
Sustainable Synthesis of a Highly Stable and Coke-Free Ni@CeO2 Catalyst for the Efficient Carbon Dioxide Reforming of Methane

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
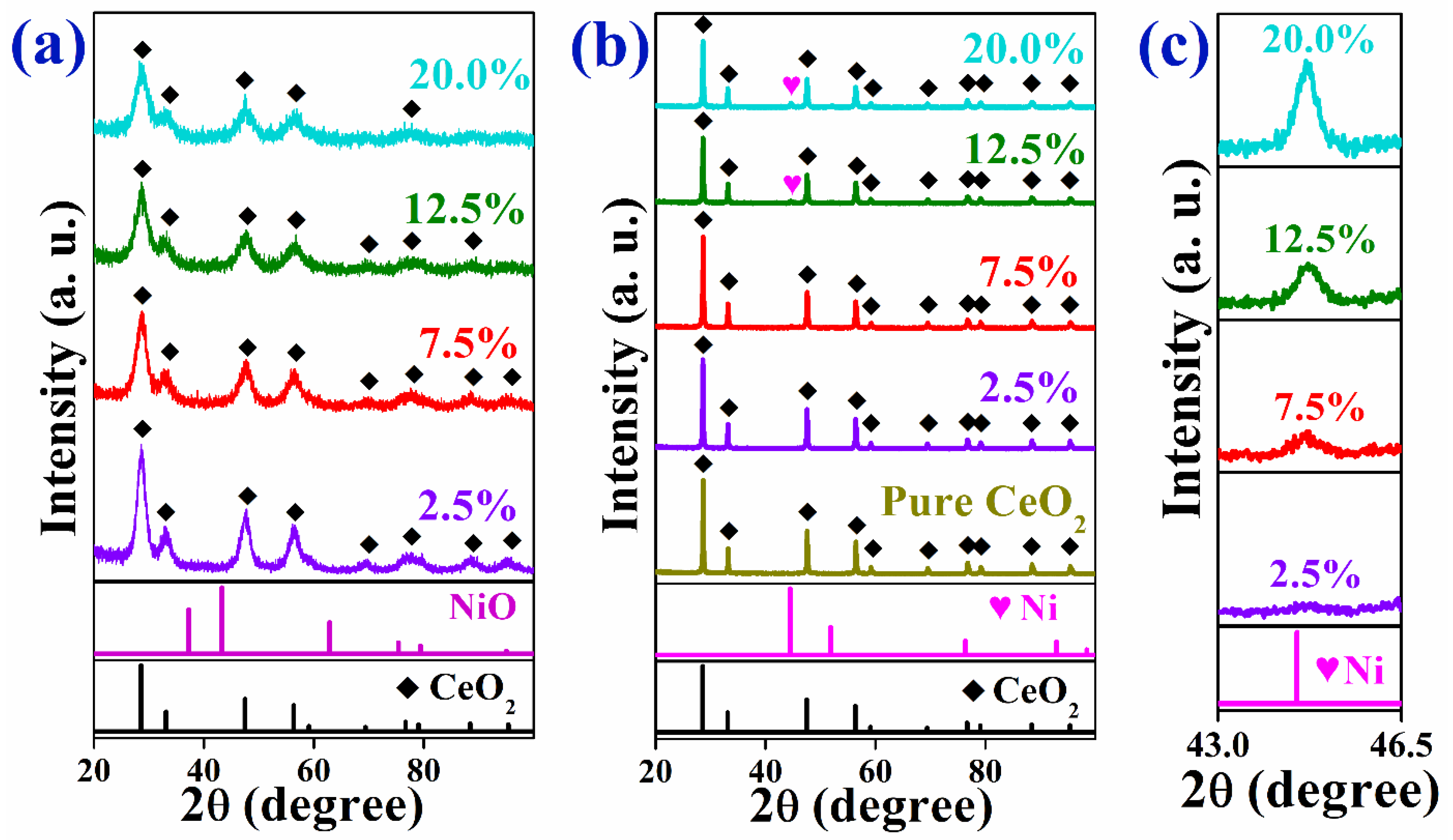
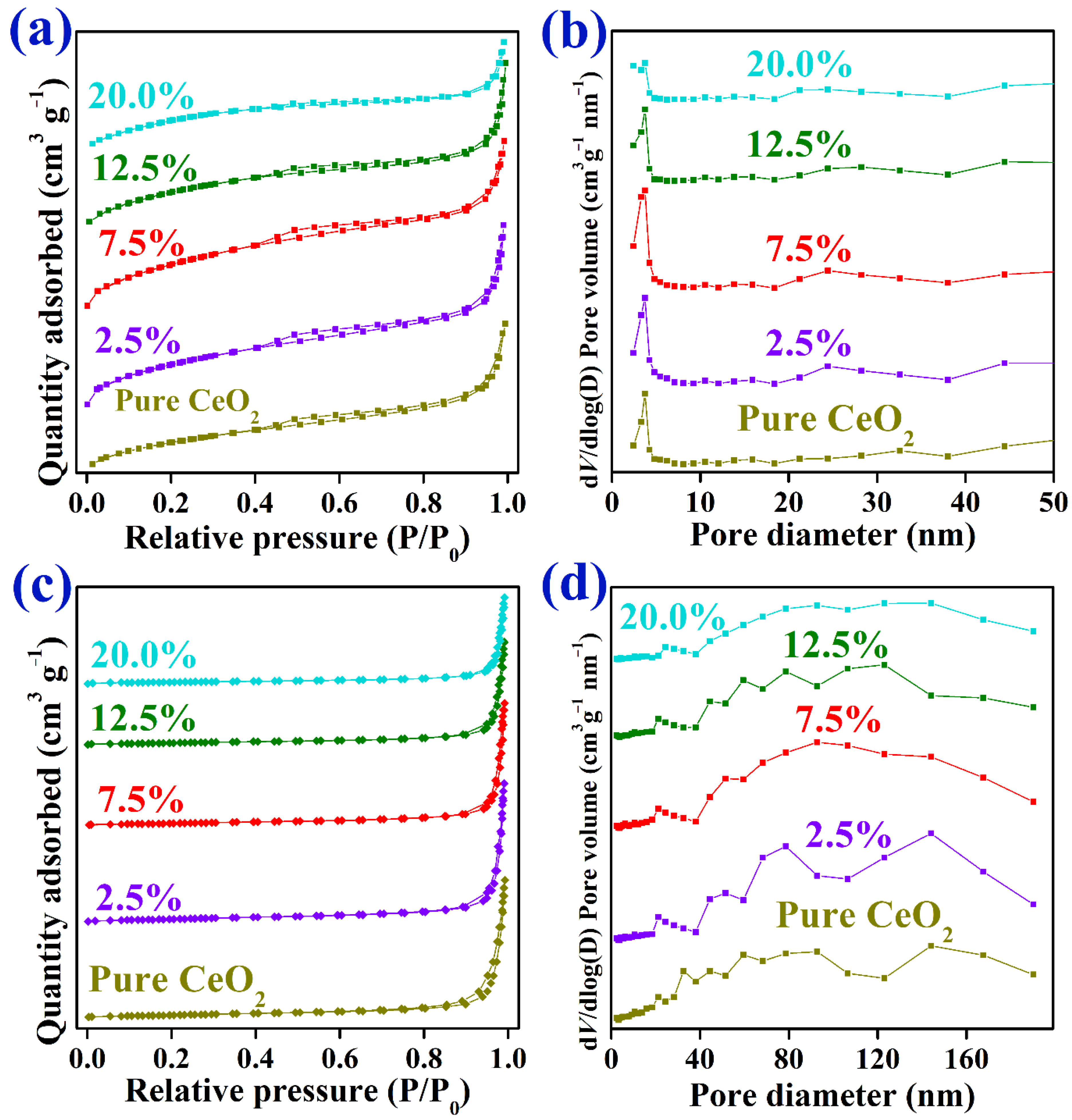
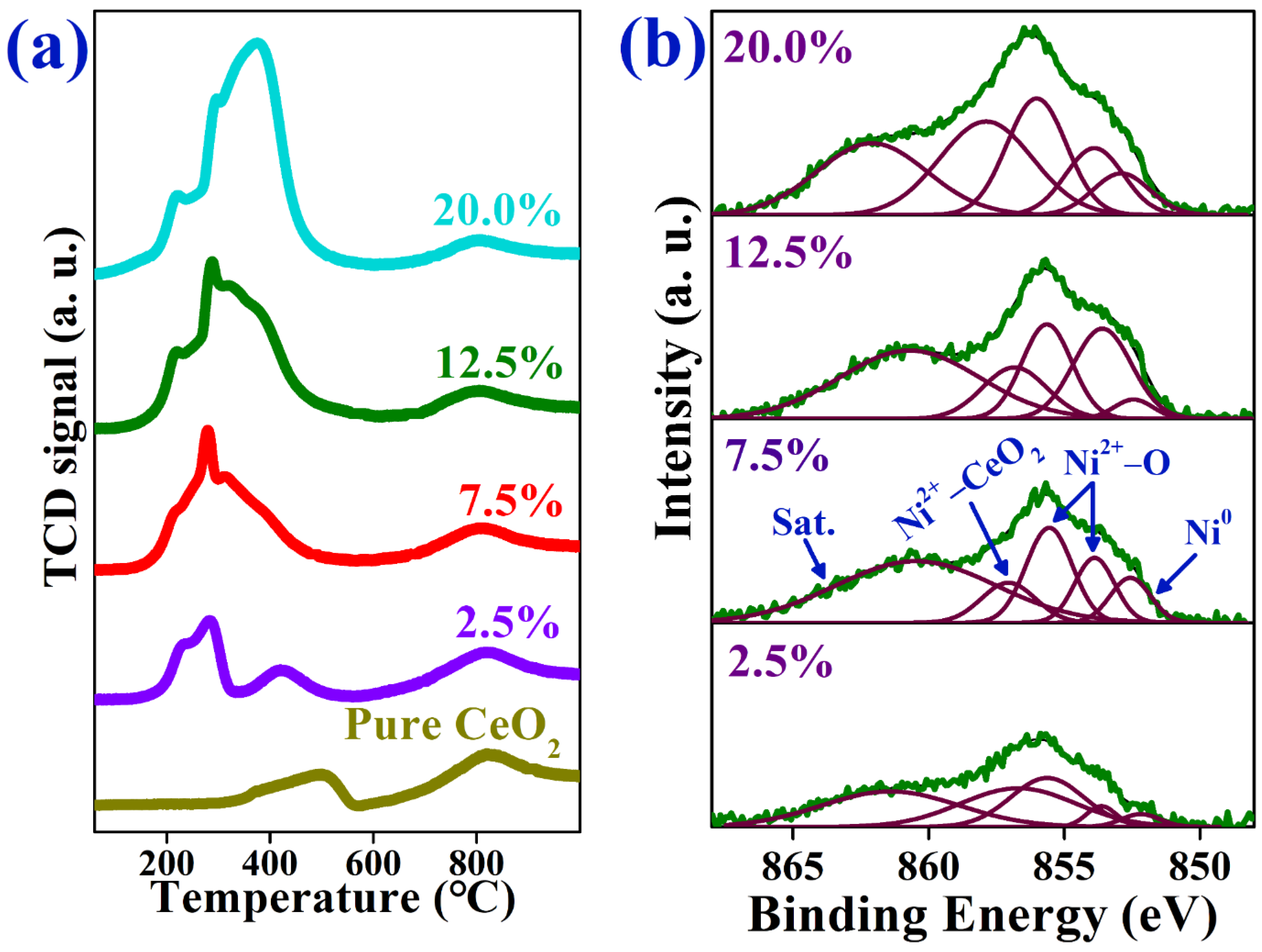
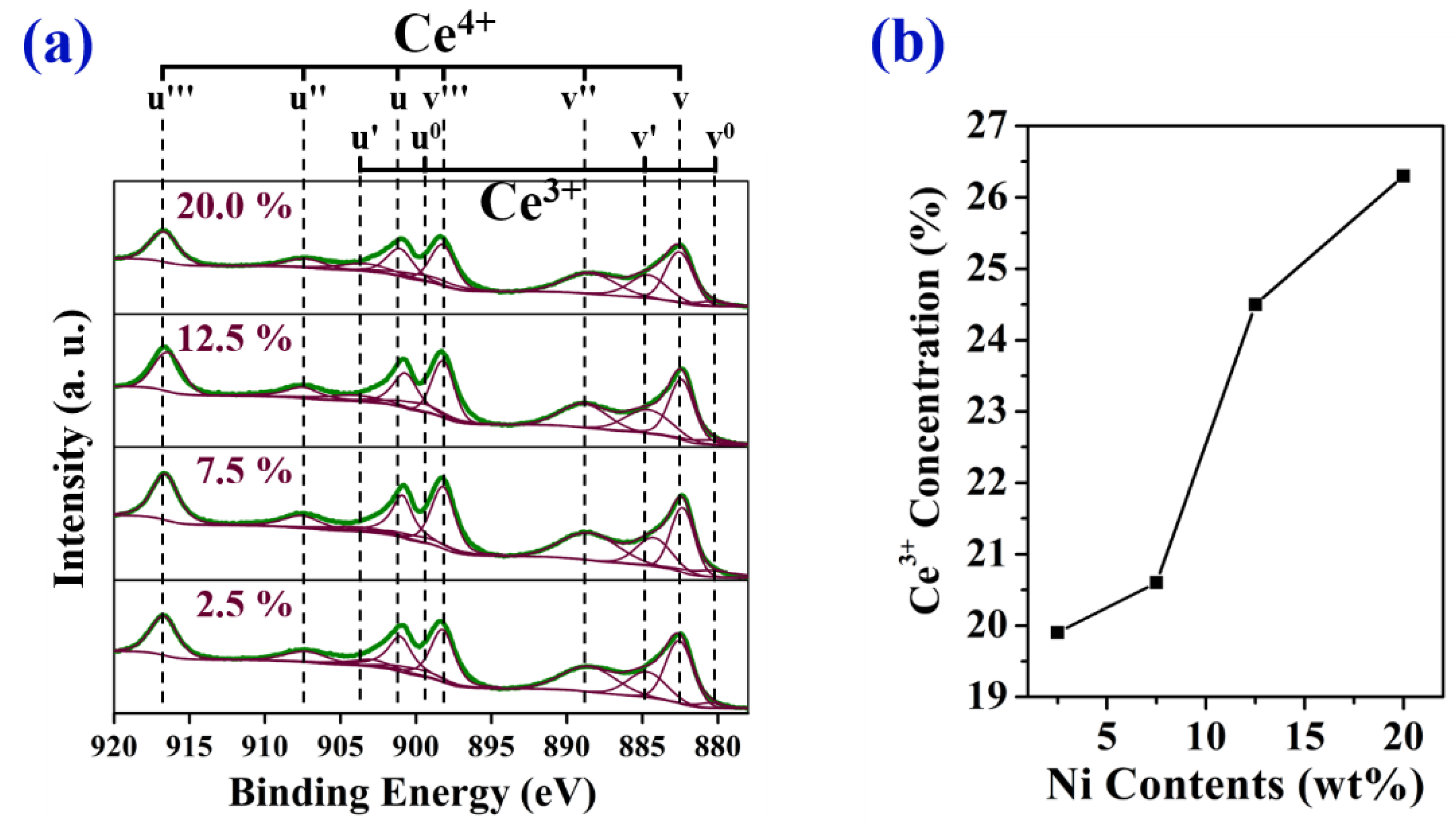
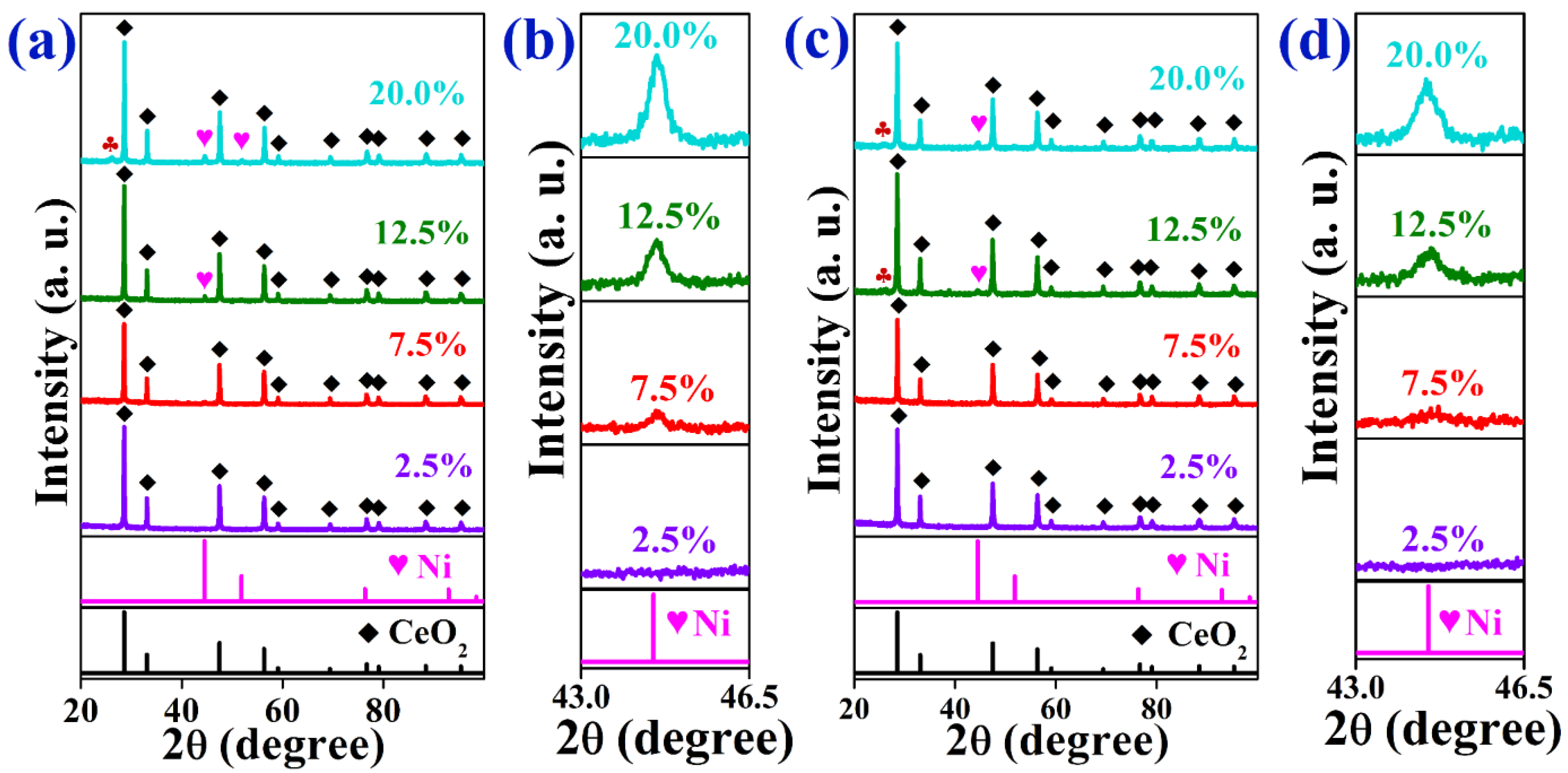
2.1. Characterization of As-Prepared Catalysts
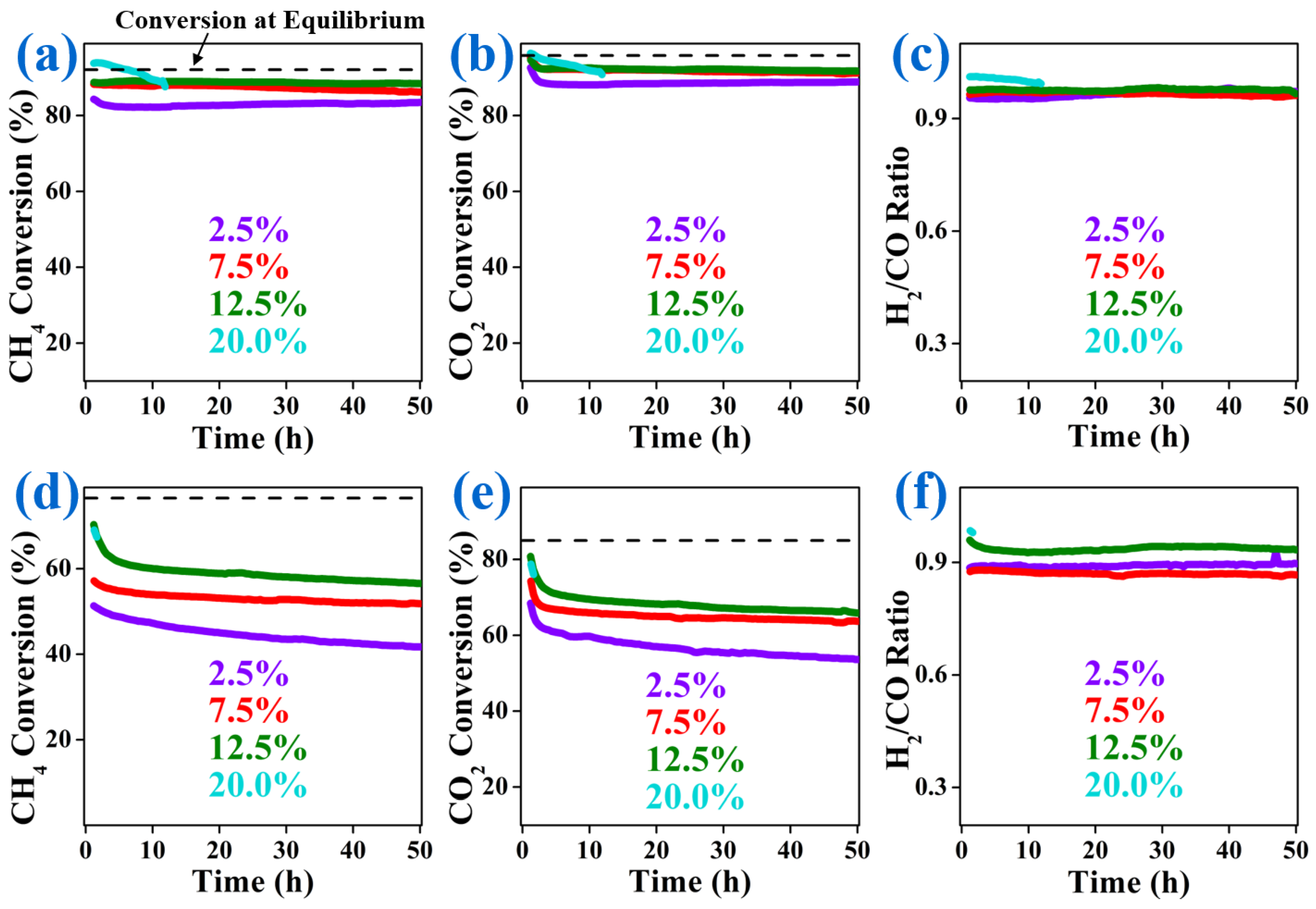
2.2. Catalytic Performance
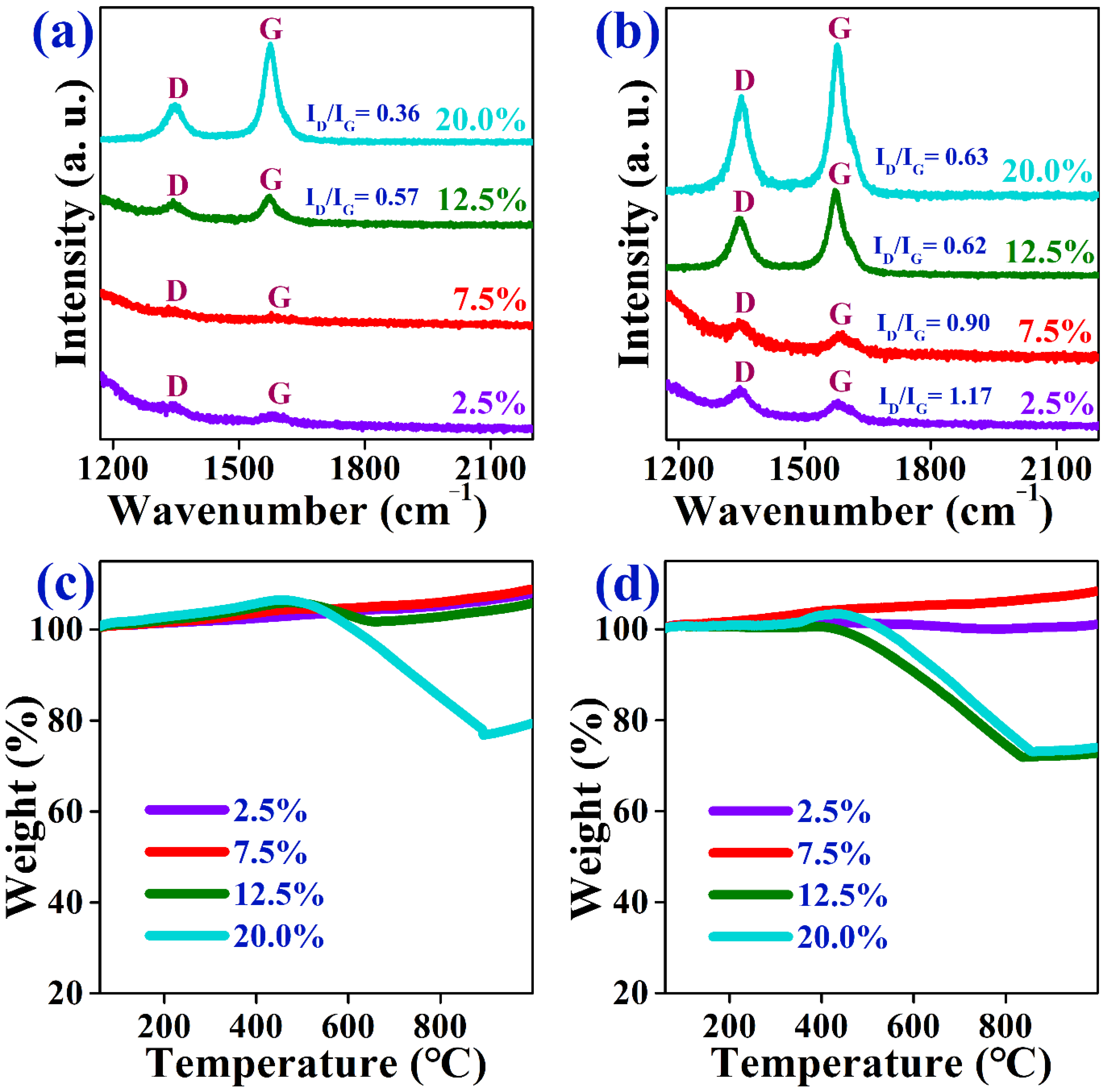
2.3. Characterization of Spent Catalysts
3. Experimental Section
3.1. Materials
3.2. Synthesis of Catalysts
3.3. Physical Characterization of Catalysts
3.4. Catalytic Activity Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jung, S.; Lee, J.; Moon, D.H.; Kim, K.-H.; Kwon, E.E. Upgrading biogas into syngas through dry reforming. Renew. Sustain. Energy Rev. 2021, 143, 110949. [Google Scholar] [CrossRef]
- Subramanian, S.; Song, Y.; Kim, D.; Yavuz, C.T. Redox and Nonredox CO2 Utilization: Dry Reforming of Methane and Catalytic Cyclic Carbonate Formation. ACS Energy Lett. 2020, 5, 1689–1700. [Google Scholar] [CrossRef]
- Li, M.; Sun, Z.; Hu, Y.H. Catalysts for CO2 reforming of CH4: A review. J. Mater. Chem. A 2021, 9, 12495–12520. [Google Scholar] [CrossRef]
- Li, M.; Sun, Z.; Hu, Y.H. Thermo-photo coupled catalytic CO2 reforming of methane: A review. Chem. Eng. J. 2022, 428, 131222. [Google Scholar] [CrossRef]
- Sheng, K.; Luan, D.; Jiang, H.; Zeng, F.; Wei, B.; Pang, F.; Ge, J. NixCoy Nanocatalyst Supported by ZrO2 Hollow Sphere for Dry Reforming of Methane: Synergetic Catalysis by Ni and Co in Alloy. ACS Appl. Mater. Interfaces 2019, 11, 24078–24087. [Google Scholar] [CrossRef]
- Liu, H.; Li, Y.; He, D. Recent Progress of Catalyst Design for Carbon Dioxide Reforming of Methane to Syngas. Energy Technol. 2020, 8, 1900493. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Panagiotopoulou, P.; Artemakis, G. A review of recent efforts to promote dry reforming of methane (DRM) to syngas production via bimetallic catalyst formulations. Appl. Catal. B Environ. 2021, 296, 120210. [Google Scholar] [CrossRef]
- Mohammadi, M.M.; Shah, C.; Dhandapani, S.K.; Chen, J.; Abraham, S.R.; Sullivan, W.; Buchner, R.D.; Kyriakidou, E.A.; Lin, H.; Lund, C.R.F.; et al. Single-Step Flame Aerosol Synthesis of Active and Stable Nanocatalysts for the Dry Reforming of Methane. ACS Appl. Mater. Interfaces 2021, 13, 17618–17628. [Google Scholar] [CrossRef]
