Catalyst Performance Studies on the Guerbet Reaction in a Continuous Flow Reactor Using Mono- and Bi-Metallic Cu-Ni Porous Metal Oxides

 ,
,  , ,
, ,  ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Characterization of the Fresh Catalysts
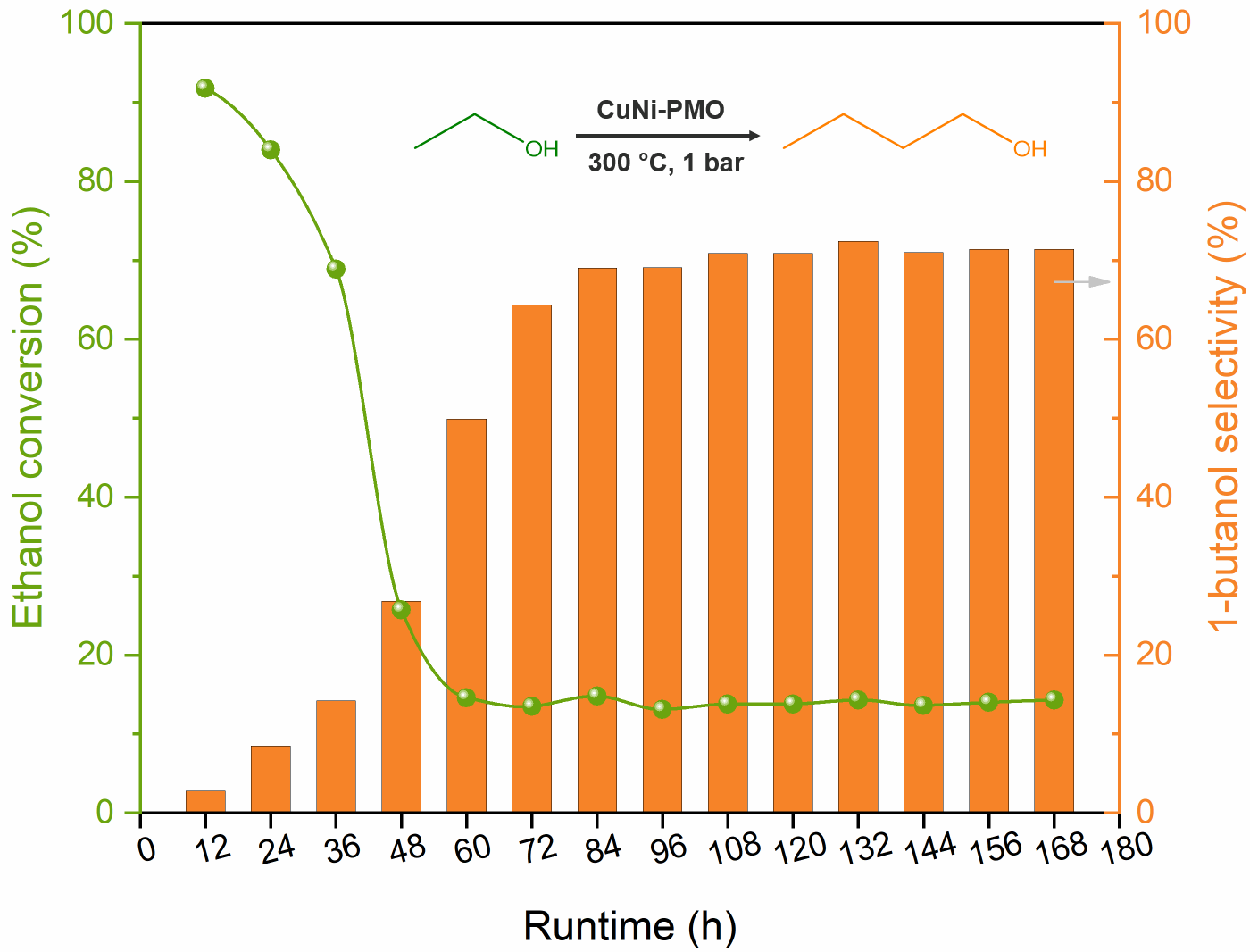
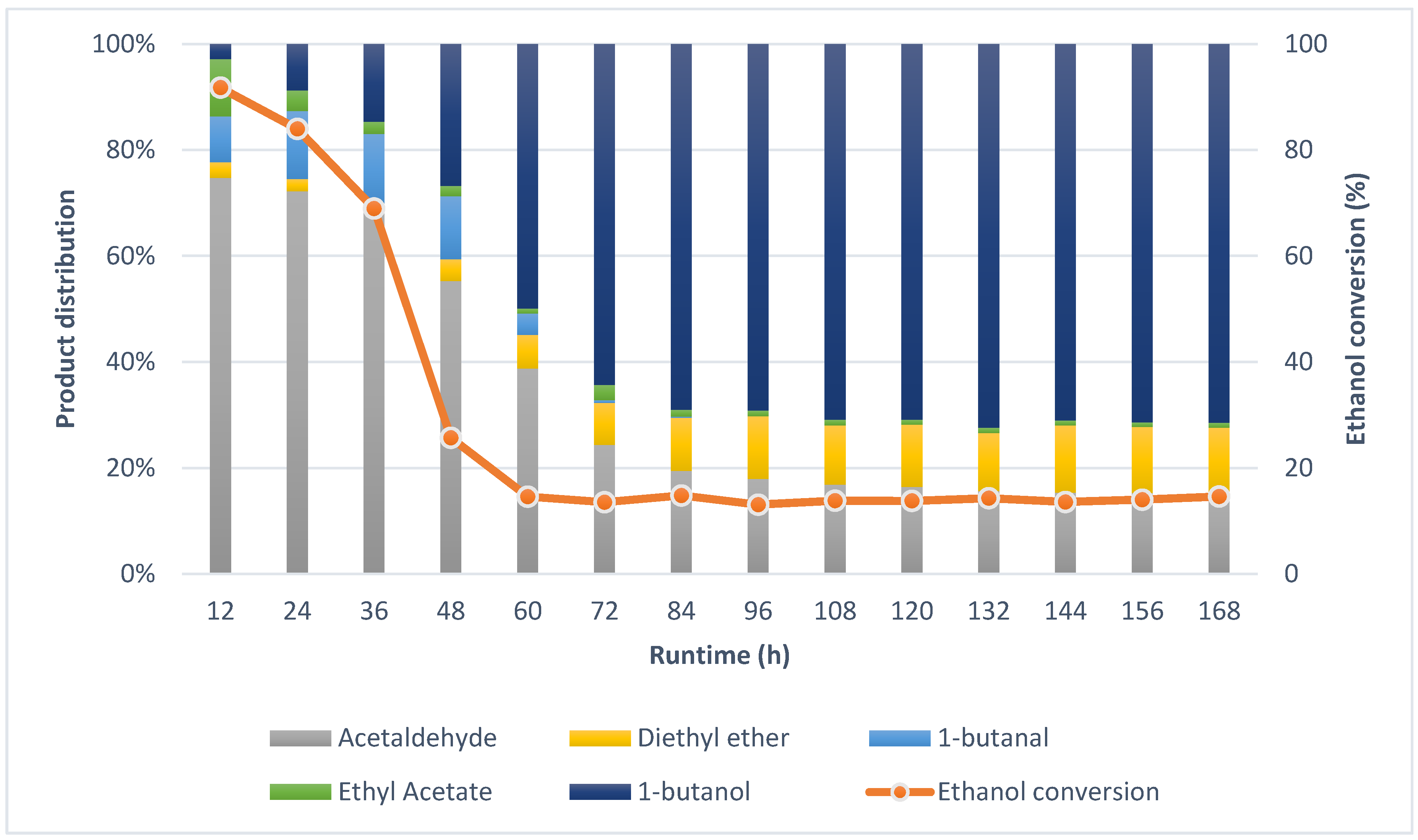
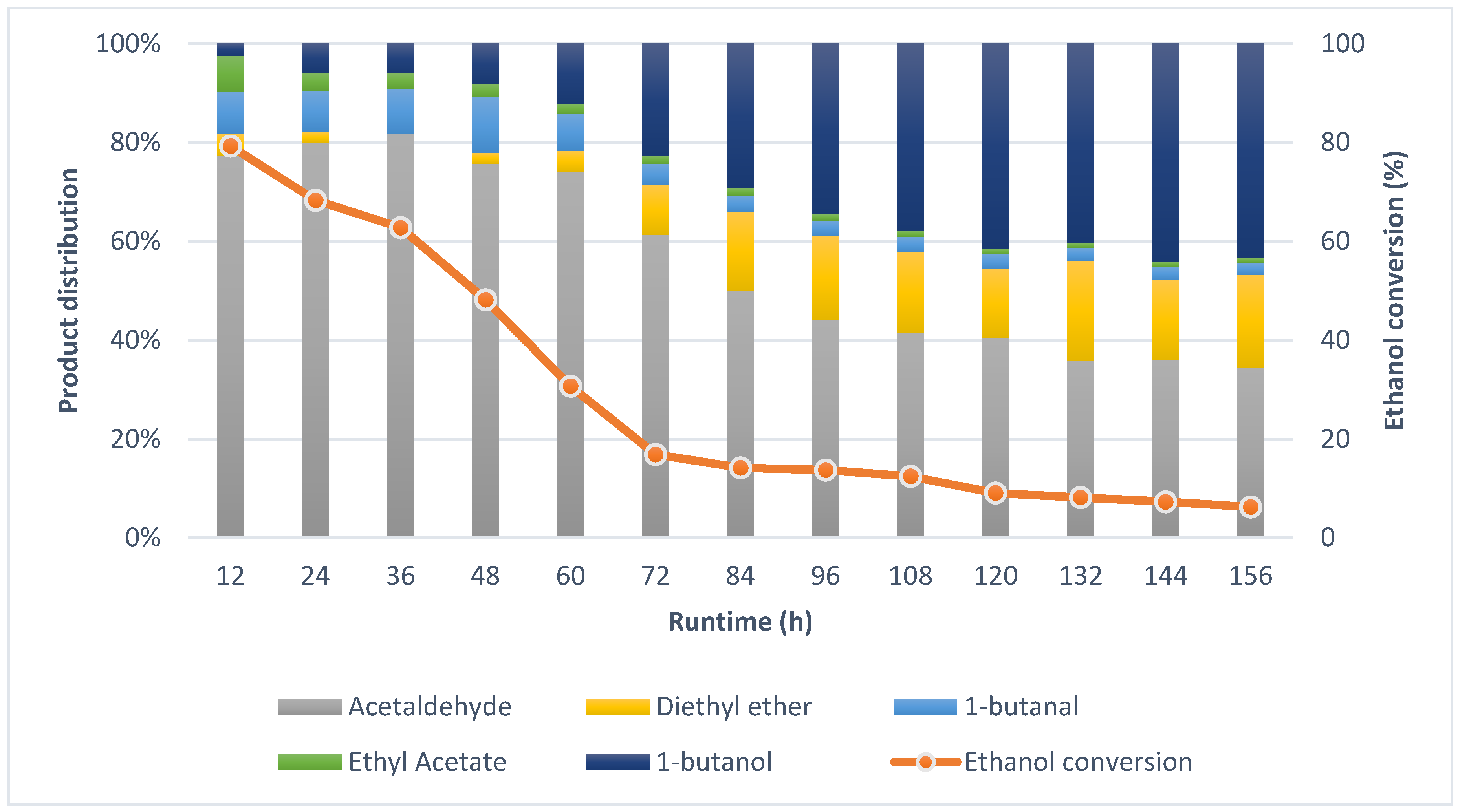
2.2. Guerbet Coupling of Ethanol in a Continuous Setup
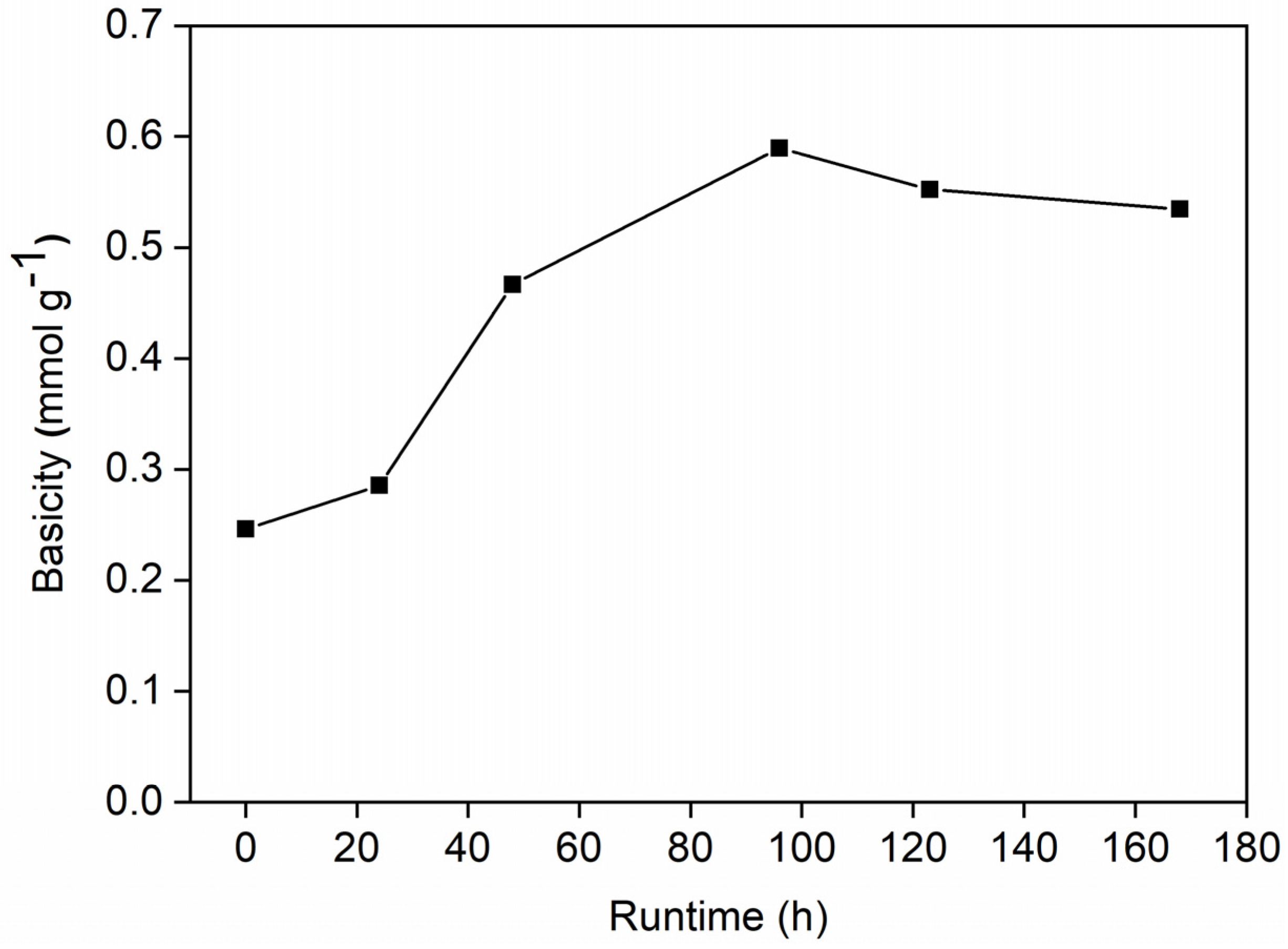
2.3. Characterization of Spent Catalysts
2.4. Effects of Reaction Conditions and Reactor Configuration
3. Experimental
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Catalytic Reactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ragauskas, A.J.; Williams, C.K.; Davison, B.H.; Britovsek, G.; Cairney, J.; Eckert, C.A.; Frederick, W.J.; Hallett, J.P.; Leak, D.J.; Liotta, C.L.; et al. The Path Forward for Biofuels and Biomaterials. Science 2006, 311, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eagan, N.M.; Kumbhalkar, M.D.; Buchanan, J.S.; Dumesic, J.A.; Huber, G.W. Chemistries and processes for the conversion of ethanol into middle-distillate fuels. Nat. Rev. Chem. 2019, 3, 223–249. [Google Scholar] [CrossRef]
- Balat, M. Production of bioethanol from lignocellulosic materials via the biochemical pathway: A review. Energy Convers. Manag. 2011, 52, 858–875. [Google Scholar] [CrossRef]
- Sun, Y.; Cheng, J. Hydrolysis of lignocellulosic materials for ethanol production: A review. Bioresour. Technol. 2002, 83, 1–11. [Google Scholar] [CrossRef]
- Trindade, W.R.D.S.; Santos, R.G.D. Review on the characteristics of butanol, its production and use as fuel in internal combustion engines. Renew. Sustain. Energy Rev. 2017, 69, 642–651. [Google Scholar] [CrossRef]
- Devarapalli, M.; Atiyeh, H.K. A review of conversion processes for bioethanol production with a focus on syngas fermentation. Biofuel Res. J. 2015, 2, 268–280. [Google Scholar] [CrossRef]
