
Characterization of Ni-Phases and Their Transformations in Fluid Catalytic Cracking (FCC) Catalysts: Comparison of Conventional Versus Boron-Based Ni-Passivation

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Catalyst Samples
2.2. Catalyst Characterization
3. Results and Discussion
3.1. Chemical Composition and Content of Nickel
3.2. Qualitative and Quantitative Determination of NiO by PXRD
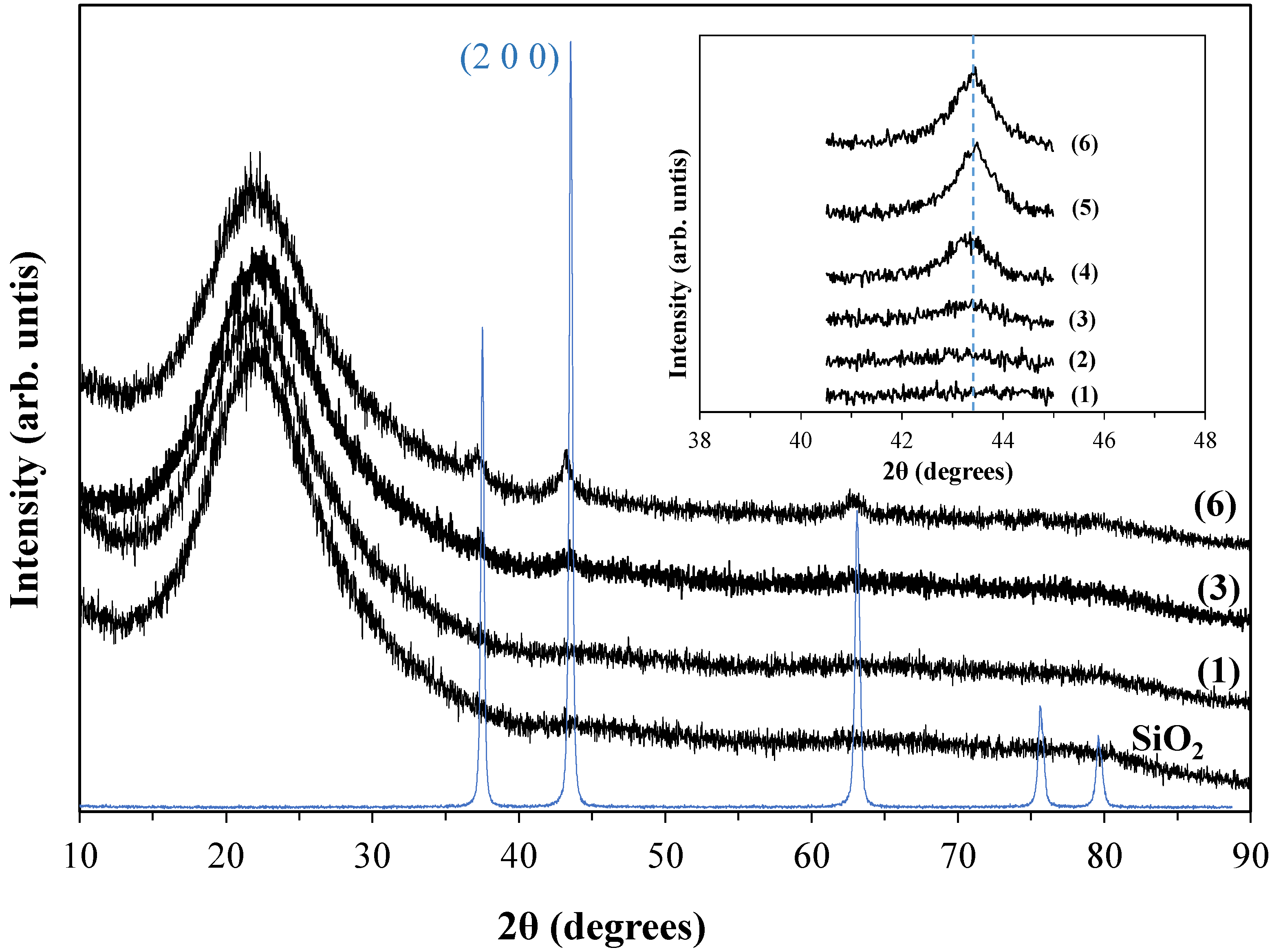
3.2.1. Ni/SiO2 Samples
3.2.2. Ni/Al2O3 and Ni/USY Samples
3.2.3. Ni-Loaded FCC and Ecat Samples
3.3. Fourier-Transform Infrared Spectroscopy (FTIR)
3.4. Diffuse Reflectance UV-Vis/Near IR
3.5. Raman Spectroscopy
3.6. X-Ray Photoelectron Spectroscopy (XPS)
3.7. Scanning Electron Microscopy—Energy Dispersive Spectroscopy (SEM-EDS)
- SEM images and point EDS microanalysis
- EDS elemental mapping of flat cross-sections
3.8. Transmission Electron Microscopy (TEM)—High resolution TEM (HRTEM)
3.9. Temperature Programmed Reduction with H2 (TPR-H2)
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vogt, E.T.C.; Weckhuysen, B.M. Fluid catalytic cracking: Recent developments on the grand old lady of zeolite catalysis. Chem. Soc. Rev. 2015, 44, 7342–7370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gholami, Z.; Gholami, F.; Tišler, Z.; Tomas, M.; Vakili, M. A Review on Production of Light Olefins via Fluid Catalytic Cracking. Energies 2021, 14, 1089. [Google Scholar] [CrossRef]
- Bai, P.; Etim, U.J.; Yan, Z.; Mintova, S.; Zhang, Z.; Zhong, Z.; Gao, X. Fluid catalytic cracking technology: Current status and recent discoveries on catalyst contamination. Catal. Rev. 2019, 61, 333–405. [Google Scholar] [CrossRef]
- Etim, U.J.; Wu, P.; Bai, P.; Xing, W.; Ullah, R.; Subhan, F.; Yan, Z. Location and Surface Species of Fluid Catalytic Cracking Catalyst Contaminants: Implications for Alleviating Catalyst Deactivation. Energy Fuels 2016, 30, 10371–10382. [Google Scholar] [CrossRef]
- Busca, G.; Riani, P.; Garbarino, G.; Ziemacki, G.; Gambino, L.; Montanari, E.; Millini, R. The state of nickel in spent Fluid Catalytic Cracking catalysts. Appl. Catal. A Gen. 2014, 486, 176–186. [Google Scholar] [CrossRef]
- Clough, M.; Pope, J.C.; Lin, L.T.X.; Komvokis, V.; Pan, S.S.; Yilmaz, B. Nanoporous materials forge a path forward to enable sustainable growth: Technology advancements in fluid catalytic cracking. Microporous Mesoporous Mater. 2017, 254, 45–58. [Google Scholar] [CrossRef]
- Corma, A.; Grande, M.S.; Iglesias, M.; del Pino, C.; Rojas, R.M. Nickel passivation on fluidised cracking catalysts with different antimony complexes. Appl. Catal. A Gen. 1992, 85, 61–71. [Google Scholar] [CrossRef]
