
Pd Supported on Pr-Rich Cerium–Zirconium–Praseodymium Mixed Oxides for Propane and CO Oxidation

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
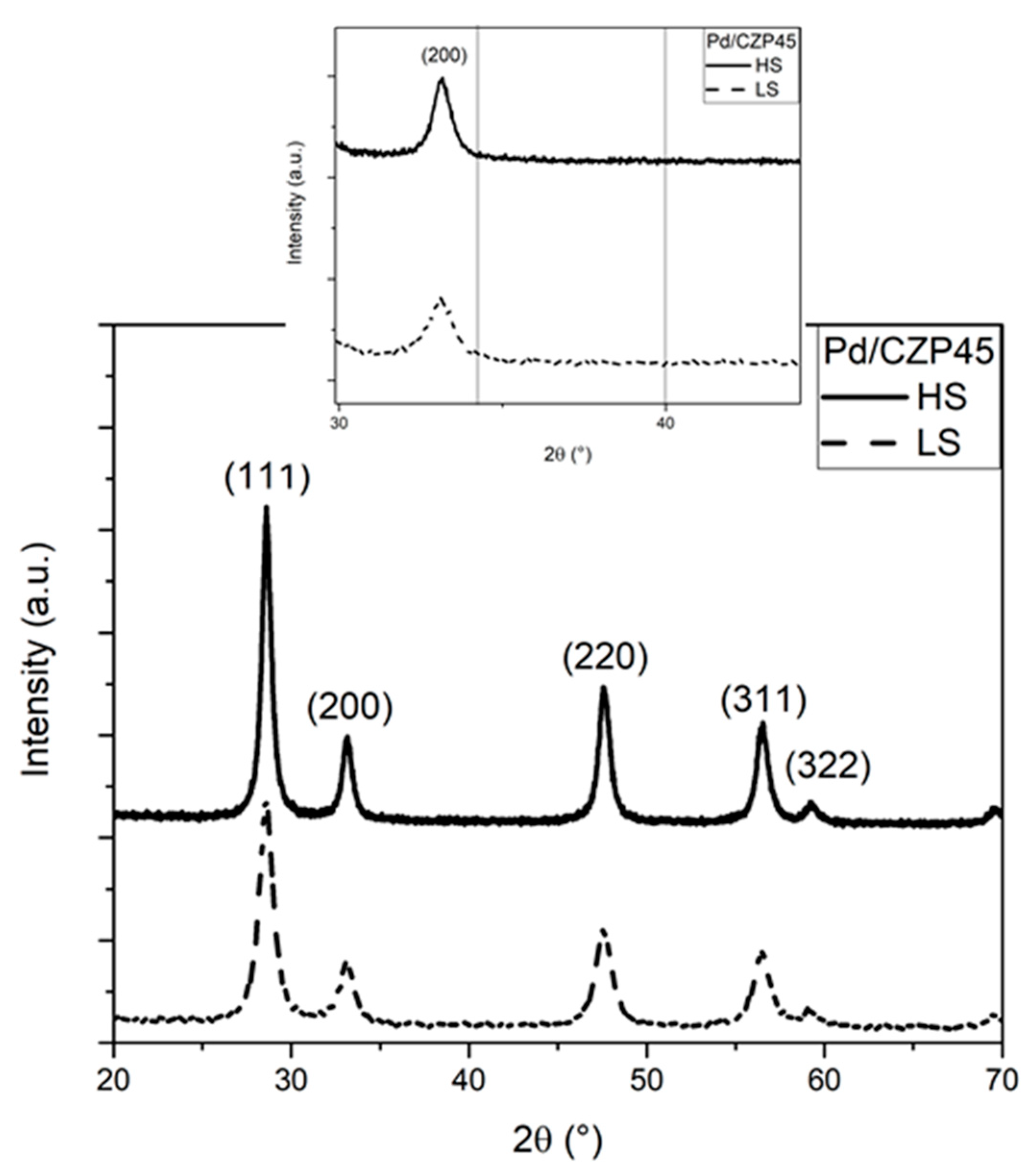
2.1. Structural and Textural Properties
2.2. Reducibility
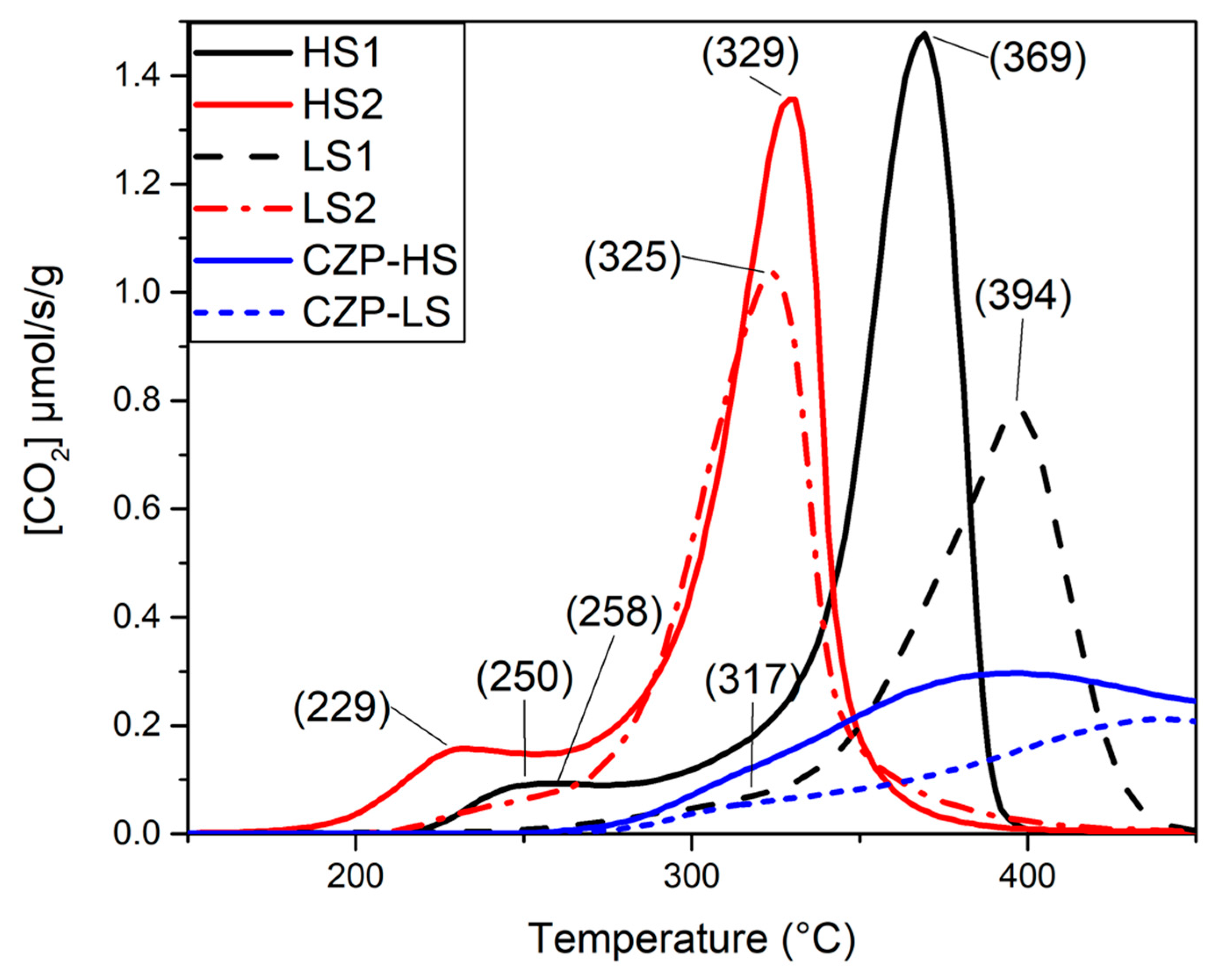
2.3. Lattice Oxygen Reactivity
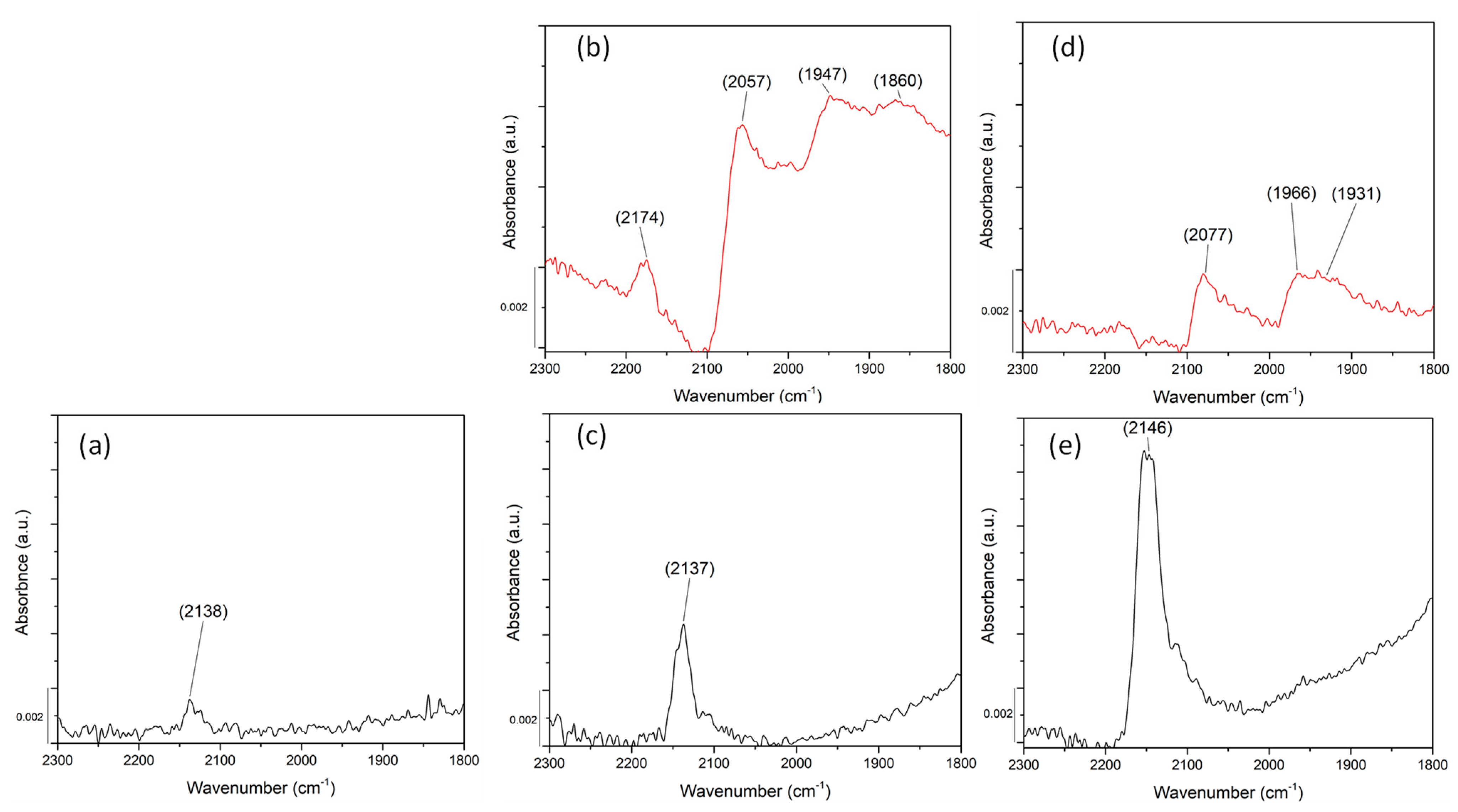
2.4. Probing Pd Sites Using CO
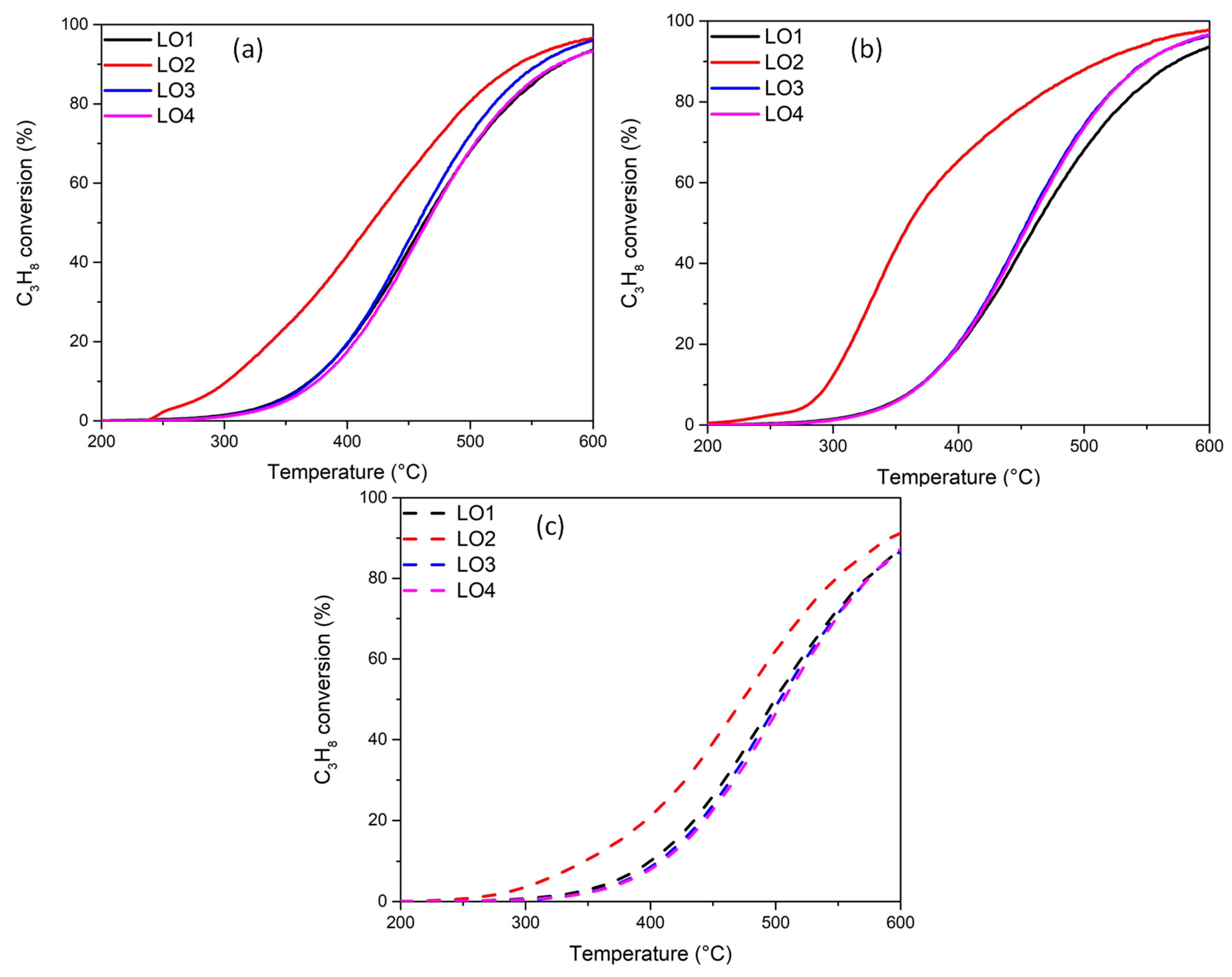
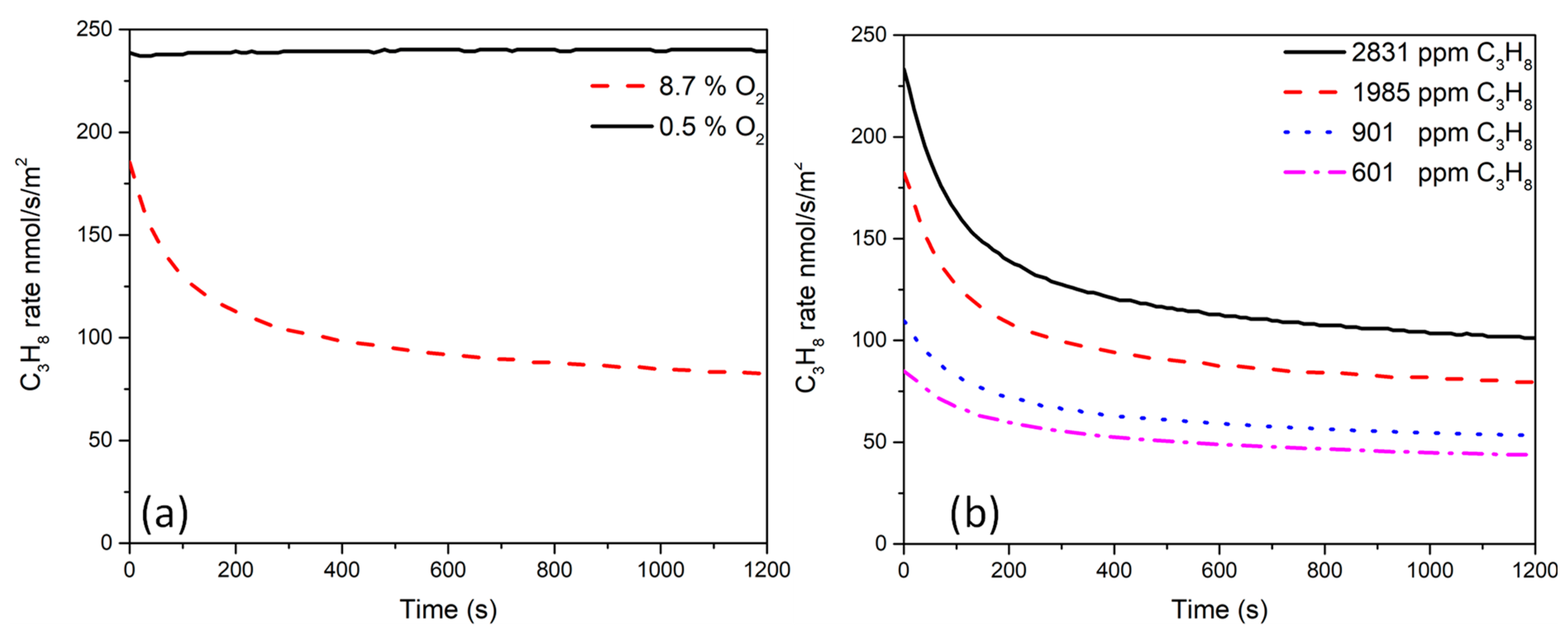
2.5. Catalytic Activity Measurements
3. Materials and Methods
3.1. Synthesis
3.2. Catalyst Characterizations
3.3. Catalytic Activity Measurement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Farrauto, R.J.; Deeba, M.; Alerasool, S. Gasoline automobile catalysis and its historical journey to cleaner air. Nat. Catal. 2019, 2, 603–613. [Google Scholar] [CrossRef]
- Au-Yeung, J.; Chen, K.; Bell, A.T.; Iglesia, E. Isotopic studies of methane oxidation pathways on PdO catalysts. J. Catal. 1999, 188, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Colussi, S.; Trovarelli, A.; Groppi, G.; Llorca, J. The effect of CeO2 on the dynamics of Pd-PdO transformation over Pd/Al2O3 combustion catalysts. Catal. Commun. 2007, 8, 1263–1266. [Google Scholar] [CrossRef]
- Lott, P.; Dolcet, P.; Casapu, M.; Grunwaldt, J.D.; Deutschmann, O. The effect of prereduction on the performance of pd/Al2O3 and Pd/CeO2 catalysts during methane oxidation. Ind. Eng. Chem. Res. 2019, 58, 12561–12570. [Google Scholar] [CrossRef]
- Wang, D.; Gong, J.; Luo, J.; Li, J.; Kamasamudram, K.; Currier, N.; Yezerets, A. Distinct reaction pathways of methane oxidation on different oxidation states over Pd-based three-way catalyst (TWC). Appl. Catal. A Gen. 2019, 572, 44–50. [Google Scholar] [CrossRef]
- Karinshak, K.A.; Lott, P.; Harold, M.P.; Deutschmann, O. In situ Activation of Bimetallic Pd−Pt Methane Oxidation Catalysts. ChemCatChem 2020, 12, 3712–3720. [Google Scholar] [CrossRef]
- Xiong, H.; Wiebenga, M.H.; Carrillo, C.; Gaudet, J.R.; Pham, H.N.; Kunwar, D.; Oh, S.H.; Qi, G.; Kim, C.H.; Datye, A.K. Design considerations for low-temperature hydrocarbon oxidation reactions on Pd based catalysts. Appl. Catal. B Environ. 2018, 236, 436–444. [Google Scholar] [CrossRef]