- Luk, H.T.; Mondelli, C.; Ferré, D.C.; Stewart, J.A.; Pérez-Ramírez, J. Status and prospects in higher alcohols synthesis from syngas. Chem. Soc. Rev. 2017, 46, 1358–1426. [Google Scholar] [CrossRef]
- Gabriëls, D.; Hernández, W.Y.; Sels, B.; Van Der Voort, P.; Verberckmoes, A. Review of catalytic systems and thermodynamics for the Guerbet condensation reaction and challenges for biomass valorization. Catal. Sci. Technol. 2015, 5, 3876–3902. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Barta, K. Cleave and couple: Toward fully sustainable catalytic conversion of lignocellulose to value added building blocks and fuels. Chem. Commun. 2018, 54, 7725–7745. [Google Scholar] [CrossRef]
- Hanspal, S.; Young, Z.D.; Prillaman, J.T.; Davis, R.J. Influence of surface acid and base sites on the Guerbet coupling of ethanol to butanol over metal phosphate catalysts. J. Catal. 2017, 352, 182–190. [Google Scholar] [CrossRef]
- Marcu, I.-C.; Tanchoux, N.; Fajula, F.; Tichit, D. Catalytic conversion of ethanol into butanol over M–Mg–Al mixed oxide catalysts (M= Pd, Ag, Mn, Fe, Cu, Sm, Yb) obtained from LDH precursors. Catal. Lett. 2013, 143, 23–30. [Google Scholar] [CrossRef]
- León, M.; Díaz, E.; Vega, A.; Ordóñez, S.; Auroux, A. Consequences of the iron–aluminium exchange on the performance of hydrotalcite-derived mixed oxides for ethanol condensation. Appl. Catal. B Environ. 2011, 102, 590–599. [Google Scholar] [CrossRef]
- Chieregato, A.; Velasquez, O.J.; Bandinelli, C.; Fornasari, G.; Cavani, F.; Mella, M. On the Chemistry of Ethanol on Basic Oxides: Revising Mechanisms and Intermediates in the Lebedev and Guerbet reactions. Chem. Sustain. Chem. 2015, 8, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, J.T.; Davis, R.J. Heterogeneous catalysts for the Guerbet coupling of alcohols. ACS Catal. 2013, 3, 1588–1600. [Google Scholar] [CrossRef]
