Hydrogen Generation from Formic Acid at Room Temperature. Angew. Chem. Int. Ed. 2013, 52, 4406–4409. [Google Scholar] [CrossRef]
- Kunal, P.; Li, H.; Dewing, B.L.; Zhang, L.; Jarvis, K.; Henkelman, G.; Humphrey, S.M. Microwave-Assisted Synthesis of PdxAu100–xAlloy Nanoparticles: A Combined Experimental and Theoretical Assessment of Synthetic and Compositional Effects upon Catalytic Reactivity. ACS Catal. 2016, 6, 4882–4893. [Google Scholar] [CrossRef]
- Seraj, S.; Kunal, P.; Henkelman, G.; Humphrey, S.M.; Werth, C.J. PdAu Alloy Nanoparticle Catalysts: Effective Candidates for Nitrite Reduction in Water. ACS Catal. 2017, 7, 3268–3276. [Google Scholar] [CrossRef]
- Goswami, A.; Rathi, A.K.; Aparicio, C.; Tomanec, O.; Petr, M.; Pocklanova, R.; Gawande, M.; Varma, R.S.; Zboril, R. In Situ Generation of Pd–Pt Core–Shell Nanoparticles on Reduced Graphene Oxide (Pd@Pt/rGO) Using Microwaves: Applications in Dehalogenation Reactions and Reduction of Olefins. ACS Appl. Mater. Interfaces 2017, 9, 2815–2824. [Google Scholar] [CrossRef]
- Pocklanova, R.; Rathi, A.K.; Gawande, M.B.; Datta, K.K.R.; Ranc, V.; Cepe, K.; Petr, M.; Varma, R.S.; Kvitek, L.; Zboril, R. Gold nanoparticle-decorated graphene oxide: Synthesis and application in oxidation reactions under benign conditions. J. Mol. Catal. A Chem. 2016, 424, 121–127. [Google Scholar] [CrossRef]
- Wei, Z.; Pan, R.; Hou, Y.; Yang, Y.; Liu, Y. Graphene-supported Pd catalyst for highly selective hydrogenation of resorcinol to 1, 3-cyclohexanedione through giant π-conjugate interactions. Sci. Rep. 2015, 5, 15664. [Google Scholar] [CrossRef] [PubMed]
- Navalon, S.; Dhakshinamoorthy, A.; Alvaro, M.; Garcia, H. Carbocatalysis by Graphene-Based Materials. Chem. Rev. 2014, 114, 6179. [Google Scholar] [CrossRef] [PubMed]
- Primo, A.; Neatu, F.; Florea, M.; Parvulescu, V.; Garcia, H. Graphenes in the absence of metals as carbocatalysts for selective acetylene hydrogenation and alkene hydrogenation. Nat. Commun. 2014, 5, 5291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piburn, G.W.; Li, H.; Kunal, P.; Henkelman, G.; Humphrey, S.M. Rapid Synthesis of Rhodium-Palladium Alloy Nanocatalysts. ChemCatChem 2018, 10, 329–333. [Google Scholar] [CrossRef]
- Dahal, N.; Garcίa, S.; Zhou, J.; Humphrey, S.M. Beneficial Effects of Microwave-Assisted Heating versus Conventional Heating in Noble Metal Nanoparticle Synthesis. ACS Nano 2012, 6, 9433–9446. [Google Scholar] [CrossRef]
- Cong, C.; Nakayama, S.; Maenosono, S.; Harada, M. Microwave-Assisted Polyol Synthesis of Pt/Pd and Pt/Rh Bimetallic Nanoparticles in Polymer Solutions Prepared by Batch and Continuous-Flow Processing. Ind. Eng. Res. 2018, 57, 179–190. [Google Scholar] [CrossRef]





























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
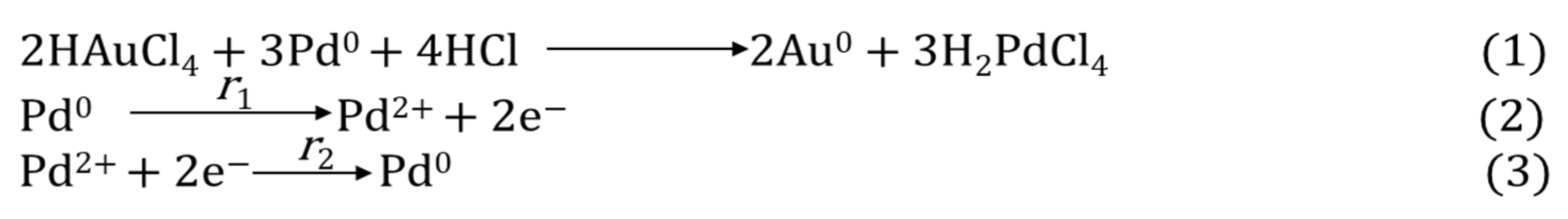{kind=link}
| Solvent | Boiling Point (°C) | Tangent Loss (tan δ) |
|---|---|---|
| Dichloromethane | 40 | 0.042 |
