Comparative Studies on the Susceptibility of (R)-2,3-Dipalmitoyloxypropylphosphonocholine (DPPnC) and Its Phospholipid Analogues to the Hydrolysis or Ethanolysis Catalyzed by Selected Lipases and Phospholipases



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
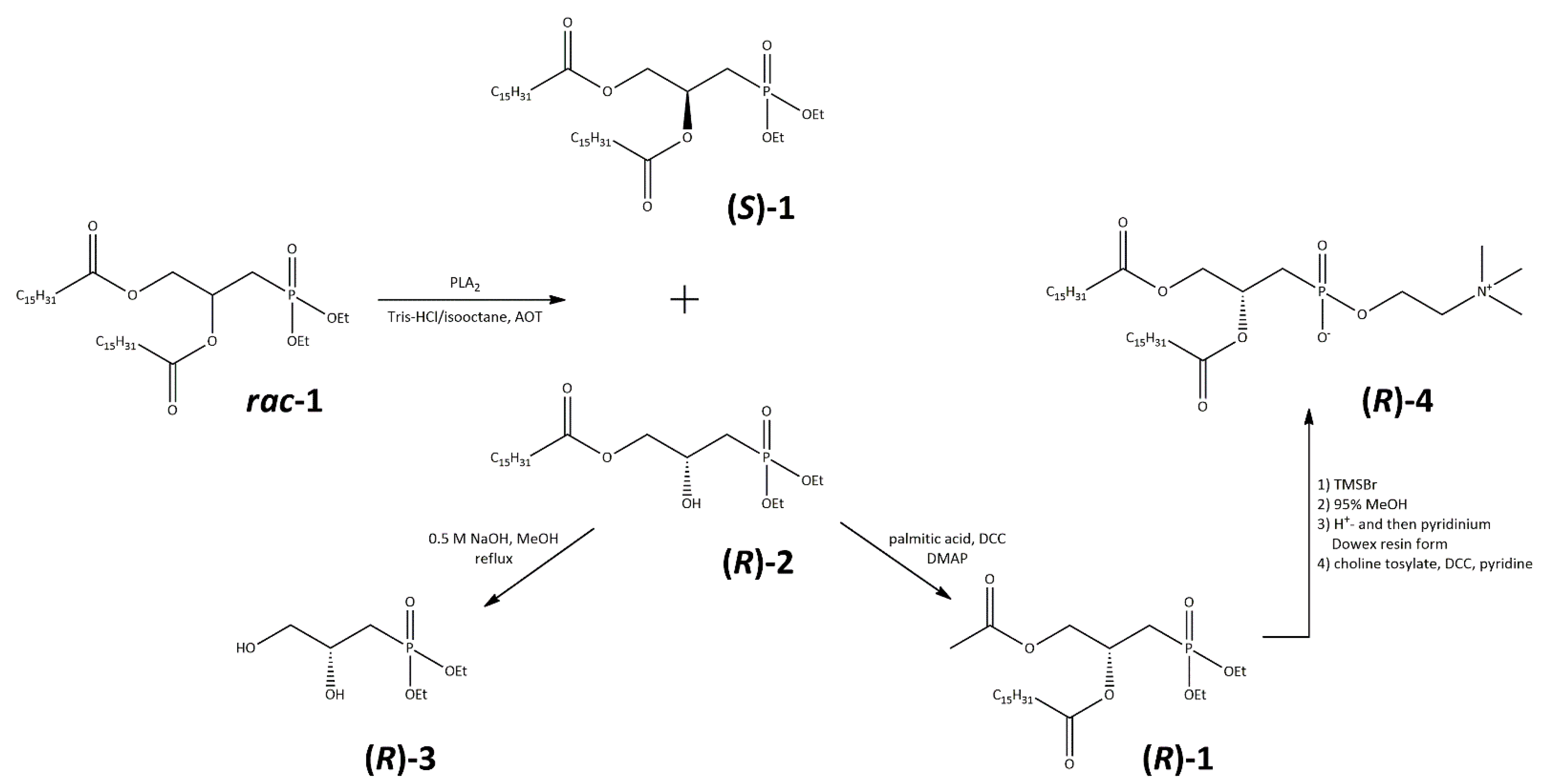
2.1. Synthesis of (R)-2,3-Dipalmitoyloxypropylphosphonocholine (DPPnC, 4)
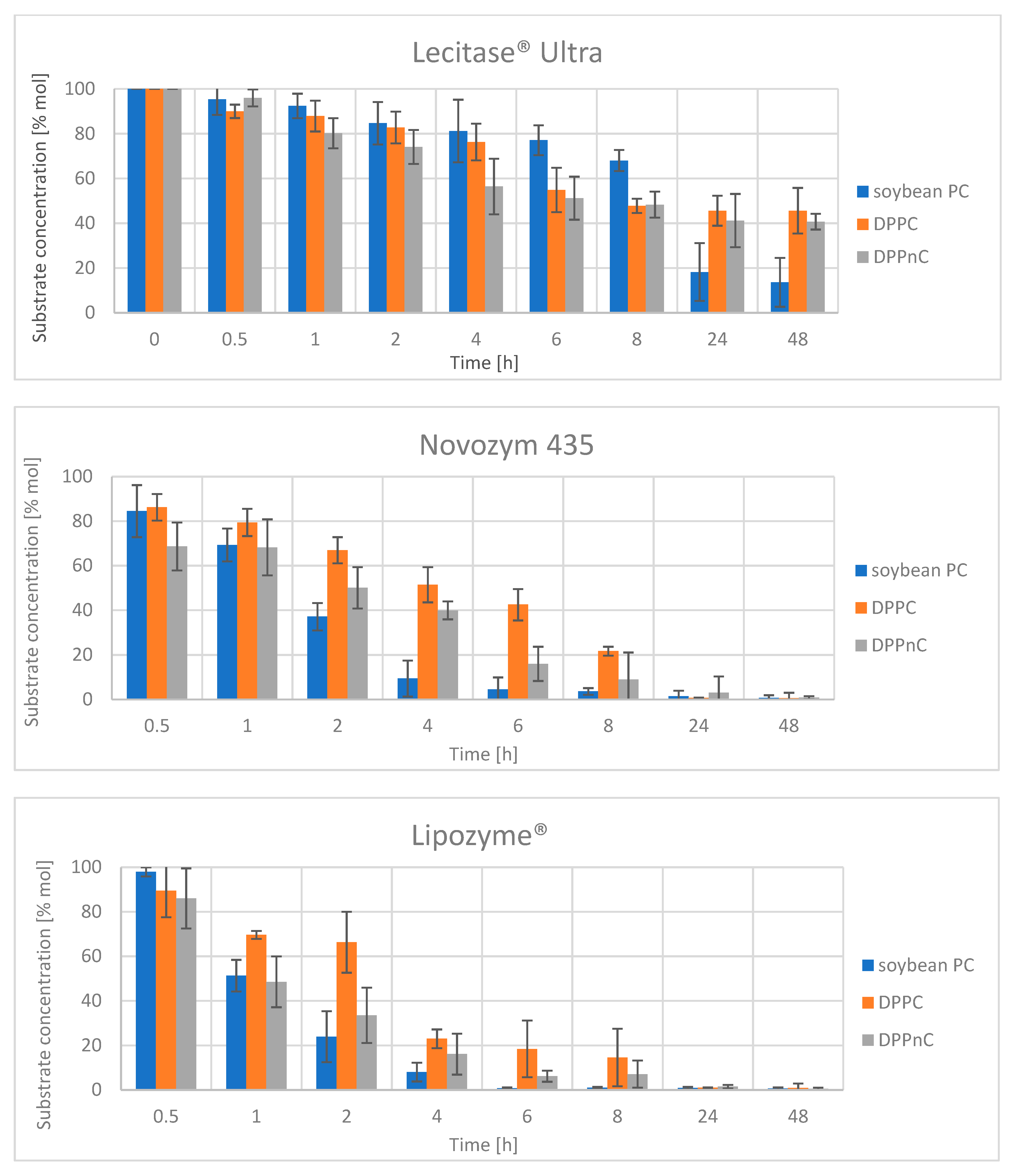
2.2. Enzymatic Reactions of Soybean PC, DPPC and DPPnC with Selected Lipases and Phospholipases
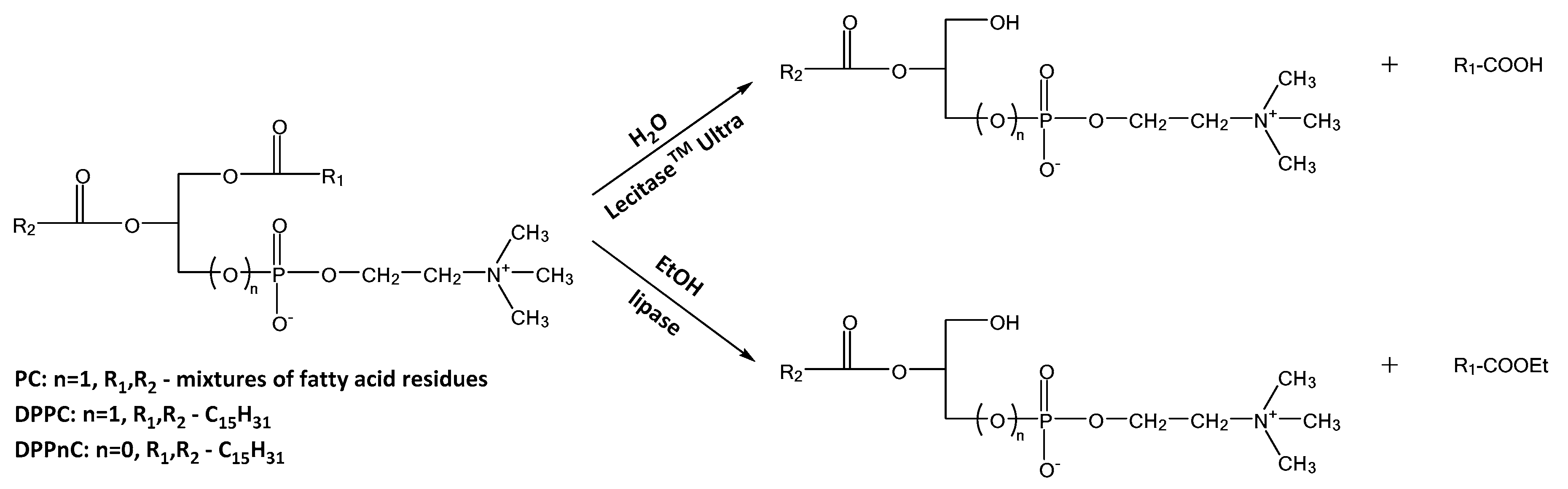
2.2.1. Reactions at sn-1 Position
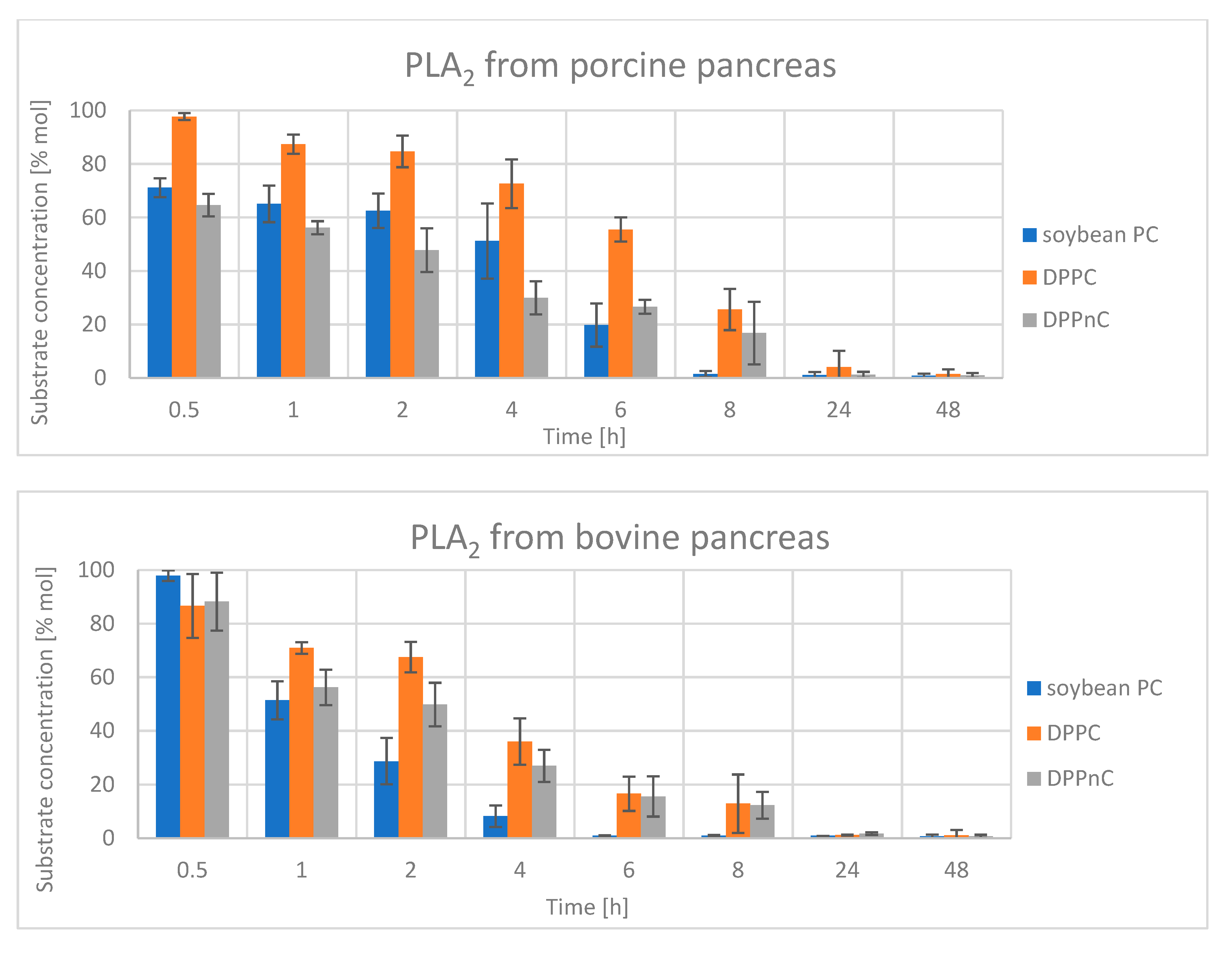
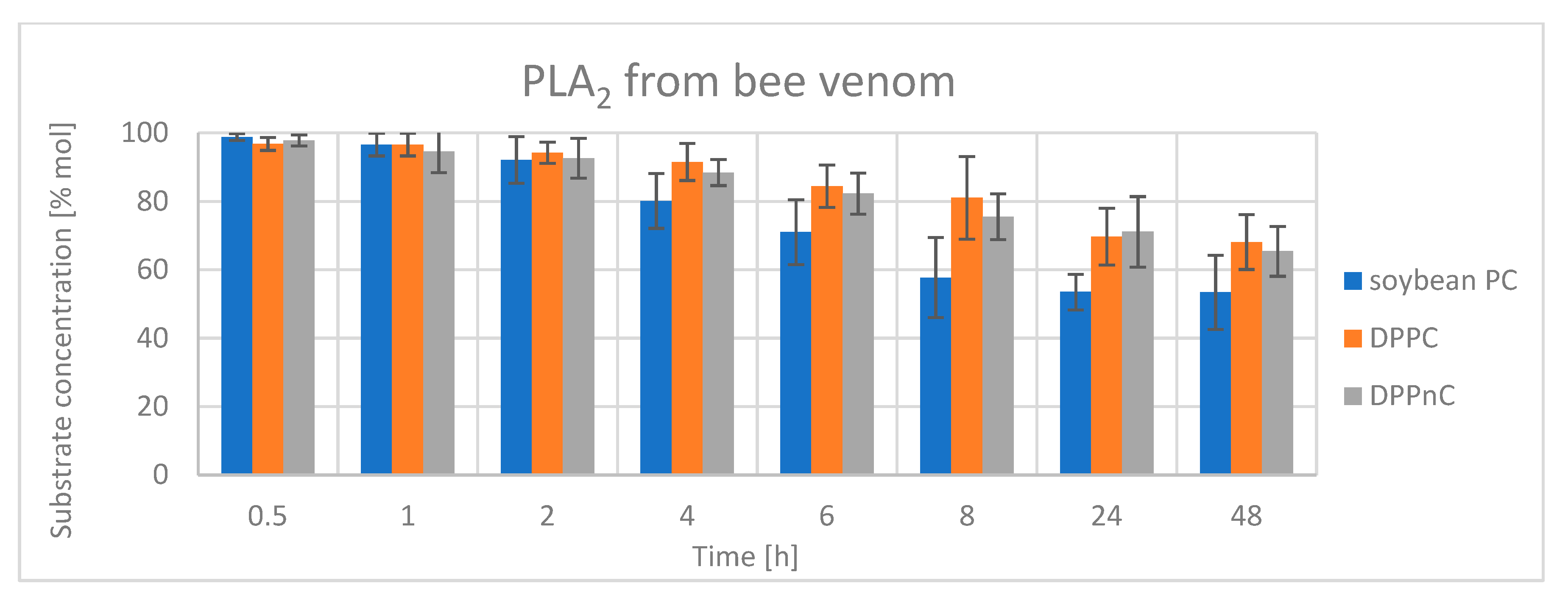
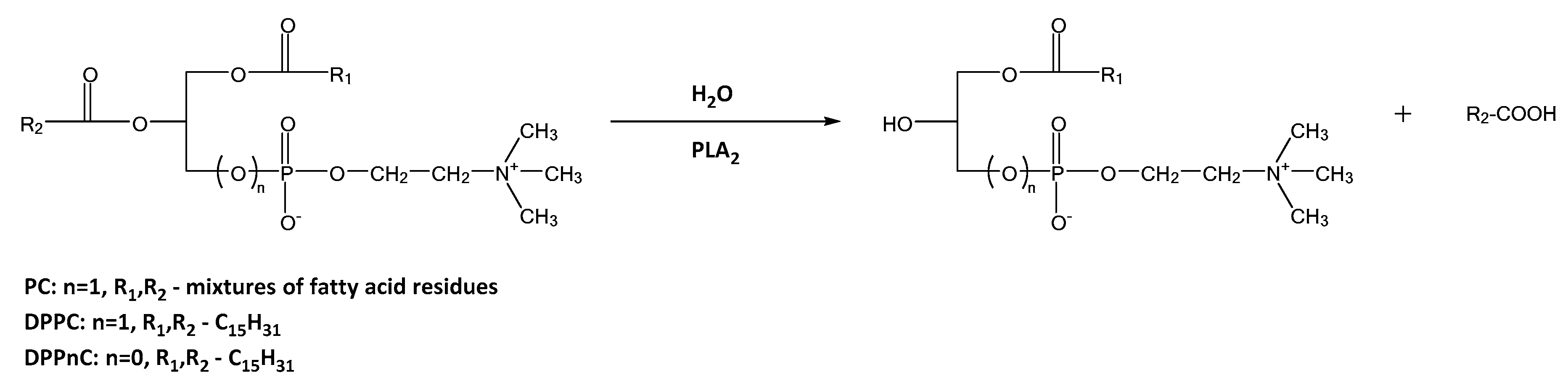
2.2.2. Hydrolysis Catalyzed by Phospholipases A2
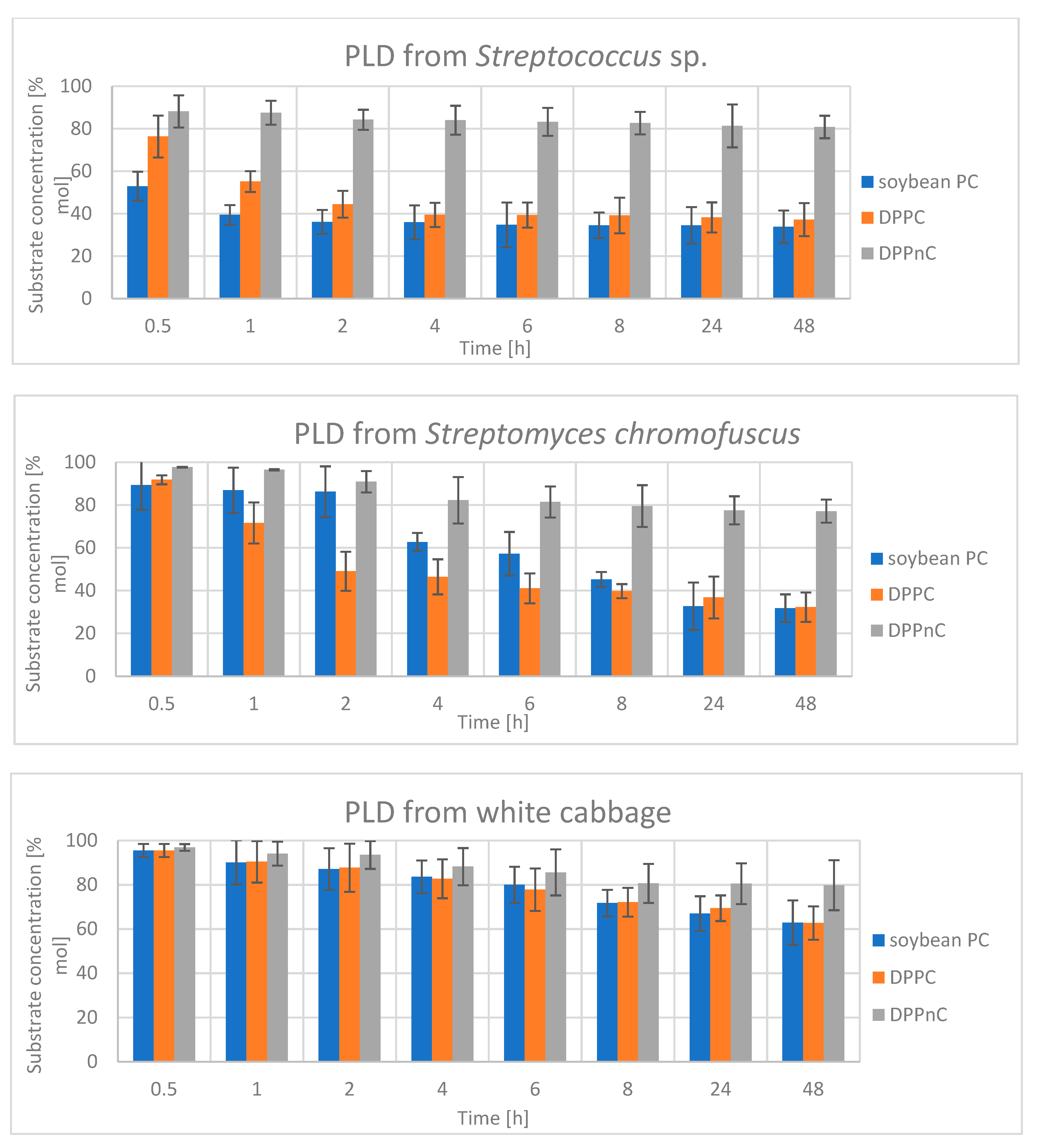
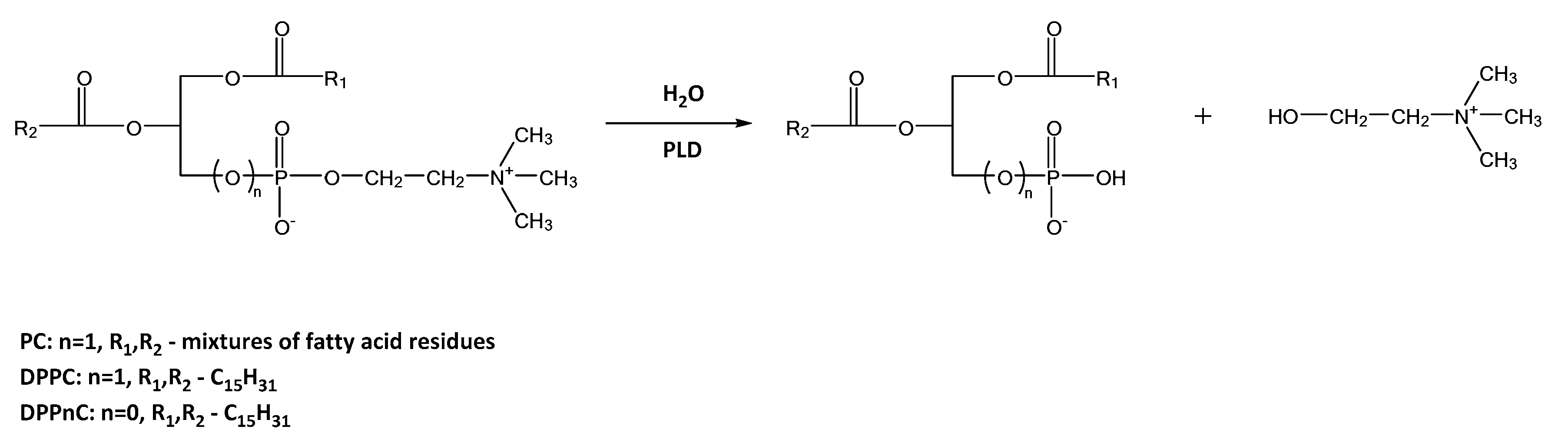
2.2.3. Hydrolysis Catalyzed by Phospholipases D
3. Materials and Methods
3.1. Solvents and Reagents
3.2. Buffers
3.3. Enzymes
3.4. Enzyme Solutions
3.5. Substrates and Products of Enzymatic Hydrolysis
3.6. Analytical Methods
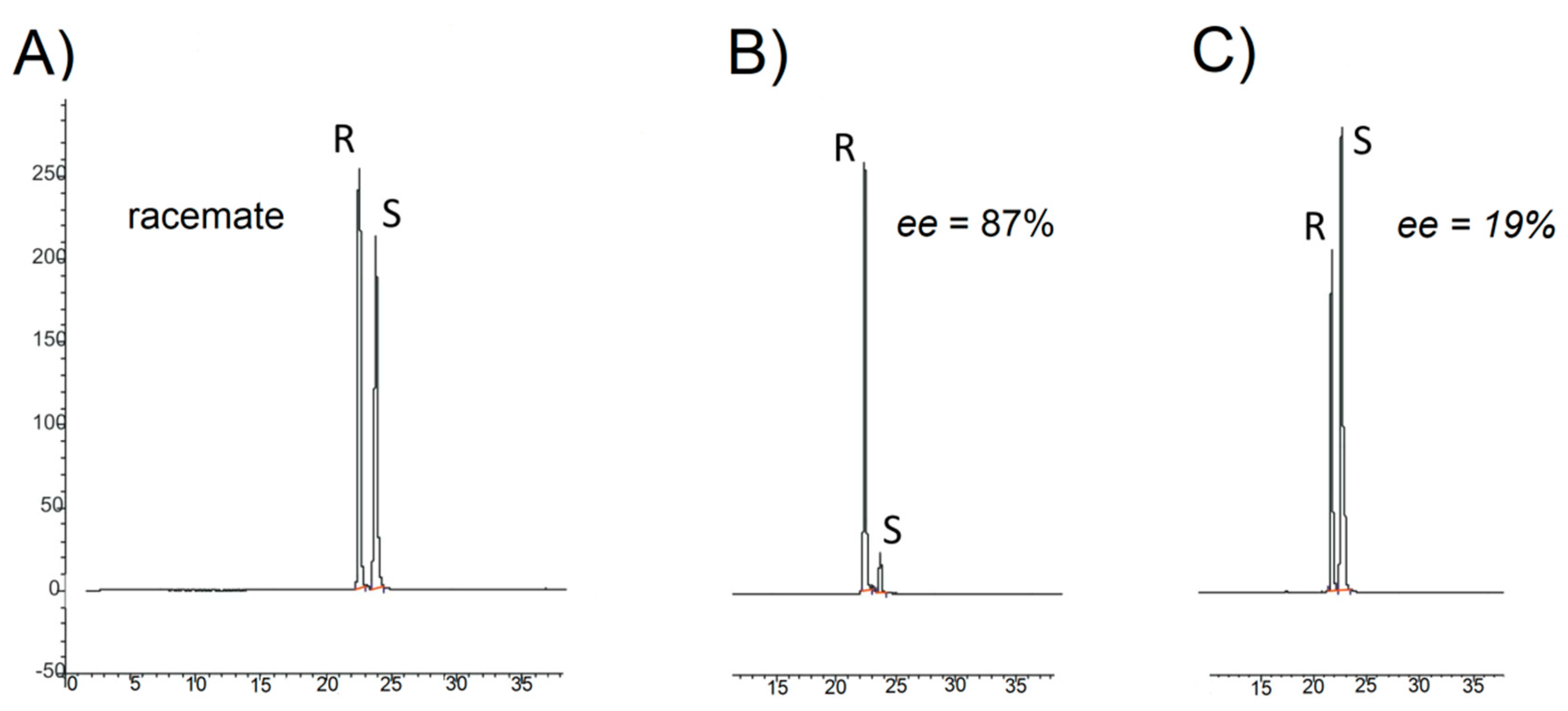
3.7. Enantioselective Hydrolysis of rac Diethyl 2,3-Dipalmitoyloxypropylphosphonate (1)
3.8. Synthesis of (R)-Diethyl 2,3-Dihydroxypropylphosphonate (3)
3.9. Synthesis of (R)-Diethyl 2,3-Dipalmitoyloxypropylphosphonate [(R)-1)]
3.10. Synthesis of (R)-2,3-Dipalmitoyloxypropylphosphonocholine (DPPnC, 4)
3.11. Enzymatic Hydrolysis of DPPC, DPPnC and Soybean PC
3.11.1. Reactions Catalyzed by Lecitase® Ultra
3.11.2. Reactions Catalyzed by Lipases