- Wu, X.; Fang, G.; Tong, Y.; Jiang, D.; Liang, Z.; Leng, W.; Liu, L.; Tu, P.; Wang, H.; Ni, J. Catalytic Upgrading of Ethanol to n-Butanol: Progress in Catalyst Development. Chem. Sustain. Chem. 2018, 11, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Di Cosimo, J.I.; Apesteguı, A.C.R.; Ginés, M.J.L.; Iglesia, E. Structural requirements and reaction pathways in condensation reactions of alcohols on MgyAlOx catalysts. J. Catal. 2000, 190, 261–275. [Google Scholar] [CrossRef] [Green Version]
- Moteki, T.; Flaherty, D.W. Mechanistic insight to C–C bond formation and predictive models for cascade reactions among alcohols on Ca-and Sr-hydroxyapatites. ACS Catal. 2016, 6, 4170–4183. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.R.; Shylesh, S.; Bell, A.T. Mechanism and kinetics of ethanol coupling to butanol over hydroxyapatite. ACS Catal. 2016, 6, 939–948. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, S.; Takagaki, A.; Ebitani, K. Characterization, synthesis and catalysis of hydrotalcite-related materials for highly efficient materials transformations. Green Chem. 2013, 15, 2026–2042. [Google Scholar] [CrossRef]
- Di Cosimo, J.I.; Dıez, V.K.; Xu, M.; Iglesia, E.; Apesteguıa, C.R. Structure and surface and catalytic properties of Mg-Al basic oxides. J. Catal. 1998, 178, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Cavani, F.; Trifiro, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Zaccheria, F.; Scotti, N.; Ravasio, N. The Role of Copper in the Upgrading of Bioalcohols. Chem. Cat. Chem. 2018, 10, 1526–1535. [Google Scholar] [CrossRef]
- Bravo-Suárez, J.J.; Subramaniam, B.; Chaudhari, R.V. Vapor-phase methanol and ethanol coupling reactions on CuMgAl mixed metal oxides. Appl. Catal. A Gen. 2013, 455, 234–246. [Google Scholar] [CrossRef]
- Cheng, F.; Guo, H.; Cui, J.; Hou, B.; Xi, H.; Jia, L.; Li, D. Coupling of methanol and ethanol over CuMgAlOx catalysts: The roles of copper species and alkalinity. React. Kinet. Mech. Catal. 2019, 126, 119–136. [Google Scholar] [CrossRef]
- Benito, P.; Vaccari, A.; Antonetti, C.; Licursi, D.; Schiarioli, N.; Rodriguez-Castellón, E.; Raspolli Galletti, A.M. Tunable copper-hydrotalcite derived mixed oxides for sustainable ethanol condensation to n-butanol in liquid phase. J. Clean. Prod. 2019, 209, 1614–1623. [Google Scholar] [CrossRef]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Bimetallic catalysts for upgrading of biomass to fuels and chemicals. Chem. Soc. Rev. 2012, 41, 8075–8098. [Google Scholar] [CrossRef]