- Kim, H.; Eissa, A.A.-S.; Kim, S.B.; Lee, H.; Kim, W.; Seo, D.J.; Lee, K.; Yoon, W.L. One-pot synthesis of a highly mesoporous Ni/MgAl2O4 spinel catalyst for efficient steam methane reforming: Influence of inert annealing. Catal. Sci. Technol. 2021, 11, 4447–4458. [Google Scholar] [CrossRef]
- Al-Swai, B.M.; Osman, N.; Alnarabiji, M.S.; Adesina, A.A.; Abdullah, B. Syngas Production via Methane Dry Reforming over Ceria–Magnesia Mixed Oxide-Supported Nickel Catalysts. Ind. Eng. Chem. Res. 2019, 58, 539–552. [Google Scholar] [CrossRef]
- Wu, J.; Qiao, L.-Y.; Zhou, Z.-F.; Cui, G.-J.; Zong, S.-S.; Xu, D.-J.; Ye, R.-P.; Chen, R.-P.; Si, R.; Yao, Y.-G. Revealing the Synergistic Effects of Rh and Substituted La2B2O7 (B = Zr or Ti) for Preserving the Reactivity of Catalyst in Dry Reforming of Methane. ACS Catal. 2019, 9, 932–945. [Google Scholar] [CrossRef]
- Lovell, E.C.; Großman, H.; Horlyck, J.; Scott, J.; Mädler, L.; Amal, R. Asymmetrical Double Flame Spray Pyrolysis-Designed SiO2/Ce0.7Zr0.3O2 for the Dry Reforming of Methane. ACS Appl. Mater. Interfaces 2019, 11, 25766–25777. [Google Scholar] [CrossRef]
- Teh, L.P.; Setiabudi, H.D.; Timmiati, S.N.; Aziz, M.A.A.; Annuar, N.H.R.; Ruslan, N.N. Recent progress in ceria-based catalysts for the dry reforming of methane: A review. Chem. Eng. Sci. 2021, 242, 116606. [Google Scholar] [CrossRef]
- Minette, F.; de Wilde, J. Multi-scale modeling and simulation of low-pressure methane bi-reforming using structured catalytic reactors. Chem. Eng. J. 2021, 407, 127218. [Google Scholar] [CrossRef]
- Liang, T.-Y.; Raja, D.S.; Chin, K.C.; Huang, C.-L.; Sethupathi, S.A.P.; Leong, L.K.; Tsai, D.-H.; Lu, S.-Y. Bimetallic Metal–Organic Framework-Derived Hybrid Nanostructures as High-Performance Catalysts for Methane Dry Reforming. ACS Appl. Mater. Interfaces 2020, 12, 15183–15193. [Google Scholar] [CrossRef]
- Li, Z.; Lin, Q.; Li, M.; Cao, J.; Liu, F.; Pan, H.; Wang, Z.; Kawi, S. Recent advances in process and catalyst for CO2 reforming of methane. Renew. Sustain. Energy Rev. 2020, 134, 110312. [Google Scholar] [CrossRef]
- Lyu, Y.; Jocz, J.; Xu, R.; Stavitski, E.; Sievers, C. Nickel Speciation and Methane Dry Reforming Performance of Ni/CexZr1–xO2 Prepared by Different Synthesis Methods. ACS Catal. 2020, 10, 11235–11252. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, Z.; Chen, X.; Rui, N.; Betancourt, L.E.; Lin, L.; Xu, W.; Sun, C.-J.; Abeykoon, A.M.M.; Rodriguez, J.A.; et al. Effects of Zr Doping into Ceria for the Dry Reforming of Methane over Ni/CeZrO2 Catalysts: In Situ Studies with XRD, XAFS, and AP-XPS. ACS Catal. 2020, 10, 3274–3284. [Google Scholar] [CrossRef]