3.11.3. Reactions Catalyzed by Phospholipases A2
3.11.4. Reactions Catalyzed by PLD from Streptomyces chromofuscus
3.11.5. Reactions Catalyzed by PLD from Streptococcus sp. and PLD from White Cabbage
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liang, C.R.; Rosenberg, H. The biosynthesis of the phosphonic analogue of cephalin in Tetrahymena. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1966, 125, 548–562. [Google Scholar] [CrossRef]
- Mason, W.T. Isolation and characterization of the lipids of the sea anemone, Metridium senile. Biochim. Biophys. Acta 1972, 280, 538–544. [Google Scholar] [CrossRef]
- Mukhamedova, K.S.; Glushenkova, A.I. Natural phosphonolipids. Chem. Nat. Compd. 2000, 36, 329–341. [Google Scholar] [CrossRef]
- Moschidis, M.; Demopoulos, C.; Kritikou, L. Isolation of hens’ phosphonolipids by thin-layer chromatography, their identification and silicic acid column chromatographic separation. J. Chromatogr. 1984, 292, 473–478. [Google Scholar] [CrossRef]
- Floch, V.; Legros, N.; Loisel, S.; Guillaume, C.; Guilbot, J.; Benvegnu, T.; Ferrieres, V.; Plusquellec, D.; Ferec, C. New biocompatible cationic amphiphiles derivative from glycine betaine: A novel family of efficient nonviral gene transfer agents. Biochem. Biophys. Res. Commun. 1998, 251, 360–365. [Google Scholar] [CrossRef]
