
Green Chemo-Enzymatic Protocols for the Synthesis of Enantiopure β-Blockers (S)-Esmolol and (S)-Penbutolol

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of Racemic Chlorohydrin 3 (for Esmolol)
2.2. Synthesis of Racemic Chlorohydrin 8 (for Penbutolol)
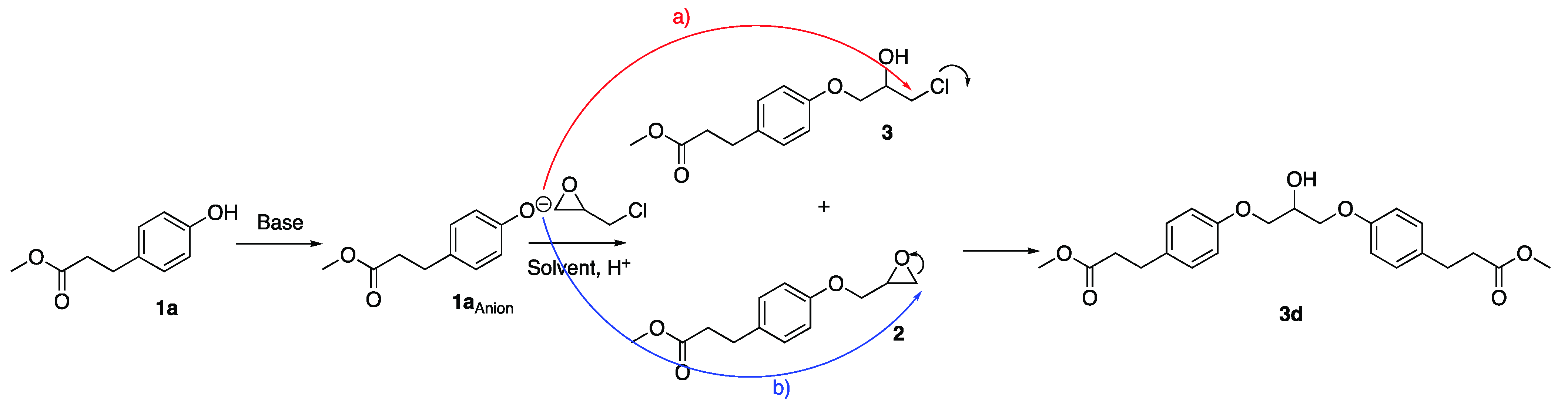
2.3. Characterisation of by-Products in Synthesis of Esmolol Precursors
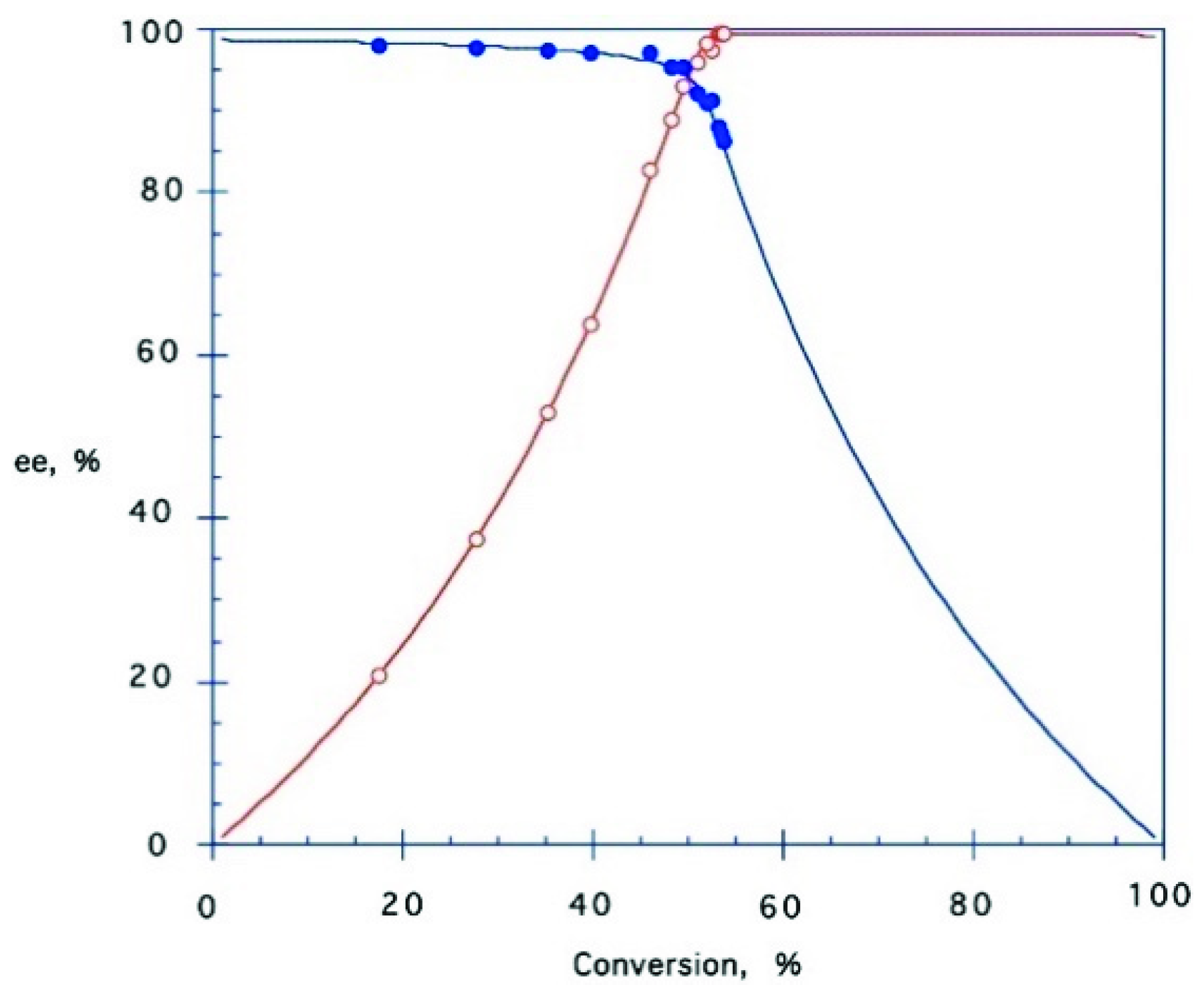
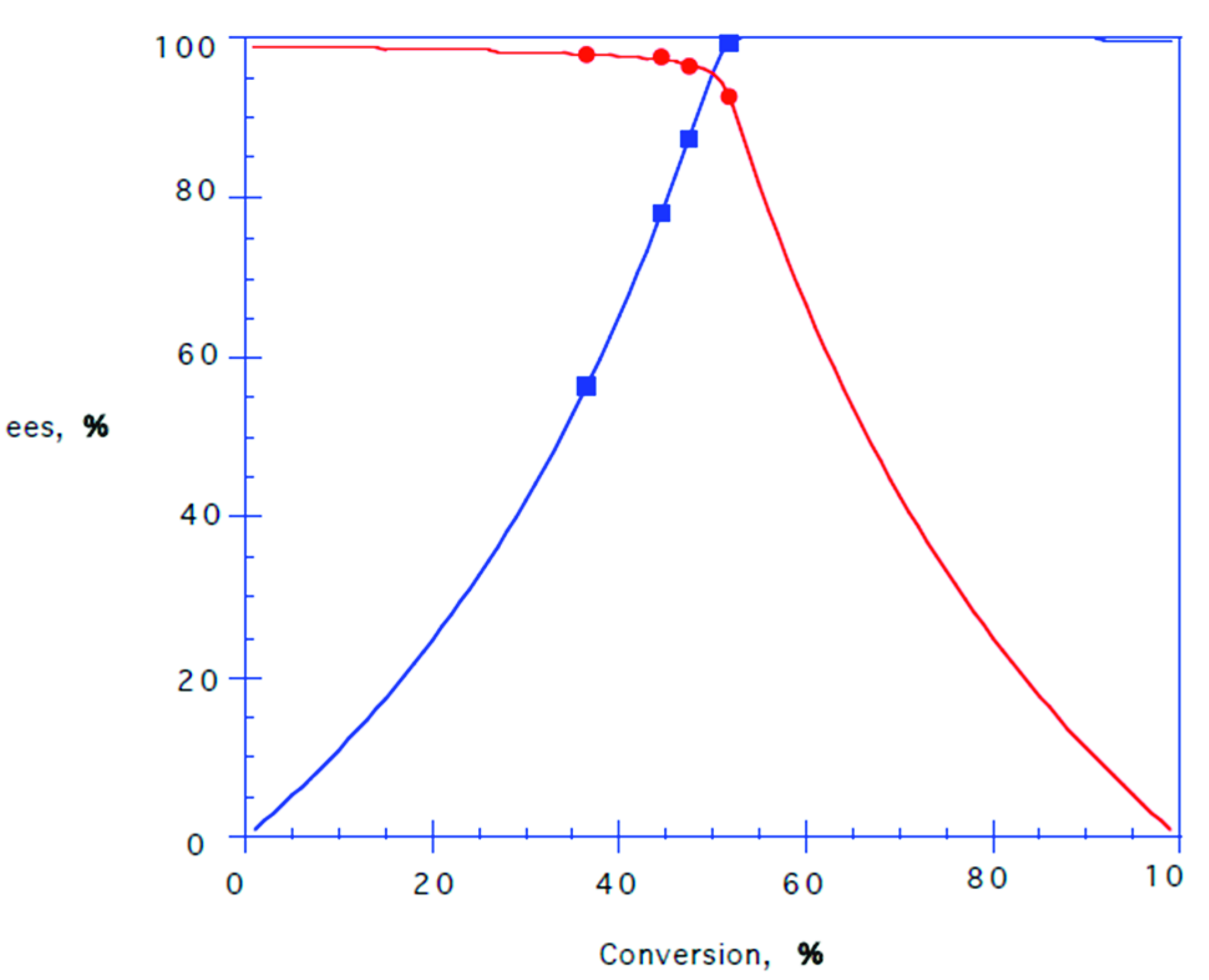
2.4. Lipase-Catalysed Kinetic Resolution of Chlorohydrins 3 and 8
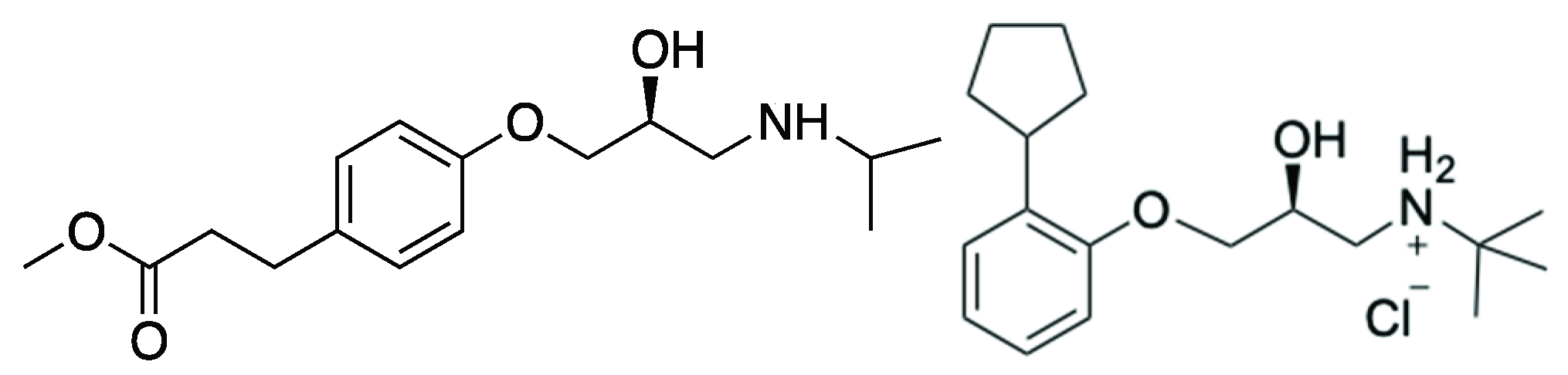
2.5. Synthesis of (S)-Esmolol ((S)-5)
2.6. Synthesis of (S)-Penbutolol ((S)-10)
2.7. Specific Rotation of Pure Enantiomers
3. Materials and Methods
3.1. Chemicals and Solvents
3.2. TLC Analyses and Column Chromatography
3.3. Enzymes
3.4. Chiral HPLC Analyses
3.5. Achiral HPLC Analysis of Dimer 3d
3.6. Liquid Chromatography-Mass Spectroscopy (LC-MS) of Esmolol Dimer 3d
3.7. Mass Spectrometry Analysis of by-Product 3d
3.8. Optical Rotation
3.9. Absolute Configurations
3.10. NMR Analyses
3.11. Synthesis Protocols
3.11.1. Methyl 3-(4-(Oxiran-2-ylmethoxy)phenyl)propanoate (2)
3.11.2. Methyl 3-(4-(3-Chloro-2-hydroxypropoxy)phenyl)propanoate (3)
3.11.3. 3,3′-(((2-Hydroxypropane-1,3-diyl)bis(oxy))bis(4,1-phenylene))dipropanoate (3d)
3.11.4. Synthesis of Chlorohydrin (R)-3 and Ester (S)-4 by CALB Catalysed Kinetic Resolution of Methyl 3-(4-(3-Chloro-2-hydroxypropoxy)phenyl)propanoate (3)