- Ciuparu, D.; Perkins, E.; Pfefferle, L. In situ DR-FTIR investigation of surface hydroxyls on γ-Al2O3 supported PdO catalysts during methane combustion. Appl. Catal. A Gen. 2004, 263, 145–153. [Google Scholar] [CrossRef]
- Chin, Y.H.; Buda, C.; Neurock, M.; Iglesia, E. Consequences of metal-oxide interconversion for C-H bond activation during CH4 reactions on Pd catalysts. J. Am. Chem. Soc. 2013, 135, 15425–15442. [Google Scholar] [CrossRef]
- Goodman, E.D.; Dai, S.; Yang, A.C.; Wrasman, C.J.; Gallo, A.; Bare, S.R.; Hoffman, A.S.; Jaramillo, T.F.; Graham, G.W.; Pan, X.; et al. Uniform Pt/Pd Bimetallic Nanocrystals Demonstrate Platinum Effect on Palladium Methane Combustion Activity and Stability. ACS Catal. 2017, 7, 4372–4380. [Google Scholar] [CrossRef]
- Nagai, Y.; Hirabayashi, T.; Dohmae, K.; Takagi, N.; Minami, T.; Shinjoh, H.; Matsumoto, S. Sintering inhibition mechanism of platinum supported on ceria-based oxide and Pt-oxide-support interaction. J. Catal. 2006, 242, 103–109. [Google Scholar] [CrossRef]
- Jones, J.; Xiong, H.; DeLaRiva, A.T.; Peterson, E.J.; Pham, H.; Challa, S.R.; Qi, G.; Oh, S.; Wiebenga, M.H.; Hernández, X.I.P.; et al. Thermally stable single-atom platinum-on-ceria catalysts via atom trapping. Science 2016, 353, 150–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, H.; Kunwar, D.; Jiang, D.; García-Vargas, C.E.; Li, H.; Du, C.; Canning, G.; Pereira-Hernandez, X.I.; Wan, Q.; Lin, S.; et al. Engineering catalyst supports to stabilize PdOx two-dimensional rafts for water-tolerant methane oxidation. Nat. Catal. 2021, 4, 830–839. [Google Scholar] [CrossRef]
- Reddy, K.P.; Choi, H.; Kim, D.; Choi, M.; Ryoo, R.; Park, J.Y. The facet effect of ceria nanoparticles on platinum dispersion and catalytic activity of methanol partial oxidation. Chem. Commun. 2021, 57, 7382–7385. [Google Scholar] [CrossRef]
- Yoo, M.; Yu, Y.S.; Ha, H.; Lee, S.; Choi, J.S.; Oh, S.; Kang, E.; Choi, H.; An, H.; Lee, K.S.; et al. A tailored oxide interface creates dense Pt single-atom catalysts with high catalytic activity. Energy Environ. Sci. 2020, 13, 1231–1239. [Google Scholar] [CrossRef]