- Rosenthal, F.; Chodsky, V.; Han, C.H.; Long, T.; Jewish, I.; Igth, M.; Park, N.H. Diether phosphinate lecithin: Chemical synthesis and effects on phospholipase C. Biochim. Biophys. Acta 1969, 187, 385–392. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, H.; Davrazou, F.; Kutateladze, T.G.; Shi, X.; Gozani, O.; Prestwich, G.D. Stabilized phosphatidylinositol-5-phosphate analogues as ligands for the nuclear protein ING2: Chemistry, biology, and molecular modeling. J. Am. Chem. Soc. 2007, 129, 6498–6505. [Google Scholar] [CrossRef] [Green Version]
- Baer, E.; Stanacev, N.Z. Phosphonolipids. I. Synthesis of a phosphonic acid analogue of cephalin. J. Biol. Chem. 1964, 239, 3209–3214. [Google Scholar] [CrossRef]
- Baer, E.; Stanacev, N.Z. Phosphonolipids. II. Synthesis of dialkyl L-alfa-glyceryl-(2-aminoethyl) phosphonates. J. Biol. Chem. 1965, 240, 44–48. [Google Scholar] [CrossRef]
- Fields, S.C. Synthesis of natural products containing a C-P bond. Tetrahedron 1999, 55, 12237–12273. [Google Scholar] [CrossRef]
- Xu, Y.; Prestwich, G.D. Synthesis of chiral (α,α-difluoroalkyl) phosphonate analogues of (Lyso) phosphatidic acid via hydrolytic kinetic resolution. Org. Lett. 2002, 4, 4021–4024. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Qian, L.; Prestwich, G.D. Synthesis of monofluorinated analogues of lysophosphatidic acid analogues were enantiospecifically prepared from chiral protected glycerol synthons, and the surprising enantiospecific and receptor-specific biological readouts, with one compound showing. J. Org. Chem. 2003, 68, 5320–5330. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Qian, L.; Prestwich, G.D. Synthesis of α-fluorinated phosphonates from α-fluorovinylphosphonates: A new route to analogues of lysophosphatidic acid. Org. Lett. 2003, 5, 2267–2270. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Aoki, J.; Shimizu, K.; Umezu-Goto, M.; Hama, K.; Takanezawa, Y. Structure—Activity relationships of fluorinated lysophosphatidic acid. J. Med. Chem. 2005, 48, 3319–3327. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, G.D.; Gajewiak, J.; Zhang, H.; Xu, X.; Yang, G.; Serban, M. Phosphatase-resistant analogues of lysophosphatidic acid: Agonists promote healing, antagonists and autotaxin inhibitors treat cancer. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2008, 1781, 588–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mituła, P.; Wawrzeńczyk, C. Synthesis of a series of new racemic [2,3-bis(acyloxy)propyl]phosphonocholines. Arkivoc 2012, 2012, 216–232. [Google Scholar] [CrossRef]
- De Maria, L.; Vind, J.; Oxenbøll, K.M.; Svendsen, A.; Patkar, S. Phospholipases and their industrial applications. Appl. Microbiol. Biotechnol. 2007, 74, 290–300. [Google Scholar] [CrossRef]
- Borrelli, G.M.; Trono, D. Recombinant lipases and phospholipases and their use as biocatalysts for industrial applications. Int. J. Mol. Sci. 2015, 16, 20774–20840. [Google Scholar] [CrossRef] [Green Version]
- Adlercreutz, P. Immobilisation and application of lipases in organic media. Chem. Soc. Rev. 2013, 42, 6406–6436. [Google Scholar] [CrossRef] [Green Version]
- Bornscheuer, U.T. Enzymes in lipid modification: Past achievements and current trends. Eur. J. Lipid Sci. Technol. 2014, 116, 1322–1331. [Google Scholar] [CrossRef]