- Hernández, W.Y.; De Vlieger, K.; Van Der Voort, P.; Verberckmoes, A. Ni−Cu Hydrotalcite-Derived Mixed Oxides as Highly Selective and Stable Catalysts for the Synthesis of β-Branched Bioalcohols by the Guerbet Reaction. Chem. Sustain. Chem. 2016, 9, 3196–3205. [Google Scholar] [CrossRef]
- Sun, Z.; Couto Vasconcelos, A.; Bottari, G.; Stuart, M.C.A.; Bonura, G.; Cannilla, C.; Frusteri, F.; Barta, K. Efficient Catalytic Conversion of Ethanol to 1-Butanol via the Guerbet Reaction over Copper- and Nickel-Doped Porous. ACS Sustain. Chem. Eng. 2017, 5, 1738–1746. [Google Scholar] [CrossRef]
- Jordison, T.L.; Peereboom, L.; Miller, D.J. Impact of water on condensed phase ethanol Guerbet reactions. Ind. Eng. Chem. Res. 2016, 55, 6579–6585. [Google Scholar] [CrossRef]
- Quesada, J.; Faba, L.; Díaz, E.; Ordóñez, S. Role of the surface intermediates in the stability of basic mixed oxides as catalyst for ethanol condensation. Appl. Catal. A Gen. 2017, 542, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Silvester, L.; Lamonier, J.-F.; Faye, J.; Capron, M.; Vannier, R.-N.; Lamonier, C.; Dubois, J.L.; Couturier, J.L.; Calais, C.; Dumeignil, F. Reactivity of ethanol over hydroxyapatite-based Ca-enriched catalysts with various carbonate contents. Catal. Sci. Technol. 2015, 5, 2994–3006. [Google Scholar] [CrossRef]
- Kannan, S.; Dubey, A.; Knozinger, H. Synthesis and characterization of CuMgAl ternary hydrotalcites as catalysts for the hydroxylation of phenol. J. Catal. 2005, 231, 381–392. [Google Scholar] [CrossRef]
- Takehira, K.; Shishido, T.; Wang, P.; Kosaka, T.; Takaki, K. Autothermal reforming of CH4 over supported Ni catalysts prepared from Mg–Al hydrotalcite-like anionic clay. J. Catal. 2004, 221, 43–54. [Google Scholar] [CrossRef]