| Acetone | 56 | 0.054 |
| Chloroform | 61 | 0.091 |
| Methanol | 65 | 0.659 |
| Tetrahydrofuran | 66 | 0.047 |
| Hexane | 69 | 0.020 |
| Ethyl acetate | 77 | 0.059 |
| Ethanol | 78 | 0.941 |
| Acetonitrile | 81 | 0.062 |
| 2-Propanol | 82 | 0.799 |
| 1,2-Dichloroethane | 84 | 0.127 |
| 2-Butanol | 99 | 0.447 |
| Water | 100 | 0.123 |
| Formic Acid | 101 | 0.722 |
| Toluene | 111 | 0.040 |
| Acetic acid | 118 | 0.174 |
| 1-Butanol | 118 | 0.571 |
| Chlorobenzene | 131 | 0.101 |
| N-N′-dimethylformamide | 154 | 0.161 |
| 1,2-Dichlorobenzene | 180 | 0.280 |
| Dimethyl Sulfoxide | 189 | 0.825 |
| Ethylene glycol | 197 | 1.350 |
| N-methyl-2-pyrrolidone | 204 | 0.275 |
| Nitrobenzene | 210 | 0.589 |
| Vessel Material | Melting Point (°C) | Tangent Loss (tan δ) |
|---|---|---|
| Plexiglass | 160 | 5.7 × 10−3 |
| Polystyrene | 240 | 3.3 × 10−4 |
| Teflon | 327 | 1.5 × 10−4 |
| Borosilicate glass | 1648 | 1.0 × 10−3 |
| Quartz | 1715 | 6.0 × 10−5 |
| Porcelain | 1840 | 1.1 × 10−3 |
| SiC | 2700 | 0.02–1.05 |
| Manuscript Section | Publication Year | Instrument-Manufacturer, Watts Used | Solvent(s) | Temperature, Time | NP Composition |
|---|---|---|---|---|---|
| 2.1. | 2004 | Domestic-Spectra, 900 W | EG | N.A., 21 s | Pd-Au |
| 2.2. | 2009 | Domestic, 1000 W | OA + OAm | N.A., Variable (30 s, 90 s) | *Pd-Ni, Pd-Cu, Pd-Ag, Pd-Ru, Pd-Rh, Pd-Pt, Pd-Au |
| 2.3. | 2010 | Domestic, 200 W | H2O | N.A., 3 min | Pd-Pt |
| 2.4. | 2011 | Domestic-LG, 850 W | Muriatic Solution | Variable; 110-130 °C, Variable (15 min-60 min) | *Pd-Au |
| 2.5. | 2014 | Domestic-Galanz, 1000 W | TEG | N.A., 100 s | Pd-Pt |
| 2.6. | 2015 | N.A., 400 W | H2O | 100 °C, Variable; 30 min-2 hr | Pd-Ag |
| 2.7. | 2016 | MARS 5- CEM Corp., 800 W | EG | 150 °C, Variable (30 s-1 hr) | Pd-Au |
| 2.8. | 2017 | NN-J155WB EPG-Panasonic, 250 W | H2O | N.A., 3 min | Pd-Rh |
| 2.9. | 2018 | MARS 5- CEM Corp., 800 W | EG | 165 °C, Variable (30 s-3 hr) | Pd-Rh |
| 2.10. | 2018 | Flow Synthesis; Microsynth Plus-Milestone General K. K., 700W | Batch synthesis: Discover SP-CEM Corp.; 300 W | EG, Glycerol | 198 °C (EG), 250 °C (Glycerol), 10 min | *Pd-Pt |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunal, P.; Toops, T.J. A Review of Microwave-Assisted Synthesis-Based Approaches to Reduce Pd-Content in Catalysts. Catalysts 2020, 10, 991. https://doi.org/10.3390/catal10090991
Kunal P, Toops TJ. A Review of Microwave-Assisted Synthesis-Based Approaches to Reduce Pd-Content in Catalysts. Catalysts. 2020; 10(9):991. https://doi.org/10.3390/catal10090991
Chicago/Turabian StyleKunal, Pranaw, and Todd J. Toops. 2020. "A Review of Microwave-Assisted Synthesis-Based Approaches to Reduce Pd-Content in Catalysts" Catalysts 10, no. 9: 991. https://doi.org/10.3390/catal10090991
APA StyleKunal, P., & Toops, T. J. (2020). A Review of Microwave-Assisted Synthesis-Based Approaches to Reduce Pd-Content in Catalysts. Catalysts, 10(9), 991. https://doi.org/10.3390/catal10090991