- Angajala, G.; Pavan, P.; Subashini, R. Lipases: An overview of its current challenges and prospectives in the revolution of biocatalysis. Biocatal. Agric. Biotechnol. 2016, 7, 257–270. [Google Scholar] [CrossRef]
- Carvalho, A.C.L.D.M.; Fonseca, T.D.S.; de Mattos, M.C.; de Oliveira, M.D.C.F.; de Lemos, T.M.L.G.; Molinari, F.; Romano, D.; Serra, I. Recent advances in lipase-mediated preparation of pharmaceuticals and their intermediates. Int. J. Mol. Sci. 2015, 16, 29682–29716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baer, E.; Basu, H. Phosphonic acid analogues of carbohydrate metabolites. I. Synthesis of L-and D-dihydroxypropylphosphonic acid. Can. J. Biochem. 1969, 47, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.C.; Fernandez-Lafuente, R. Lipase from Rhizomucor miehei as an industrial biocatalyst in chemical process. J. Mol. Catal. B Enzym. 2010, 64, 1–22. [Google Scholar] [CrossRef]
- Ortiz, C.; Ferreira, M.L.; Barbosa, O.; Dos Santos, J.C.S.; Rodrigues, R.C.; Berenguer-Murcia, Á.; Briand, L.E.; Fernandez-Lafuente, R. Novozym 435: The “perfect” lipase immobilized biocatalyst? Catal. Sci. Technol. 2019, 9, 2380–2420. [Google Scholar] [CrossRef] [Green Version]
- Virgen-Ortíz, J.J.; dos Santos, J.C.S.; Ortiz, C.; Berenguer-Murcia, Á.; Barbosa, O.; Rodrigues, R.C.; Fernandez-Lafuente, R. Lecitase ultra: A phospholipase with great potential in biocatalysis. Mol. Catal. 2019, 473, 110405. [Google Scholar] [CrossRef] [Green Version]
- Adlercreutz, D.; Wehtje, E. An enzymatic method for the synthesis of mixed-acid phosphatidylcholine. JAOCS J. Am. Oil Chem. Soc. 2004, 81, 553–557. [Google Scholar] [CrossRef]