- Das, S.; Sengupta, M.; Bag, A.; Shah, M.; Bordoloi, A. Facile synthesis of highly disperse Ni–Co nanoparticles over mesoporous silica for enhanced methane dry reforming. Nanoscale 2018, 10, 6409–6425. [Google Scholar] [CrossRef]
- de la Cruz-Flores, V.G.; Martinez-Hernandez, A.; Gracia-Pinilla, M.A. Deactivation of Ni-SiO2 catalysts that are synthetized via a modified direct synthesis method during the dry reforming of methane. Appl. Catal. A Gen. 2020, 594, 117455. [Google Scholar] [CrossRef]
- Shi, L.-Y.; Li, Y.-X.; Xue, D.-M.; Tan, P.; Jiang, Y.; Liu, X.-Q.; Sun, L.-B. Fabrication of highly dispersed nickel in nanoconfined spaces of as-made SBA-15 for dry reforming of methane with carbon dioxide. Chem. Eng. J. 2020, 390, 124491. [Google Scholar] [CrossRef]
- Niu, J.; Liland, S.E.; Yang, J.; Rout, K.R.; Ran, J.; Chen, D. Effect of oxide additives on the hydrotalcite derived Ni catalysts for CO2 reforming of methane. Chem. Eng. J. 2019, 377, 119763. [Google Scholar] [CrossRef]
- Yang, E.; Nam, E.; Lee, J.; Lee, H.; Park, E.D.; Lim, H.; An, K. Al2O3-Coated Ni/CeO2 nanoparticles as coke-resistant catalyst for dry reforming of methane. Catal. Sci. Technol. 2020, 10, 8283–8294. [Google Scholar] [CrossRef]
- Rezaei, R.; Moradi, G.; Sharifnia, S. Dry Reforming of Methane over Ni-Cu/Al2O3 Catalyst Coatings in a Microchannel Reactor: Modeling and Optimization Using Design of Experiments. Energy Fuels 2019, 33, 6689–6706. [Google Scholar] [CrossRef]
- Guo, Y.; Li, Y.; Ning, Y.; Liu, Q.; Tian, L.; Zhang, R.; Fu, Q.; Wang, Z.-J. CO2 Reforming of Methane over a Highly Dispersed Ni/Mg–Al–O Catalyst Prepared by a Facile and Green Method. Ind. Eng. Chem. Res. 2020, 59, 15506–15514. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, J.; Wu, Y.; Pan, J.; Zhang, Q.; Tan, Y.; Han, Y. Insight into the effects of the oxygen species over Ni/ZrO2 catalyst surface on methane reforming with carbon dioxide. Appl. Catal. B Environ. 2019, 244, 427–437. [Google Scholar] [CrossRef]
- Seo, J.-C.; Kim, H.; Lee, Y.-L.; Nam, S.; Roh, H.-S.; Lee, K.; Park, S.B. One-Pot Synthesis of Full-Featured Mesoporous Ni/Al2O3 Catalysts via a Spray Pyrolysis-Assisted Evaporation-Induced Self-Assembly Method for Dry Reforming of Methane. ACS Sustain. Chem. Eng. 2021, 9, 894–904. [Google Scholar] [CrossRef]
- Sokefun, Y.O.; Joseph, B.; Kuhn, J.N. Impact of Ni and Mg Loadings on Dry Reforming Performance of Pt/Ceria-Zirconia Catalysts. Ind. Eng. Chem. Res. 2019, 58, 9322–9330. [Google Scholar] [CrossRef]