- Gänzler, A.M.; Casapu, M.; Maurer, F.; Störmer, H.; Gerthsen, D.; Ferré, G.; Vernoux, P.; Bornmann, B.; Frahm, R.; Murzin, V.; et al. Tuning the Pt/CeO2 Interface by in Situ Variation of the Pt Particle Size. ACS Catal. 2018, 8, 4800–4811. [Google Scholar] [CrossRef]
- Ferré, G.; Aouine, M.; Bosselet, F.; Burel, L.; Aires, F.J.C.S.; Geantet, C.; Ntais, S.; Maurer, F.; Casapu, M.; Grunwaldt, J.; et al. Exploiting the dynamic properties of Pt on ceria for low-temperature CO oxidation. Catal. Sci. Technol. 2020, 10, 3904–3917. [Google Scholar] [CrossRef]
- Colussi, S.; Gayen, A.; Camellone, M.F.; Boaro, M.; Llorca, J.; Fabris, S.; Trovarelli, A. Nanofaceted Pd—O Sites in Pd—Ce Surface Superstructures: Enhanced Activity in Catalytic Combustion of Methane. Angew. Chem. Int. Ed. 2009, 48, 8481–8484. [Google Scholar] [CrossRef]
- Bruix, A.; Rodriguez, J.A.; Ramírez, P.J.; Senanayake, S.D.; Evans, J.; Park, J.B.; Stacchiola, D.; Liu, P.; Hrbek, J.; Illas, F. A new type of strong metal-support interaction and the production of H2 through the transformation of water on Pt/CeO2(111) and Pt/CeOx/TiO2(110) catalysts. J. Am. Chem. Soc. 2012, 134, 8968–8974. [Google Scholar] [CrossRef]
- You, R.; Li, Z.; Cao, T.; Nan, B.; Si, R.; Huang, W. Synthesis in a Glovebox: Utilizing Surface Oxygen Vacancies To Enhance the Atomic Dispersion of Palladium on Ceria for Carbon Monoxide Oxidation and Propane Combustion. ACS Appl. Nano Mater. 2018, 1, 4988–4997. [Google Scholar] [CrossRef]
- Spezzati, G.; Su, Y.; Hofmann, J.P.; Benavidez, A.D.; DeLaRiva, A.T.; McCabe, J.; Datye, A.K.; Hensen, E.J.M. Atomically dispersed Pd-O species on CeO2(111) as highly active sites for low-temperature CO oxidation. ACS Catal. 2017, 7, 6887–6891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.; Wan, G.; García-Vargas, C.E.; Li, L.; Pereira-Hernández, X.I.; Wang, C.; Wang, Y. Elucidation of the Active Sites in Single-Atom Pd1/CeO2 Catalysts for Low-Temperature CO Oxidation. ACS Catal. 2020, 10, 11356–11364. [Google Scholar] [CrossRef]