- Tuza, P.V.; Manfro, R.L.; Ribeiro, N.F.P.; Souza, M.M.V.M. Production of renewable hydrogen by aqueous-phase reforming of glycerol over Ni–Cu catalysts derived from hydrotalcite precursors. Renew. Energy 2013, 50, 408–414. [Google Scholar] [CrossRef]
- Tanasoi, S.; Tanchoux, N.; Urdă, A.; Tichit, D.; Săndulescu, I.; Fajula, F.; Marcu, I.C. New Cu-based mixed oxides obtained from LDH precursors, catalysts for methane total oxidation. Appl. Catal. A Gen. 2009, 363, 135–142. [Google Scholar] [CrossRef]
- Han, J.; Zeng, H.Y.; Xu, S.; Chen, C.R.; Liu, X.J. Catalytic properties of CuMgAlO catalyst and degradation mechanism in CWPO of methyl orange. Appl. Catal. A Gen. 2016, 527, 72–80. [Google Scholar] [CrossRef]
- Yu, X.-P.; Chu, W.; Wang, N.; Ma, F. Hydrogen Production by Ethanol Steam Reforming on NiCuMgAl Catalysts Derived from Hydrotalcite-Like Precursors. Catal. Lett. 2011, 141, 1228–1236. [Google Scholar] [CrossRef]
- Li, Q.; Yi, H.; Tang, X.; Zhao, S.; Zhao, B.; Liu, D.; Gao, F. Preparation and characterization of Cu/Ni/Fe hydrotalcite-derived compounds as catalysts for the hydrolysis of carbon disulfide. Chem. Eng. J. 2016, 284, 103–111. [Google Scholar] [CrossRef]
- Li, D.; Koike, M.; Chen, J.; Nakagawa, Y.; Tomishige, K. Preparation of Ni–Cu/Mg/Al catalysts from hydrotalcite-like compounds for hydrogen production by steam reforming of biomass tar. Int. J. Hydrogen Energy 2014, 39, 10959–10970. [Google Scholar] [CrossRef]
- Montanari, B.; Vaccari, A.; Gazzano, M.; Käßner, P.; Papp, H.; Pasel, J.; Dziembaj, R.; Makowski, W.; Lojewski, T. Characterization and activity of novel copper-containing catalysts for selective catalytic reduction of NO with NH3. Appl. Catal. B Environ. 1997, 13, 205–217. [Google Scholar] [CrossRef]
- Ashok, J.; Subrahmanyam, M.; Venugopal, A. Hydrotalcite structure derived Ni–Cu–Al catalysts for the production of H2 by CH4 decomposition. Int. J. Hydrogen Energy 2008, 33, 2704–2713. [Google Scholar] [CrossRef]