3.11.5. Esmolol (5)
3.11.6. (S)-Esmolol ((S)-5)
3.11.7. 1-Chloro-3-(2-cyclopentylphenoxy)propan-2-ol (8)
3.11.8. Synthesis of Chlorohydrin (R)-8 by CALB-Catalysed Kinetic Resolution of 1-Chloro-3-(2-cyclopentylphenoxy)propan-2-ol (8)
3.11.9. (S)-Penbutolol ((S)-10)
3.11.10. (S)-Penbutolol Hydrochloride ((S)-10∙HCl)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lund, I.T.; Bøckmann, P.L.; Jacobsen, E.E. Highly enantioselective CALB-catalyzed kinetic resolution of building blocks for β-blocker atenolol. Tetrahedron 2016, 72, 7288–7292. [Google Scholar] [CrossRef]
- Blindheim, F.H.; Hansen, M.B.; Evjen, S.; Zhu, W.; Jacobsen, E.E. Chemoenzymatic Synthesis of Synthons as Precursors for Enantiopure Clenbuterol and Other β2-Agonists. Catalysts 2018, 8, 516. [Google Scholar] [CrossRef]
- Gundersen, M.A.; Austli, G.B.; Løvland, S.S.; Hansen, M.B.; Rødseth, M.; Jacobsen, E.E. Lipase Catalyzed Synthesis of Enantiopure Precursors and Derivatives for β-Blockers Practolol, Pindolol and Carteolol. Catalysts 2021, 11, 503. [Google Scholar] [CrossRef]
- Verho, O.; Bäckvall, J.-E. Chemoenzymatic Dynamic Kinetic Resolution: A Powerful Tool for the Preparation of Enantiomerically Pure Alcohols and Amines. J. Am. Chem. Soc. 2015, 137, 3996–4009. [Google Scholar] [CrossRef] [PubMed]
- Maisel, W.H.; Friedman, P.L. Esmolol and Other Intravenous Beta-Blockers. Card. Electrophysiol. Rev. 2000, 4, 240–242. [Google Scholar] [CrossRef]