- Jeong, H.; Kwon, O.; Kim, B.S.; Bae, J.; Shin, S.; Kim, H.E.; Kim, J.; Lee, H. Highly durable metal ensemble catalysts with full dispersion for automotive applications beyond single-atom catalysts. Nat. Catal. 2020, 3, 368–375. [Google Scholar] [CrossRef]
- Baek, K.; Min, K.; Kim, K.; Ha, Y. Propane combustion over supported Pd catalysts. Res. Chem. Intermed. 2010, 36, 603–611. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, D.; Couble, J.; Aouine, M.; Massin, L.; Mascunan, P.; Díez-Ramírez, J.; Klotz, M.; Tardivat, C.; Vernoux, P. Effect of the Reduction Step on the Catalytic Performance of Pd–CeMO2 Based Catalysts (M = Gd, Zr) for Propane Combustion. Top Catal. 2016, 59, 1638–1650. [Google Scholar] [CrossRef]
- Deng, Y.; Tian, P.; Liu, S.; He, H.; Wang, Y.; Ouyang, L.; Yuan, S. Enhanced catalytic performance of atomically dispersed Pd on Pr-doped CeO2 nanorod in CO oxidation. J. Hazard. Mater. 2021, 426, 127793. [Google Scholar] [CrossRef]
- Frizon, V.; Bassat, J.M.; Pollet, M.; Durand, E.; Hernandez, J.; Pajot, K.; Vernoux, P.; Demourgues, A. Tuning the Pr Valence State to Design High Oxygen Mobility, Redox and Transport Properties in the CeO2-ZrO2-PrOx Phase Diagram. J. Phys. Chem. C 2019, 123, 6351–6362. [Google Scholar] [CrossRef] [Green Version]
- Fahed, S. Étude Des Oxydes Mixtes Cérium-Zirconium-Praséodyme Pour L’Oxydation Des Composés Imbrûlés Emis Par Les Moteurs à Essence et Diesel. Ph.D. Thesis. 2022. Available online: https://www.theses.fr/2022NORMC211 (accessed on 7 July 2022).
- Colussi, S.; Trovarelli, A.; Cristiani, C.; Lietti, L.; Groppi, G. The influence of ceria and other rare earth promoters on palladium-based methane combustion catalysts. Catal. Today 2012, 180, 124–130. [Google Scholar] [CrossRef]
- Luo, M.F.; Hou, Z.Y.; Yuan, X.X.; Zheng, X.M. Characterization study of CeO2 supported Pd catalyst for low-temperature carbon monoxide oxidation. Catal. Lett. 1998, 50, 205–209. [Google Scholar] [CrossRef]