- Shi, C.; Wang, S.; Ge, X.; Deng, S.; Chen, B.; Shen, J. A review of different catalytic systems for dry reforming of methane: Conventional catalysis-alone and plasma-catalytic system. J. CO2 Util. 2021, 46, 101462. [Google Scholar] [CrossRef]
- Bian, Z.; Das, S.; Wai, M.H.; Hongmanorom, P.; Kawi, S. A Review on Bimetallic Nickel-Based Catalysts for CO2 Reforming of Methane. Chemphyschem 2017, 18, 3117–3134. [Google Scholar] [CrossRef] [Green Version]
- Lian, Z.; Olanrele, S.O.; Si, C.; Yang, M.; Li, B. Critical Role of Interfacial Sites between Nickel and CeO2 Support in Dry Reforming of Methane: Revisit of Reaction Mechanism and Origin of Stability. J. Phys. Chem. C 2020, 124, 5118–5124. [Google Scholar] [CrossRef]
- Yan, X.; Hu, T.; Liu, P.; Li, S.; Zhao, B.; Zhang, Q.; Jiao, W.; Chen, S.; Wang, P.; Lu, J.; et al. Highly efficient and stable Ni/CeO2-SiO2 catalyst for dry reforming of methane: Effect of interfacial structure of Ni/CeO2 on SiO2. Appl. Catal. B: Environ. 2019, 246, 221–231. [Google Scholar] [CrossRef]
- Omoregbe, O.; Danh, H.T.; Abidin, S.Z.; Setiabudi, H.D.; Abdullah, B.; Vu, K.B.; Vo, D.-V.N. Influence of Lanthanide Promoters on Ni/SBA-15 Catalysts for Syngas Production by Methane Dry Reforming. Procedia Eng. 2016, 148, 1388–1395. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Han, K.; Yu, W.; Zhao, L.; Wang, Y.; Wang, X.; Yu, H.; Shi, W. Low Temperature CO2 Reforming with Methane Reaction over CeO2-Modified Ni@SiO2 Catalysts. ACS Appl. Mater. Interfaces 2020, 12, 35022–35034. [Google Scholar] [CrossRef]
- Wang, J.B.; Tai, Y.-L.; Dow, W.-P.; Huang, T.-J. Study of ceria-supported nickel catalyst and effect of yttria doping on carbon dioxide reforming of methane. Appl. Catal. A Gen. 2001, 218, 69–79. [Google Scholar] [CrossRef]
- Liu, Z.; Grinter, D.C.; Lustemberg, P.G.; Nguyen-Phan, T.-D.; Zhou, Y.; Luo, S.; Waluyo, I.; Crumlin, E.J.; Stacchiola, D.J.; Zhou, J.; et al. Dry Reforming of Methane on a Highly-Active Ni-CeO2 Catalyst: Effects of Metal-Support Interactions on C−H Bond Breaking. Angew. Chem. Int. 2016, 55, 7455–7459. [Google Scholar] [CrossRef] [Green Version]
- Safavinia, B.; Wang, Y.; Jiang, C.; Roman, C.; Darapaneni, P.; Larriviere, J.; Cullen, D.A.; Dooley, K.M.; Dorman, J.A. Enhancing CexZr1–xO2 Activity for Methane Dry Reforming Using Subsurface Ni Dopants. ACS Catal. 2020, 10, 4070–4079. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, Y.; Akhtamberdinova, Z.; Chen, X.; Jiang, G.; Liu, D. Dry Reforming of Shale Gas and Carbon Dioxide with Ni-Ce-Al2O3 Catalyst: Syngas Production Enhanced over Ni-CeOx Formation. ChemCatChem 2018, 10, 4689–4698. [Google Scholar] [CrossRef]