- Chmielarz, L.; Kuśtrowski, P.; Rafalska-Łasocha, A.; Dziembaj, R. Selective oxidation of ammonia to nitrogen on transition metal containing mixed metal oxides. Appl. Catal. B Environ. 2005, 58, 235–244. [Google Scholar] [CrossRef]
- Dragoi, B.; Ungureanu, A.; Chirieac, A.; Ciotonea, C.; Rudolf, C.; Royer, S.; Dumitriu, E. Structural and catalytic properties of mono-and bimetallic nickel–copper nanoparticles derived from MgNi (Cu) Al-LDHs under reductive conditions. Appl. Catal. A Gen. 2015, 504, 92–102. [Google Scholar] [CrossRef]
- Parida, K.; Das, J. Mg/Al hydrotalcites: Preparation, characterisation and ketonisation of acetic acid. J. Mol. Catal. A Chem. 2000, 151, 185–192. [Google Scholar] [CrossRef]
- Delidovich, I.; Palkovits, R. Structure–performance correlations of Mg–Al hydrotalcite catalysts for the isomerization of glucose into fructose. J. Catal. 2015, 327, 1–9. [Google Scholar] [CrossRef]
- Abelló, S.; Medina, F.; Tichit, D.; Pérez-Ramírez, J.; Groen, J.C.; Sueiras, J.E.; Salagre, P.; Cesteros, Y. Aldol condensations over reconstructed Mg–Al hydrotalcites: Structure–activity relationships related to the rehydration method. Chem. A Eur. J. 2005, 11, 728–739. [Google Scholar] [CrossRef]
- Lee, G.; Jeong, Y.; Takagaki, A.; Jung, J.C. Sonication assisted rehydration of hydrotalcite catalyst for isomerization of glucose to fructose. J. Mol. Catal. A Chem. 2014, 393, 289–295. [Google Scholar] [CrossRef]
- Satta, A.; Shamiryan, D.; Baklanov, M.R.; Whelan, C.M.; Le, Q.T.; Beyer, G.P.; Vantomme, A.; Maex, K. The removal of copper oxides by ethyl alcohol monitored in situ by spectroscopic ellipsometry. J. Electrochem. Soc. 2003, 150, G300–G306. [Google Scholar] [CrossRef]
- Coskun, F.; Cetinkaya, S.; Eroglu, S. Reduction of Nickel Oxide with Ethanol. JOM 2017, 69, 987–992. [Google Scholar] [CrossRef]
- Macala, G.S.; Matson, T.D.; Johnson, C.L.; Lewis, R.S.; Iretskii, A.V.; Ford, P.C. Hydrogen transfer from supercritical methanol over a solid base catalyst: A model for lignin depolymerization. Chem. Sustain. Energy Mater. 2009, 2, 215–217. [Google Scholar] [CrossRef]