- Hiley, C.I.; Fisher, J.M.; Thompsett, D.; Kashtiban, R.J.; Sloan, J.; Walton, R.I. Incorporation of square-planar Pd2+ in fluorite CeO2: Hydrothermal preparation, local structure, redox properties and stability. J. Mater. Chem. A 2015, 3, 13072–13079. [Google Scholar] [CrossRef] [Green Version]
- Madier, Y.; Descorme, C.; Le Govic, A.M.; Duprez, D. Oxygen mobility in CeO2 and CexZr(1−x)O2 compounds: Study by CO transient oxidation and 18O/16O isotopic exchange. J. Phys. Chem. B 1999, 103, 10999–11006. [Google Scholar] [CrossRef]
- Spezzati, G.; Benavidez, A.D.; DeLaRiva, A.T.; Su, Y.; Hofmann, J.P.; Asahina, S.; Olivier, E.J.; Neethling, J.H.; Miller, J.T.; Datye, A.K.; et al. CO oxidation by Pd supported on CeO2(100) and CeO2(111) facets. Appl. Catal. B Environ. 2019, 243, 36–46. [Google Scholar] [CrossRef]
- Muravev, V.; Spezzati, G.; Su, Y.Q.; Parastaev, A.; Chiang, F.K.; Longo, A.; Escudero, C.; Kosinov, N.; Hensen, E.J.M. Interface dynamics of Pd–CeO2 single-atom catalysts during CO oxidation. Nat. Catal. 2021, 4, 469–478. [Google Scholar] [CrossRef]
- Bazin, P.; Saur, O.; Lavalley, J.C.; Daturi, M.; Blanchard, G. FT-IR study of CO adsorption on Pt/CeO2: Characterisation and structural rearrangement of small Pt particles. Phys. Chem. Chem. Phys. 2005, 7, 187–194. [Google Scholar] [CrossRef]
- Craciun, R.; Daniell, W.; Knözinger, H. The effect of CeO2 structure on the activity of supported Pd catalysts used for methane steam reforming. Appl. Catal. A Gen. 2002, 230, 153–168. [Google Scholar] [CrossRef]
- Colinot, F.; Meunier, F.; Daturi, M. Low temperature oxidation catalysis for diesel engine emission control by FT-IR. Spectrosc. Eur. 2009, 21, 9–12. [Google Scholar]
- Bensalem, A.; Muller, J.; Tessier, D.; Bozon-verduraz, F. Spectroscopic study of CO adsorption on palladiurn-ceria catalysts. J. Chem. Soc. 1996, 92, 3233–3237. [Google Scholar]
- Binet, C.; Jadi, A.; Lavalley, J.C.; Boutonnet-Kizling, M. Metal-support interaction in Pd/CeO2 catalysts: Fourier-transform infrared studies of the effects of the reduction temperature and metal loading. Part 1.—Catalysts prepared by the microemulsion technique. J. Chem. Soc. Faraday Trans. 1992, 88, 2079–2084. [Google Scholar] [CrossRef]