- Esmolol. Available online: https://www.legemiddelhandboka.no (accessed on 1 August 2022).
- Banoth, L.; Banerjee, U.C. New chemical and chemo-enzymatic synthesis of (RS)-, (R)-, and (S)-esmolol. Arab. J. Chem. 2017, 10, S3603–S3613. [Google Scholar] [CrossRef]
- Narsaiah, A.V.; Kumar, J.K. Novel Asymmetric Synthesis of (S)-Esmolol Using Hydrolytic Kinetic Resolution. Synth. Commun. 2011, 41, 1603–1608. [Google Scholar] [CrossRef]
- Byrne, F.P.; Jin, S.; Paggiola, G.; Petchey, T.H.M.; Clark, J.H.; Farmer, T.J.; Hunt, A.J.; McElroy, C.R.; Sherwood, J. Tools and techniques for solvent selection: Green solvent selection guides. Sustain. Chem. Process. 2016, 4, 7. [Google Scholar] [CrossRef]
- Hamaguchi, S.; Asada, M.; Hasegawa, J.; Watanabe, K. Asymmetric hydrolysis of racemic 2-oxazolidinone esters with lipases. Agric. Biol. Chem. 1984, 48, 2331–2337. [Google Scholar] [CrossRef]
- Ader, U.; Schneider, M.P. Enzyme assisted preparation of enantiomerically pure β-adrenergic blockers III. Optically active chlorohydrin derivatives and their conversion. Tetrahedron Asymmetry 1992, 3, 521–524. [Google Scholar] [CrossRef]
- Phukan, P.; Sudalai, A. Regioselective alkylation of phenol with cyclopentanol over montmorillonite k10: An efficient synthesis of 1-(2-cyclopentylphenoxy)-3-[(1,1-dimethylethyl) amino] propan-2-ol {(S)-penbutolol}. J. Chem. Soc. Perkin Trans. 1999, 20, 3015–3018. [Google Scholar] [CrossRef]