- Chein, R.-Y.; Fung, W.-Y. Syngas production via dry reforming of methane over CeO2 modified Ni/Al2O3 catalysts. Int. J. Hydrog. Energy 2019, 44, 14303–14315. [Google Scholar] [CrossRef]
- Yao, L.; Galvez, M.E.; Hu, C.; da Costa, P. Synthesis Gas Production via Dry Reforming of Methane over Manganese Promoted Nickel/Cerium–Zirconium Oxide Catalyst. Ind. Eng. Chem. Res. 2018, 57, 16645–16656. [Google Scholar] [CrossRef]
- Wolfbeisser, A.; Sophiphun, O.; Bernardi, J.; Wittayakun, J.; Föttinger, K.; Rupprechter, G. Methane dry reforming over ceria-zirconia supported Ni catalysts. Catal. Today 2016, 277, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Wang, R.; Zhao, Y.; Wang, S.; Ma, X. Effect of Ce doping on the catalytic performance of xNiCeOy@SiO2 catalysts for dry reforming of methane. Asia-Pac. J. Chem. Eng. 2021, 16, e2678. [Google Scholar] [CrossRef]
- Rodriguez-Gomez, A.; Lopez-Martin, A.; Ramirez, A.; Gascon, J.; Caballero, A. Elucidating the Promotional Effect of Cerium in the Dry Reforming of Methane. ChemCatChem 2021, 13, 553–563. [Google Scholar] [CrossRef]
- Eissa, A.A.; Kim, N.H.; Lee, J.H. Rational design of a highly mesoporous Fe–N–C/Fe3C/C–S–C nanohybrid with dense active sites for superb electrocatalysis of oxygen reduction. J. Mater. Chem. A 2020, 8, 23436–23454. [Google Scholar] [CrossRef]
- Cychosz, K.A.; Guillet-Nicolas, R.; García-Martínez, J.; Thommes, M. Recent advances in the textural characterization of hierarchically structured nanoporous materials. Chem. Soc. Rev. 2017, 46, 389–414. [Google Scholar] [CrossRef]
- Venkataswamy, P.; Rao, K.N.; Jampaiah, D.; Reddy, B.M. Nanostructured manganese doped ceria solid solutions for CO oxidation at lower temperatures. Appl. Catal. B: Environ. 2015, 162, 122–132. [Google Scholar] [CrossRef]
- Sagar, V.T.; Pintar, A. Enhanced surface properties of CeO2 by MnOx doping and their role in mechanism of methane dry reforming deduced by means of in-situ DRIFTS. Appl. Catal. A Gen. 2020, 599, 117603. [Google Scholar] [CrossRef]
- Tao, K.; Zhang, Y.; Terao, S.; Tsubaki, N. Development of platinum-based bimodal pore catalyst for CO2 reforming of CH4. Catal. Today 2010, 153, 150–155. [Google Scholar] [CrossRef]
- Deng, G.; Zhang, G.; Zhu, X.; Guo, Q.; Liao, X.; Chen, X.; Li, K. Optimized Ni-based catalysts for methane reforming with O2-containing CO2. Appl. Catal. B: Environ. 2021, 289, 120033. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Meloni, E.; Ricca, A. Influence of Catalytic Formulation and Operative Conditions on Coke Deposition over CeO2-SiO2 Based Catalysts for Ethanol Reforming. Energies 2017, 10, 1030. [Google Scholar] [CrossRef] [Green Version]