- Badri, A.; Binet, C.; Lavalley, J.C. Metal-support interaction in Pd/CeO2 catalysts. Part 2.—Ceria textural effects. J. Chem. Soc. Faraday Trans. 1996, 92, 1603–1608. [Google Scholar] [CrossRef]
- Tereshchenko, A.; Polyakov, V.; Guda, A.; Lastovina, T.; Pimonova, Y.; Bulgakov, A.; Tarasov, A.; Kustov, L.; Butova, V.; Trigub, A.; et al. Ultra-Small Pd Nanoparticles on Ceria as an Advanced Catalyst for CO Oxidation. Catalysts 2019, 9, 385. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Liu, X.; Meng, D.; Guo, Y.; Guo, Y.; Lu, G. Effect of Ceria Crystal Plane on the Physicochemical and Catalytic Properties of Pd/Ceria for CO and Propane Oxidation. ACS Catal. 2016, 6, 2265–2279. [Google Scholar] [CrossRef]
- Wang, G.; Meng, M.; Zha, Y.; Ding, T. High-temperature close coupled catalysts Pd/Ce-Zr-M/Al2O3 (M = Y, Ca or Ba) used for the total oxidation of propane. Fuel 2010, 89, 2244–2251. [Google Scholar] [CrossRef]
- Li, M.; Weng, D.; Wu, X.; Wan, J.; Wang, B. Importance of re-oxidation of palladium by interaction with lanthana for propane combustion over Pd/Al2O3 catalyst. Catal. Today 2013, 201, 19–24. [Google Scholar] [CrossRef]
- Luo, J.Y.; Meng, M.; Yao, J.S.; Li, X.G.; Zha, Y.Q.; Wang, X.; Zhang, T.Y. One-step synthesis of nanostructured Pd-doped mixed oxides MOx-CeO2 (M = Mn, Fe, Co, Ni, Cu) for efficient CO and C3H8 total oxidation. Appl. Catal. B Environ. 2009, 87, 92–103. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Composition | Pd wt.% a | Pd µmol/g | SSA (m²/g) | Pore Volume (cm3/g) | Lattice Parameter (Å) | Average Crystal Size (nm) b |
|---|---|---|---|---|---|---|---|
| Pd-LS | Pd/Ce0.45Zr0.10Pr0.45O2−x | 0.17 | 16 | 6 | 0.016 | 5.409 | 11.6 ± 0.5 |
| Pd-HS | Pd/Ce0.45Zr0.10Pr0.45O2−x | 0.14 | 13 | 27 | 0.093 | 5.398 | 8.1 ± 0.4 |
| First TPR (LS1 and HS1) | Second TPR (LS2 and HS2) | ||
|---|---|---|---|
| Catalysts | H2 Uptake (µmol/g) T < 500 °C | H2 Uptake (µmol/g) T < 500 °C | Estimation of Initial Pr4+/Pr3+ |
| Pd-HS | 1509 | 1150 | 81/19 |
| Pd-LS | 1472 | 1105 | 78/22 |
| Catalysts | After Oxidation (LS1, HS1) | After a Redox Sequence (LS2, HS2) | ||||