- Petti, T.F.; Tomczak, D.; Pereira, C.J.; Cheng, W.-C. Investigation of nickel species on commercial FCC equilibrium catalysts-implications on catalyst performance and laboratory evaluation. Appl. Catal. A Gen. 1998, 169, 95–109. [Google Scholar] [CrossRef]
- Fu, H.; Chen, Y.; Liu, T.; Zhu, X.; Yang, Y.; Song, H. Research on Hazardous Waste Removal Management: Identification of the Hazardous Characteristics of Fluid Catalytic Cracking Spent Catalysts. Molecules 2021, 26, 2289. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Wang, C.; Li, L.-L.; Chen, Y.; He, C.-H.; Zheng, L. Assessment of ecotoxicity of spent fluid catalytic cracking (FCC) refinery catalysts on Raphidocelis subcapitata and predictive models for toxicity. Ecotoxicol. Environ. Saf. 2021, 222, 112466. [Google Scholar] [CrossRef]
- Zhang, D.; Fang, S.; Zhang, H.; Liu, Z.; Zhang, Z.; Zhang, S. Utilization of Spent FCC Catalyst as Fine Aggregate in Non-sintered Brick: Alkali Activation and Environmental Risk Assessment. Front. Chem. 2021, 9, 240. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, A.; Emami, S.M.; Nemat, S. Reuse of spent FCC catalyst, waste serpentine and kiln rollers waste for synthesis of cordierite and cordierite-mullite ceramics. J. Hazard. Mater. 2017, 338, 177–185. [Google Scholar] [CrossRef]
- Ferella, F.; D’Adamo, I.; Leone, S.; Innocenzi, V.; de Michelis, I.; Vegliò, F. Spent FCC E-Cat: Towards a Circular Approach in the Oil Refining Industry. Sustainability 2019, 11, 113. [Google Scholar] [CrossRef] [Green Version]
- Komvokis, V.; Keeley, C.; Challis, S. Bottoms up! Part One. Hydrocarb. Eng. 2013, 18, 64–66, 68, 70. [Google Scholar]
- Pan, S.; Shackleford, A.; McGuire, R., Jr.; Smith, G.; Yilmaz, B. Creative Catalysis. Hydrocarb. Eng. 2015, 20, 46–52. [Google Scholar]
- Bare, S.R.; Modica, F.S.; Ringwelski, A.Z. In situ Ni K-edge XANES study of the reducibility of Ni in f.c.c. catalysts. J. Synchrotron Radiat. 1999, 6, 436–438. [Google Scholar] [CrossRef]
- Stöcker, M.; Tangstad, E.; Aas, N.; Myrstad, T. Quantitative determination of Ni and V in FCC catalysts monitored by ESR spectroscopy. Catal. Lett. 2000, 69, 223–229. [Google Scholar] [CrossRef]
- Mitchell, B.R. Metal Contamination of Cracking Catalysts. 1. Synthetic Metals Deposition on Fresh Catalysts. Ind. Eng. Chem. Prod. Res. Dev. 1980, 19, 209–213. [Google Scholar] [CrossRef]
- Kallithrakas-Kontos, N.G.; Xarchoulakos, D.C.; Boultadaki, P.; Potiriadis, C.; Kehagia, K. Selective Membrane Complexation and Uranium Isotopes Analysis in Tap Water and Seawater Samples. Anal. Chem. 2018, 90, 4611–4615. [Google Scholar] [CrossRef]