- Kumar, A.; Cross, A.; Manukyan, K.; Bhosale, R.R.; van den Broeke, L.J.P.; Miller, J.T.; Mukasyan, A.S.; Wolf, E.E. Combustion synthesis of copper–nickel catalysts for hydrogen production from ethanol. Chem. Eng. J. 2015, 278, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Wu, X.; Mao, J.; Ni, J.; Li, X. Continuous catalytic upgrading of ethanol to n-butanol over Cu–CeO 2/AC catalysts. Chem. Commun. 2016, 52, 13749–13752. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Sinnathambi, C.M.; Subbarao, D. Kinetics of de-coking of spent reforming catalyst. J. Appl. Sci. 2011, 11, 1225–1230. [Google Scholar] [CrossRef] [Green Version]
- Behera, B.; Ray, S.S. Structural changes of FCC catalyst from fresh to regeneration stages and associated coke in a FCC refining unit: A multinuclear solid state NMR approach. Catal. Today 2009, 141, 195–204. [Google Scholar] [CrossRef]
- Kosinov, N.; Uslamin, E.A.; Coumans, F.J.A.G.; Wijpkema, A.S.G.; Rohling, R.Y.; Hensen, E.J.M. Structure and evolution of confined carbon species during methane dehydroaromatization over Mo/ZSM-5. ACS Catal. 2018, 8, 8459–8467. [Google Scholar] [CrossRef] [Green Version]
- Prinetto, F.; Ghiotti, G.; Durand, R.; Tichit, D. Investigation of Acid−Base Properties of Catalysts Obtained from Layered Double Hydroxides. J. Phys. Chem. B 2000, 104, 11117–11126. [Google Scholar] [CrossRef]
- Marcu, I.-C.; Tichit, D.; Fajula, F.; Tanchoux, N. Catalytic valorization of bioethanol over Cu-Mg-Al mixed oxide catalysts. Catal. Today 2009, 147, 231–238. [Google Scholar] [CrossRef]
- Carlini, C.; Flego, C.; Marchionna, M.; Noviello, M.; Galletti, A.M.R.; Sbrana, G.; Basile, F.; Vaccari, A. Guerbet condensation of methanol with n-propanol to isobutyl alcohol over heterogeneous copper chromite/Mg–Al mixed oxides catalysts. J. Mol. Catal. A Chem. 2004, 220, 215–220. [Google Scholar] [CrossRef]
- Mears, D.E. Tests for transport limitations in experimental catalytic reactors. Ind. Eng. Chem. Process. Des. Dev. 1971, 10, 541–547. [Google Scholar] [CrossRef]