- Klunder, J.M.; Onami, T.; Sharpless, K.B. Arenesulfonate derivatives of homochiral glycidol: Versatile chiral building blocks for organic synthesis. J. Org. Chem. 1989, 54, 1295–1304. [Google Scholar] [CrossRef]
- Kan, K.; Miyama, A.; Hamaguchi, S.; Ohashi, T.; Watanabe, K. Synthesis of (S)-β-Blockers from (S)-5-Hydroxymethyl-3-tert butyl-2-oxazolidinone or (S)-5-Hydroxymethyl-3-isopropyl-2-oxazolidinone. Agric. Biol. Chem. 1985, 49, 207–210. [Google Scholar] [CrossRef]
- Bevinakatti, H.S.; Banerji, A.A. Lipase Catalysis in Organic Solvents. Application to the Synthesis of (R)- and (S)-Atenolol. J. Org. Chem. 1992, 57, 6003–6005. [Google Scholar] [CrossRef]
- Anthonsen, H.W.; Hoff, B.H.; Anthonsen, T. Calculation of enantiomer ratio and equilibrium constants in biocatalytic ping-pong bi-bi resolutions. Tetrahedron Asymmetry 1996, 7, 2633–2638. [Google Scholar] [CrossRef]
- Jacobsen, E.E.; Hoff, B.H.; Anthonsen, T. Enantiopure derivatives of 1,2-alkanediols: Substrate requirements of lipase B from Candida antarctica. Chirality 2000, 12, 654–659. [Google Scholar] [CrossRef]
- Moen, A.R.; Hoff, B.H.; Hansen, L.K.; Anthonsen, T.; Jacobsen, E.E. Absolute configurations of monoesters produced by enz-yme catalyzed hydrolysis of diethyl 3-hydroxyglutarate. Tetrahedron Asymmetry 2004, 15, 1551–1554. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chloro-Hydrin | E-Value | Chlorohydrin ee, Yield, % | Specific Rotation | Ester, ee, Yield, % | Specific Rotation | Drug, ee, Yield, % | Specific Rotation |