- McCarthy, T.J.; Ngeyi, S.P.; Liao, J.H.; DeGroot, D.C.; Hogan, T.; Kannewurf, C.R.; Kanatzidis, M.G. Molten salt synthesis and properties of three new solid-state ternary bismuth chalcogenides, .beta.-CsBiS2, .gamma.-CsBiS2, and K2Bi8Se13. Chem. Mater. 1993, 5, 331–340. [Google Scholar] [CrossRef]
- Wallenstein, D.; Harding, R.H.; Nee, J.R.D.; Boock, L.T. Recent advances in the deactivation of FCC catalysts by cyclic propylene steaming (CPS) in the presence and absence of contaminant metals. Appl. Catal. A Gen. 2000, 204, 89–106. [Google Scholar] [CrossRef]
- Wallenstein, D.; Roberie, T.; Bruhin, T. Review on the deactivation of FCC catalysts by cyclic propylene steaming. Catal. Today 2007, 127, 54–69. [Google Scholar] [CrossRef]
- Bayraktar, O.; Kugler, E.L. Temperature-programmed reduction of metal-contaminated fluid catalytic cracking (FCC) catalysts. Appl. Catal. A Gen. 2004, 260, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Psarras, A.C.; Iliopoulou, E.F.; Nalbandian, L.; Lappas, A.A.; Pouwels, C. Study of the accessibility effect on the irreversible deactivation of FCC catalysts from contaminant feed metals. Catal. Today 2007, 127, 44–53. [Google Scholar] [CrossRef]
- Vincz, C.; Rath, R.; Smith, G.M.; Yilmaz, B.; McGuire, R. Dendritic nickel porphyrin for mimicking deposition of contaminant nickel on FCC catalysts. Appl. Catal. A Gen. 2015, 495, 39–44. [Google Scholar] [CrossRef]
- Ferella, F.; Innocenzi, V.; Maggiore, F. Oil refining spent catalysts: A review of possible recycling technologies. Resour. Conserv. Recycl. 2016, 108, 10–20. [Google Scholar] [CrossRef]
- Zhang, C.C.; Shi, J.; Hartlaub, S.; Palamara, J.P.; Petrovic, I.; Yilmaz, B. In-situ diffuse reflective infrared Fourier transform spectroscopy (DRIFTS) study on Ni passivation in FCC catalysts from boron-based technology. Catal. Commun. 2021, 150, 106273. [Google Scholar] [CrossRef]
- Heracleous, E.; Lee, A.F.; Wilson, K.; Lemonidou, A.A. Investigation of Ni-based alumina-supported catalysts for the oxidative dehydrogenation of ethane to ethylene: Structural characterization and reactivity studies. J. Catal. 2005, 231, 159–171. [Google Scholar] [CrossRef]
- Samain, L.; Jaworski, A.; Edén, M.; Ladd, D.M.; Seo, D.-K.; Garcia-Garcia, F.J.; Häussermann, U. Structural analysis of highly porous γ-Al2O3. J. Solid State Chem. 2014, 217, 1–8. [Google Scholar] [CrossRef]
- Ragupathi, C.; Vijaya, J.J.; Surendhar, P.; Kennedy, L.J. Comparative investigation of nickel aluminate (NiAl2O4) nano and microstructures for the structural, optical and catalytic properties. Polyhedron 2014, 72, 1–7. [Google Scholar] [CrossRef]
- Garbarino, G.; Riani, P.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Busca, G. On the detectability limits of nickel species on NiO/γ-Al2O3 catalytic materials. Appl. Catal. A Gen. 2016, 525, 180–189. [Google Scholar] [CrossRef]