|---|---|---|---|---|---|---|
| Low/High Temperature Peak/°C | CO2 µmol/g | Low/High Temperature Peak/°C | CO2 µmol/g | 1st Peak µmol O/m−2 | CO µmol/g | |
| Pd-HS | 250/369 | 309 | 229/328 | 312 | 2.9 | 347 |
| Pd-LS | 317/394 | 231 | 258/329 | 254 | 4.9 | 288 |
| CZP45-HS * | - | 368 | ||||
| CZP45-LS * | - | 210 | ||||
| Catalysts | Reaction Mixture | GHSV/L·g−1·h−1 | r CO/mol·g−1 Pd·s−1 × 10−5 | Ref | ||
|---|---|---|---|---|---|---|
| 50 °C | 130 °C | 200 °C | ||||
| Pd-HS | CO/O2: 1910 ppm/9 vol% | 120 | 12.5 | 156 | 196 | This study |
| Pd-LS | CO/O2: 1910 ppm/9 vol% | 120 | 1.02 | 59 | 152 | This study |
| 1% Pd/CeO2 SAC | 2 vol% CO/0.1 vol%C3H6/ 0.1 vol% C3H8 /500 ppm NO/ 1.75 vol% O2/ 5 vol% H2O | 200 | - | - | 89 | [23] |
| 1% Pd/CeO2 SAC | CO/O2: 2 vol%/8 vol% | 300 | 34 | 682 | - | [22] |
| 1% Pd/CeO2-Rod | CO/O2: 1 vol%/20 vol% | 300 | 20 | - | - | [42] |
| 0.5% Pd/Pr-CeO2 5% | CO/O2: 1 vol%/20 vol% | 72 | - | 32 | 164 | [26] |
| Catalysts | LO1 | LO2 | LO3/LO4 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| T10 | T50 | Ea kJ/mol | Reduction at 500 °C | T10 | T50 | Ea kJ/mol | T10 | T50 | Ea kJ/mol | |
| Pd-HS | 370 | 460 | 81 | 100 vol%H2 | 302 | 419 | 63 | 370 | 458 | 81 |
| 370 | 460 | 81 | 1 vol% CO | 296 | 360 | 45 | 371 | 455 | 81 | |
| Pd-LS | 400 | 500 | 85 | 100 vol%H2 | 347 | 474 | 75 | 406 | 502 | 85 |
| Catalysts | Reaction Mixture | GHSV/L·g−1·h−1 | Pre-Treatment | r C3H8/mol·g−1 Pd·s−1 × 10−5 | Ref | |
|---|---|---|---|---|---|---|
| 320 °C | 350 °C | |||||
| Pd-HS | C3H8/O2: 2000 ppm/9 vol% | 120 | CO reduction, 500 °C, 1 h | 46 | 84 | This study |
| Pd-HS | C3H8/O2: 2000 ppm/9 vol% | 120 | H2 reduction, 500 °C, 1 h | 28 | 45 | This study |
| Pd-LS | C3H8/O2: 2000 ppm/9 vol% | 120 | H2 reduction, 500 °C, 1 h | 9.7 | 16 | This study |
| 0.3% Pd/CeO2 | C3H8/O2: 2000 ppm/2 vol% | 30 | Ar, 200 °C, 0.5 h | 12 | 22 | [20] |
| 0.3% Pd/CeO2 SAC | C3H8/O2: 2000 ppm/2 vol% | 30 | Ar, 200 °C, 0.5 h | 3.8 | 9 | [20] |
| 1% Pd/CeO2-O (octahedron) | C3H8/O2: 2000 ppm/2 vol% | 300 | H2 reduction, 200 °C, 0.5 h | 10 | - | [42] |