|---|---|---|---|---|---|---|---|
| (R)-3 | 157 | (R)-3a, 97, 43 | (c 1.6, i-PrOH) | (S)-4, 87, 41 | (c 1.4, i-PrOH) | (S)-5, 97, 92 | (c 1.03, CHCl3) |
| (R)-8 | 183 | (R)-8, 99, 39 | = −14.00 (c 1.6, MeOH), | (S)-10∙HCl, 99, 89 | (c 1.0, MeOH), | ||
| (S)-10, 99, 82 | (c 1.0, MeOH), |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Troøyen, S.H.; Bocquin, L.; Tennfjord, A.L.; Klungseth, K.; Jacobsen, E.E. Green Chemo-Enzymatic Protocols for the Synthesis of Enantiopure β-Blockers (S)-Esmolol and (S)-Penbutolol. Catalysts 2022, 12, 980. https://doi.org/10.3390/catal12090980
Troøyen SH, Bocquin L, Tennfjord AL, Klungseth K, Jacobsen EE. Green Chemo-Enzymatic Protocols for the Synthesis of Enantiopure β-Blockers (S)-Esmolol and (S)-Penbutolol. Catalysts. 2022; 12(9):980. https://doi.org/10.3390/catal12090980
Chicago/Turabian StyleTroøyen, Susanne Hansen, Lucas Bocquin, Anna Lifen Tennfjord, Kristoffer Klungseth, and Elisabeth Egholm Jacobsen. 2022. "Green Chemo-Enzymatic Protocols for the Synthesis of Enantiopure β-Blockers (S)-Esmolol and (S)-Penbutolol" Catalysts 12, no. 9: 980. https://doi.org/10.3390/catal12090980
APA StyleTroøyen, S. H., Bocquin, L., Tennfjord, A. L., Klungseth, K., & Jacobsen, E. E. (2022). Green Chemo-Enzymatic Protocols for the Synthesis of Enantiopure β-Blockers (S)-Esmolol and (S)-Penbutolol. Catalysts, 12(9), 980. https://doi.org/10.3390/catal12090980