- Weisz, P.B.; Prater, C.D. Interpretation of measurements in experimental catalysis. Adv. Catal. 1954, 6, 60390–60399. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
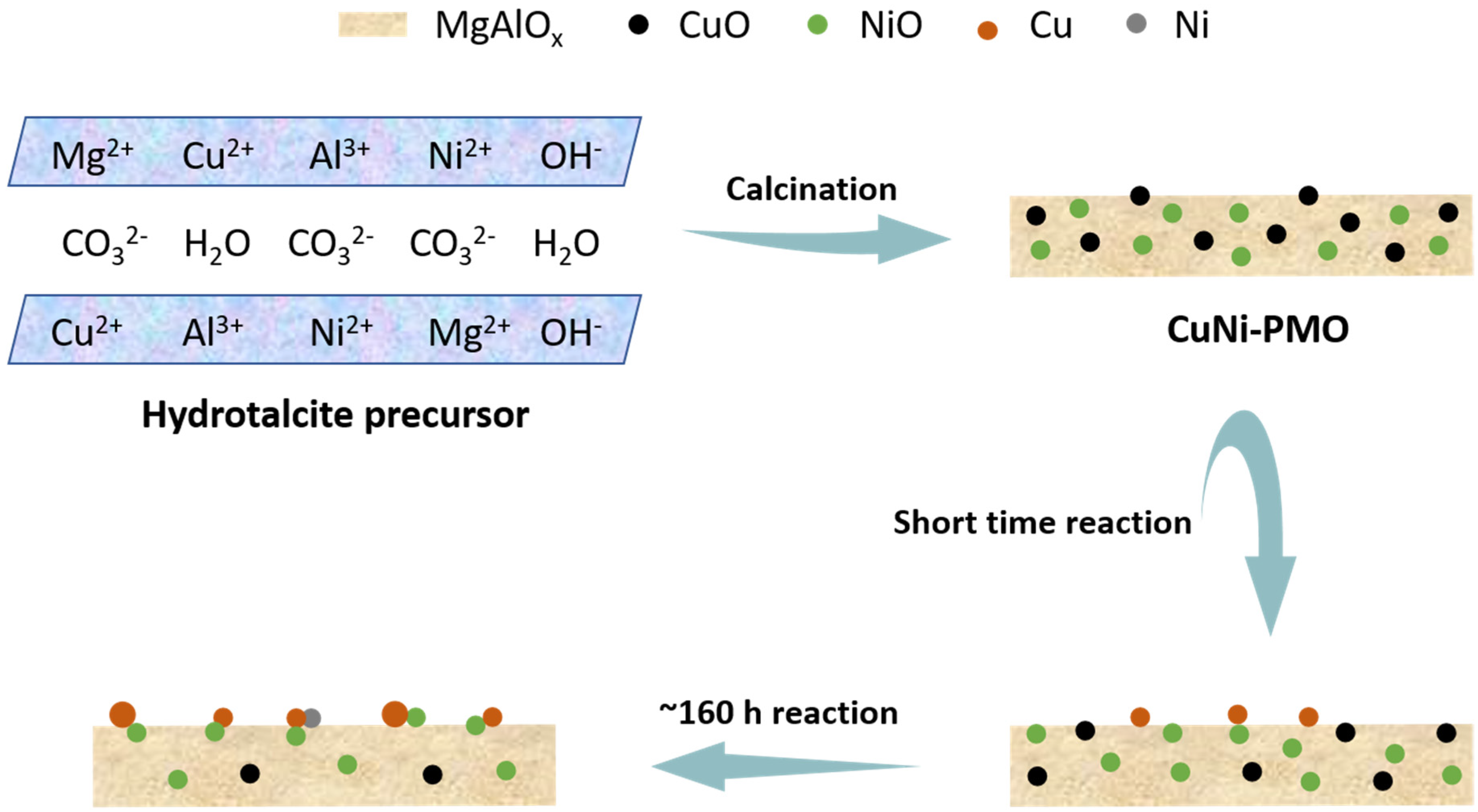{kind=link}
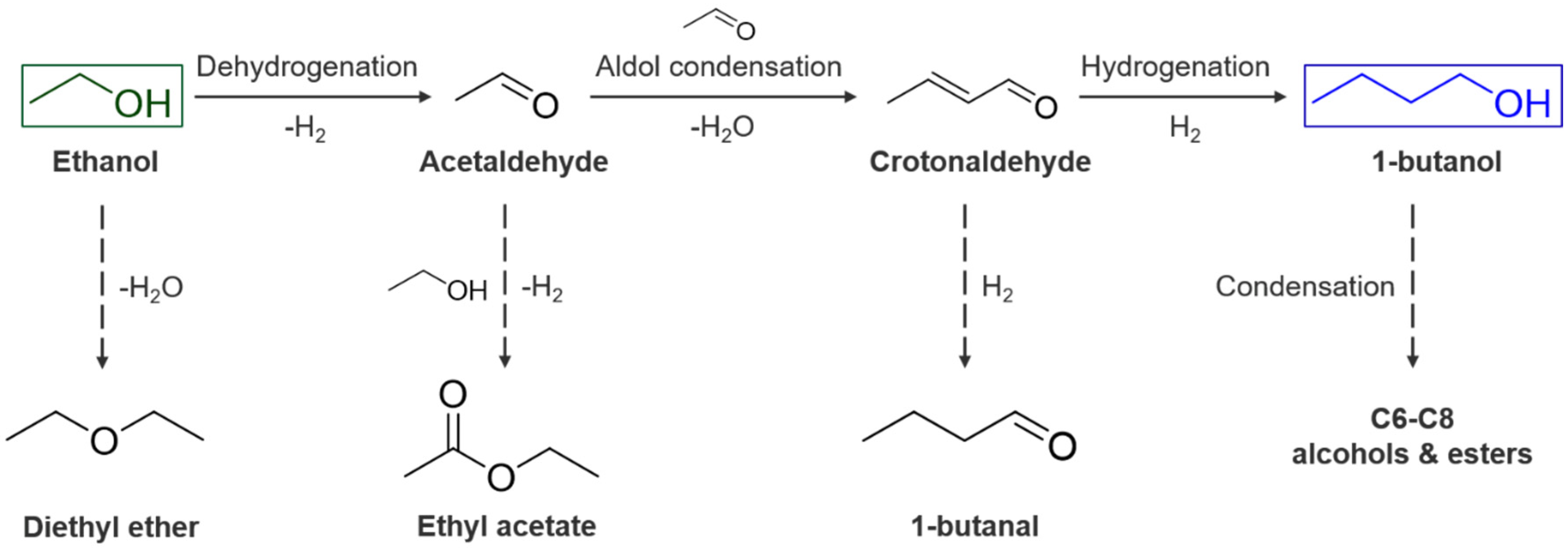{kind=link}
| Temperature (°C) | Initial Ethanol Conversion (%) | Ethanol Conversion (%) in Regime 2 | 1-Butanol Selectivity (%) in Regime 2 | Run Time (h) to Reach Regime 2 |
|---|---|---|---|---|
| 300 | 85.0 | 3.0 | 59.0 | 144 |
| 320 | 91.8 | 14.6 | 71.4 | 60 |
| 360 | 68.0 | 26.2 | 57.6 | 24 |
| Reaction Time (h) | Ethanol Conversion (%) | 1-Butanol Yield (%) |
|---|---|---|
| 2 | 39.5 | 16.4 |
| 6 | 47.9 | 21.1 |
| 10 | 63.4 | 20.8 |
| 18 | 69.4 | 20.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xi, X.-Y.; Sun, Z.-H.; Cao, H.-T.; Pei, Y.-T.; ten Brink, G.H.; Deuss, P.J.; Barta, K.; Heeres, H.J. Catalyst Performance Studies on the Guerbet Reaction in a Continuous Flow Reactor Using Mono- and Bi-Metallic Cu-Ni Porous Metal Oxides. Catalysts 2020, 10, 996. https://doi.org/10.3390/catal10090996
Xi X-Y, Sun Z-H, Cao H-T, Pei Y-T, ten Brink GH, Deuss PJ, Barta K, Heeres HJ. Catalyst Performance Studies on the Guerbet Reaction in a Continuous Flow Reactor Using Mono- and Bi-Metallic Cu-Ni Porous Metal Oxides. Catalysts. 2020; 10(9):996. https://doi.org/10.3390/catal10090996
Chicago/Turabian StyleXi, Xiao-Ying, Zhuo-Hua Sun, Hua-Tang Cao, Yu-Tao Pei, Gert H. ten Brink, Peter J. Deuss, Katalin Barta, and Hero J. Heeres. 2020. "Catalyst Performance Studies on the Guerbet Reaction in a Continuous Flow Reactor Using Mono- and Bi-Metallic Cu-Ni Porous Metal Oxides" Catalysts 10, no. 9: 996. https://doi.org/10.3390/catal10090996
APA StyleXi, X. -Y., Sun, Z. -H., Cao, H. -T., Pei, Y. -T., ten Brink, G. H., Deuss, P. J., Barta, K., & Heeres, H. J. (2020). Catalyst Performance Studies on the Guerbet Reaction in a Continuous Flow Reactor Using Mono- and Bi-Metallic Cu-Ni Porous Metal Oxides. Catalysts, 10(9), 996. https://doi.org/10.3390/catal10090996