| 1% Pd/CeO2-cube | C3H8/O2: 2000 ppm/2 vol% | 300 | H2 reduction, 200 °C, 0.5 h | 1.6 | - | [42] |
| 1% Pd/CeO2-Rod | C3H8/O2: 2000 ppm/2 vol% | 300 | H2 reduction, 200 °C, 0.5 h | 0.8 | - | [42] |
| 2% Pd/CeZr/AL2O3 | C3H8/O2: 3000 ppm/3 vol% O2 | 30 | Calcination 900 °C, 2h | 0.7 | 2.9 | [43] |
| 2% Pd/Ce-Zr-Y/AL2O3 | C3H8/O2: 3000 ppm/3 vol% | 30 | Calcination 900 °C, 2h | 0.7 | 2.9 | [43] |
| 3% Pd/Al2O3 | C3H8/O2: 1750 ppm/2 vol% O2 | 300 | N2, 500 °C, 0.5 h | 9.9 | 14 | [44] |
| 1.1% Pd/Al2O3 | C3H8/O2/H2O 500 ppm/5 vol%/ 5 vol% | 768 | H2 reduction, 250 °C, 0.5 h | 1.1 | 1.9 | [7] |
| 0.5% Pd/CeO2 | C3H8/O2: 5000 ppm/5 vol% | 15 | Calcination 500 °C, 4h | 3.4 | 5.7 | [45] |
| 1% Pd/GDC (Ce0.8Gd0.2O2) | C3H8/O2: 2000 ppm/1 vol% | 360 | H2 reduction, 500 °C, 1 h | 12.5 | 28 | [25] |
| 1% Pd/CZ (Ce0.62Zr0.38O2) | C3H8/O2: 2000 ppm/1 vol% | 360 | H2 reduction, 500 °C, 1 h | 10 | 18 | [25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fahed, S.; Pointecouteau, R.; Aouine, M.; Boreave, A.; Gil, S.; Bazin, P.; Demourgues, A.; Daturi, M.; Vernoux, P. Pd Supported on Pr-Rich Cerium–Zirconium–Praseodymium Mixed Oxides for Propane and CO Oxidation. Catalysts 2022, 12, 827. https://doi.org/10.3390/catal12080827
Fahed S, Pointecouteau R, Aouine M, Boreave A, Gil S, Bazin P, Demourgues A, Daturi M, Vernoux P. Pd Supported on Pr-Rich Cerium–Zirconium–Praseodymium Mixed Oxides for Propane and CO Oxidation. Catalysts. 2022; 12(8):827. https://doi.org/10.3390/catal12080827
Chicago/Turabian StyleFahed, Simon, Rémy Pointecouteau, Mimoun Aouine, Antoinette Boreave, Sonia Gil, Philippe Bazin, Alain Demourgues, Marco Daturi, and Philippe Vernoux. 2022. "Pd Supported on Pr-Rich Cerium–Zirconium–Praseodymium Mixed Oxides for Propane and CO Oxidation" Catalysts 12, no. 8: 827. https://doi.org/10.3390/catal12080827
APA StyleFahed, S., Pointecouteau, R., Aouine, M., Boreave, A., Gil, S., Bazin, P., Demourgues, A., Daturi, M., & Vernoux, P. (2022). Pd Supported on Pr-Rich Cerium–Zirconium–Praseodymium Mixed Oxides for Propane and CO Oxidation. Catalysts, 12(8), 827. https://doi.org/10.3390/catal12080827