- Lim, C.W.; Kim, B.H.; Kim, I.H.; Lee, M.W. Modeling and optimization of phospholipase A1-catalyzed hydrolysis of phosphatidylcholine using response surface methodology for lysophosphatidylcholine production. Biotechnol. Prog. 2015, 31, 35–41. [Google Scholar] [CrossRef]
- Morgado, M.A.P.; Cabral, J.M.S.; Prazeres, D.M.F. Hydrolysis of lecithin by phospholipase A2 in mixed reversed micelles of lecithin and sodium dioctyl sulphosuccinate. J. Chem. Technol. Biotechnol. 1995, 63, 181–189. [Google Scholar] [CrossRef]
- Kiełbowicz, G.; Gładkowski, W.; Chojnacka, A.; Wawrzeńczyk, C. A simple method for positional analysis of phosphatidylcholine. Food Chem. 2012, 135, 2542–2548. [Google Scholar] [CrossRef]
- Kiełbowicz, G.; Chojnacka, A.; Gliszczyńska, A.; Gładkowski, W.; Kłobucki, M.; Niezgoda, N.; Wawrzeńczyk, C. Positional analysis of phosphatidylcholine and phosphatidylethanolamine via LC with a charged aerosol detector. Talanta 2015, 141, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Florin-Christensen, J.; Narvaez-Vasquez, J.; Florin-Christensen, M.; Ryan, C.A. A method for distinguishing 1-acyl from 2-acyl lysophosphatidylcholines generated in biological systems. Anal. Biochem. 1999, 276, 13–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drescher, M.; Li, Y.F.; Hammerschmidt, F. Enzymes in organic chemistry, part 2: Lipase-catalysed hydrolysis of 1-acyloxy-2-arylethylphosphonates and synthesis of phosphonic acid analogues of L-phenylalanine and L-tyrosine. Tetrahedron 1995, 51, 4933–4946. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Yuan, C. Candida rugosa lipase-catalyzed enantioselective hydrolysis in organic solvents. Convenient preparation of optically pure 2-hydroxy-2-arylethanephosphonates. Tetrahedron Lett. 2002, 43, 3247–3249. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Enzymatic synthesis of organophosphorus compounds. Russ. Chem. Rev. 2011, 80, 883–910. [Google Scholar] [CrossRef]
- Majewska, P.; Serafin, M.; Klimek-Ochab, M.; Brzezińska-Rodak, M.; Żymańczyk-Duda, E. Lipases and whole cell biotransformations of 2-hydroxy-2-(ethoxyphenylphosphinyl)acetic acid and its ester. Bioorg. Chem. 2016, 66, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, M.; Shimatani, T.; Kanada, T.; Inoue, Y.; Takahashi, K. Conversion to docosahexaenoic acid-containing phosphatidylserine from squid skin lecithin by phospholipase D-mediated transphosphatidylation. J. Agric. Food Chem. 2000, 48, 4550–4554. [Google Scholar] [CrossRef]
- Smuga, D.A.; Smuga, M.; Świzdor, A.; Panek, A.; Wawrzeńczyk, C. Synthesis of dehydroepiandrosterone analogues modified with phosphatidic acid moiety. Steroids 2010, 75, 1146–1152. [Google Scholar] [CrossRef]
- Baer, E.; Stanacev, N.Z. Phosphonolipids VI. Chemical and enzymatic degradation for study of structure. Can. J. Biochem. 1966, 44, 893–897. [Google Scholar] [CrossRef]