- Sutradhar, N.; Sinhamahapatra, A.; Pahari, S.; Jayachandran, M.; Subramanian, B.; Bajaj, H.C.; Panda, A.B. Facile Low-Temperature Synthesis of Ceria and Samarium-Doped Ceria Nanoparticles and Catalytic Allylic Oxidation of Cyclohexene. J. Phys. Chem. C 2011, 115, 7628–7637. [Google Scholar] [CrossRef]
- Li, H.; Lu, G.; Dai, Q.; Wang, Y.; Guo, Y.; Guo, Y. Hierarchical Organization and Catalytic Activity of High-Surface-Area Mesoporous Ceria Microspheres Prepared Via Hydrothermal Routes. ACS Appl. Mater. Interfaces 2010, 2, 838–846. [Google Scholar] [CrossRef]
- Palma, V.; Ruocco, C.; Meloni, E.; Ricca, A. Renewable Hydrogen from Ethanol Reforming over CeO2-SiO2 Based Catalysts. Catalysts 2017, 7, 226. [Google Scholar] [CrossRef]
- Li, M.; van Veen, A.C. Tuning the catalytic performance of Ni-catalysed dry reforming of methane and carbon deposition via Ni-CeO2-x interaction. Appl. Catal. B Environ. 2018, 237, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, M.; Guan, J.; Wang, X.; Williams, C.T.; Liang, C. Nickel–Silicon Intermetallics with Enhanced Selectivity in Hydrogenation Reactions of Cinnamaldehyde and Phenylacetylene. Ind. Eng. Chem. Res. 2012, 51, 3604–3611. [Google Scholar] [CrossRef]
- Kim, M.-J.; Youn, J.-R.; Kim, H.J.; Seo, M.W.; Lee, D.; Go, K.S.; Lee, K.B.; Jeon, S.G. Effect of surface properties controlled by Ce addition on CO2 methanation over Ni/Ce/Al2O3 catalyst. Int. J. Hydrog. Energy 2020, 45, 24595–24603. [Google Scholar] [CrossRef]
- Jang, W.-J.; Kim, H.-M.; Shim, J.-O.; Yoo, S.-Y.; Jeon, K.-W.; Na, H.-S.; Lee, Y.-L.; Jeong, D.-W.; Bae, J.W.; Nah, I.W.; et al. Key properties of Ni–MgO–CeO2, Ni–MgO–ZrO2, and Ni–MgO–Ce(1−x)Zr(x)O2 catalysts for the reforming of methane with carbon dioxide. Green Chem. 2018, 20, 1621–1633. [Google Scholar] [CrossRef]
- Jang, W.-J.; Shim, J.-O.; Kim, H.-M.; Yoo, S.-Y.; Roh, H.-S. A review on dry reforming of methane in aspect of catalytic properties. Catal. Today 2019, 324, 15–26. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, J.; Zhou, Z.; Chen, S.; Zhang, T.; Song, F.; Zhang, Q.; Tsubaki, N.; Tan, Y.; Han, Y. Effects of the surface adsorbed oxygen species tuned by rare-earth metal doping on dry reforming of methane over Ni/ZrO2 catalyst. Appl. Catal. B Environ. 2020, 264, 118522. [Google Scholar] [CrossRef]
- Eissa, A.A.; Peera, S.G.; Kim, N.H.; Lee, J.H. g-C3N4 templated synthesis of the Fe3C@NSC electrocatalyst enriched with Fe–Nx active sites for efficient oxygen reduction reaction. J. Mater. Chem. A 2019, 7, 16920–16936. [Google Scholar] [CrossRef]
- Rood, S.C.; Ahmet, H.B.; Gomez-Ramon, A.; Torrente-Murciano, L.; Reina, T.R.; Eslava, S. Enhanced ceria nanoflakes using graphene oxide as a sacrificial template for CO oxidation and dry reforming of methane. Appl. Catal. B: Environ. 2019, 242, 358–368. [Google Scholar] [CrossRef]
- Marinho, A.L.A.; Rabelo-Neto, R.C.; Epron, F.; Bion, N.; Toniolo, F.S.; Noronha, F.B. Embedded Ni nanoparticles in CeZrO2 as stable catalyst for dry reforming of methane. Appl. Catal. B: Environ. 2020, 268, 118387. [Google Scholar] [CrossRef]
- Pappacena, A.; Razzaq, R.; De Leitenburg, C.; Boaro, M.; Trovarelli, A. The Role of Neodymium in the Optimization of a Ni/CeO2 and Ni/CeZrO2 Methane Dry Reforming Catalyst. Inorganics 2018, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhang, F.; Rui, N.; Li, X.; Lin, L.; Betancourt, L.E.; Su, D.; Xu, W.; Cen, J.; Attenkofer, K.; et al. Highly Active Ceria-Supported Ru Catalyst for the Dry Reforming of Methane: In Situ Identification of Ruδ+–Ce3+ Interactions for Enhanced Conversion. ACS Catal. 2019, 9, 3349–3359. [Google Scholar] [CrossRef]
- Horváth, A.; Németh, M.; Beck, A.; Maróti, B.; Sáfrán, G.; Pantaleo, G.; Liotta, L.F.; Venezia, A.M.; La Parola, V. Strong impact of indium promoter on Ni/Al2O3 and Ni/CeO2-Al2O3 catalysts used in dry reforming of methane. Appl. Catal. A Gen. 2021, 621, 118174. [Google Scholar] [CrossRef]
- Li, B.; Yuan, X.; Li, B.; Wang, X. Ceria-modified nickel supported on porous silica as highly active and stable catalyst for dry reforming of methane. Fuel 2021, 301, 121027. [Google Scholar] [CrossRef]
- da Fonseca, R.O.; Rabelo-Neto, R.C.; Simões, R.C.C.; Mattos, L.V.; Noronha, F.B. Pt supported on doped CeO2/Al2O3 as catalyst for dry reforming of methane. Int. J. Hydrogen Energy 2020, 45, 5182–5191. [Google Scholar] [CrossRef]
- Tu, P.H.; Le, D.N.; Dao, T.D.; Tran, Q.-T.; Doan, T.C.D.; Shiratori, Y.; Dang, C.M. Paper-structured catalyst containing CeO2–Ni flowers for dry reforming of methane. Int. J. Hydrogen Energy 2020, 45, 18363–18375. [Google Scholar] [CrossRef]
- Marinho, A.L.A.; Toniolo, F.S.; Noronha, F.B.; Epron, F.; Duprez, D.; Bion, N. Highly active and stable Ni dispersed on mesoporous CeO2-Al2O3 catalysts for production of syngas by dry reforming of methane. Appl. Catal. B Environ. 2021, 281, 119459. [Google Scholar] [CrossRef]
- Muñoz, M.A.; Calvino, J.J.; Rodríguez-Izquierdo, J.M.; Blanco, G.; Arias, D.C.; Pérez-Omil, J.A.; Hernández-Garrido, J.C.; González-Leal, J.M.; Cauqui, M.A.; Yeste, M.P. Highly stable ceria-zirconia-yttria supported Ni catalysts for syngas production by CO2 reforming of methane. Appl. Surf. Sci. 2017, 426, 864–873. [Google Scholar] [CrossRef]