- Ryu, E.K.; Ross, R.J.; Matsushita, T.; MacCoss, M.; Hong, C.I.; West, C.R. Phospholipid-nucleoside conjugates. 3. Syntheses and preliminary biological evaluation of 1-β-D-arabinofuranosylcytosine 5′-monophosphate-L-1,2-dipalmitin and selected 1-β-D-arabinofuranosylcytosine 5′-diphosphate-L-1,2-diacylglycerols. J. Med. Chem. 1982, 25, 1322–1329. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mituła, P.; Wawrzeńczyk, C.; Gładkowski, W. Comparative Studies on the Susceptibility of (R)-2,3-Dipalmitoyloxypropylphosphonocholine (DPPnC) and Its Phospholipid Analogues to the Hydrolysis or Ethanolysis Catalyzed by Selected Lipases and Phospholipases. Catalysts 2021, 11, 129. https://doi.org/10.3390/catal11010129
Mituła P, Wawrzeńczyk C, Gładkowski W. Comparative Studies on the Susceptibility of (R)-2,3-Dipalmitoyloxypropylphosphonocholine (DPPnC) and Its Phospholipid Analogues to the Hydrolysis or Ethanolysis Catalyzed by Selected Lipases and Phospholipases. Catalysts. 2021; 11(1):129. https://doi.org/10.3390/catal11010129
Chicago/Turabian StyleMituła, Paweł, Czesław Wawrzeńczyk, and Witold Gładkowski. 2021. "Comparative Studies on the Susceptibility of (R)-2,3-Dipalmitoyloxypropylphosphonocholine (DPPnC) and Its Phospholipid Analogues to the Hydrolysis or Ethanolysis Catalyzed by Selected Lipases and Phospholipases" Catalysts 11, no. 1: 129. https://doi.org/10.3390/catal11010129
APA StyleMituła, P., Wawrzeńczyk, C., & Gładkowski, W. (2021). Comparative Studies on the Susceptibility of (R)-2,3-Dipalmitoyloxypropylphosphonocholine (DPPnC) and Its Phospholipid Analogues to the Hydrolysis or Ethanolysis Catalyzed by Selected Lipases and Phospholipases. Catalysts, 11(1), 129. https://doi.org/10.3390/catal11010129