- Padi, S.P.; Shelly, L.; Komarala, E.P.; Schweke, D.; Hayun, S.; Rosen, B.A. Coke-free methane dry reforming over nano-sized NiO-CeO2 solid solution after exsolution. Catal. Commun. 2020, 138, 105951. [Google Scholar] [CrossRef]
- Araiza, D.G.; Arcos, D.G.; Gómez-Cortés, A.; Díaz, G. Dry reforming of methane over Pt-Ni/CeO2 catalysts: Effect of the metal composition on the stability. Catal. Today 2021, 360, 46–54. [Google Scholar] [CrossRef]
- Sadeq Al-Fatesh, A.; Olajide Kasim, S.; Aidid Ibrahim, A.; Hamza Fakeeha, A.; Elhag Abasaeed, A.; Alrasheed, R.; Ashamari, R.; Bagabas, A. Combined Magnesia, Ceria and Nickel catalyst supported over γ-Alumina Doped with Titania for Dry Reforming of Methane. Catalysts 2019, 9, 188. [Google Scholar] [CrossRef] [Green Version]
- Damaskinos, C.M.; Zavašnik, J.; Djinović, P.; Efstathiou, A.M. Dry reforming of methane over Ni/Ce0.8Ti0.2O2-δ: The effect of Ni particle size on the carbon pathways studied by transient and isotopic techniques. Appl. Catal. B Environ. 2021, 296, 120321. [Google Scholar] [CrossRef]
- Luisetto, I.; Tuti, S.; Romano, C.; Boaro, M.; Di Bartolomeo, E.; Kesavan, J.K.; Kumar, S.S.; Selvakumar, K. Dry reforming of methane over Ni supported on doped CeO2: New insight on the role of dopants for CO2 activation. J. CO2 Util. 2019, 30, 63–78. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.B.; Eissa, A.A.-S.; Kim, M.-J.; Goda, E.S.; Youn, J.-R.; Lee, K. Sustainable Synthesis of a Highly Stable and Coke-Free Ni@CeO2 Catalyst for the Efficient Carbon Dioxide Reforming of Methane. Catalysts 2022, 12, 423. https://doi.org/10.3390/catal12040423
Kim SB, Eissa AA-S, Kim M-J, Goda ES, Youn J-R, Lee K. Sustainable Synthesis of a Highly Stable and Coke-Free Ni@CeO2 Catalyst for the Efficient Carbon Dioxide Reforming of Methane. Catalysts. 2022; 12(4):423. https://doi.org/10.3390/catal12040423
Chicago/Turabian StyleKim, Seung Bo, Ahmed Al-Shahat Eissa, Min-Jae Kim, Emad S. Goda, Jae-Rang Youn, and Kyubock Lee. 2022. "Sustainable Synthesis of a Highly Stable and Coke-Free Ni@CeO2 Catalyst for the Efficient Carbon Dioxide Reforming of Methane" Catalysts 12, no. 4: 423. https://doi.org/10.3390/catal12040423
APA StyleKim, S. B., Eissa, A. A. -S., Kim, M. -J., Goda, E. S., Youn, J. -R., & Lee, K. (2022). Sustainable Synthesis of a Highly Stable and Coke-Free Ni@CeO2 Catalyst for the Efficient Carbon Dioxide Reforming of Methane. Catalysts, 12(4), 423. https://doi.org/10.3390/catal12040423