- Komvokis, V.G.; Karakoulia, S.; Iliopoulou, E.F.; Papapetrou, M.C.; Vasalos, I.A.; Lappas, A.A.; Triantafyllidis, K.S. Upgrading of Fischer–Tropsch synthesis bio-waxes via catalytic cracking: Effect of acidity, porosity and metal modification of zeolitic and mesoporous aluminosilicate catalysts. Catal. Today 2012, 196, 42–55. [Google Scholar] [CrossRef]
- Pu, X.; Liu, N.-w.; Shi, L. Acid properties and catalysis of USY zeolite with different extra-framework aluminum concentration. Microporous Mesoporous Mater. 2015, 201, 17–23. [Google Scholar] [CrossRef]
- Nishimura, Y. Development of Catalytic Cracking Process and Catalysts. Adv. Porous Mater. 2017, 5, 17–25. [Google Scholar] [CrossRef]
- Scherzer, J. Octane-Enhancing, Zeolitic FCC Catalysts: Scientific and Technical Aspects. Catal. Rev. 1989, 31, 215–354. [Google Scholar] [CrossRef]
- Blay, V.; Louis, B.; Miravalles, R.; Yokoi, T.; Peccatiello, K.A.; Clough, M.; Yilmaz, B. Engineering Zeolites for Catalytic Cracking to Light Olefins. ACS Catal. 2017, 7, 6542–6566. [Google Scholar] [CrossRef]
- Farshi, A.; Abri, H.R. The Addition of ZSM-5 to a Fluid Catalytic Cracking Catalyst for Increasing Olefins in Fluid Catalytic Cracking Light Gas. Pet. Sci. Technol. 2012, 30, 1285–1295. [Google Scholar] [CrossRef]
- Costa, T.M.H.; Gallas, M.R.; Benvenutti, E.V.; da Jornada, J.A.H. Study of Nanocrystalline γ-Al2O3 Produced by High-Pressure Compaction. J. Phys. Chem. B 1999, 103, 4278–4284. [Google Scholar] [CrossRef]
- Darmakkolla, S.R.; Tran, H.; Gupta, A.; Rananavare, S.B. A method to derivatize surface silanol groups to Si-alkyl groups in carbon-doped silicon oxides. RSC Adv. 2016, 6, 93219–93230. [Google Scholar] [CrossRef]
- El-Kemary, M.; Nagy, N.; El-Mehasseb, I. Nickel oxide nanoparticles: Synthesis and spectral studies of interactions with glucose. Mater. Sci. Semicond. Process. 2013, 16, 1747–1752. [Google Scholar] [CrossRef]
- Li, J.; Ren, Y.; Yue, B.; He, H. Ni/Al2O3 catalysts derived from spinel NiAl2O4 for low-temperature hydrogenation of maleic anhydride to succinic anhydride. Chin. J. Catal. 2017, 38, 1166–1173. [Google Scholar] [CrossRef]
- Domingo, A.; Rodríguez-Fortea, A.; Swart, M.; de Graaf, C.; Broer, R. Ab initio absorption spectrum of NiO combining molecular dynamics with the embedded cluster approach in a discrete reaction field. Phys. Rev. B 2012, 85, 155143. [Google Scholar] [CrossRef]
- Huang, W.; Ding, S.; Chen, Y.; Hao, W.; Lai, X.; Peng, J.; Tu, J.; Cao, Y.; Li, X. 3D NiO hollow sphere/reduced graphene oxide composite for high-performance glucose biosensor. Sci. Rep. 2017, 7, 5220. [Google Scholar] [PubMed] [Green Version]
- Mrabet, C.; Amor, M.B.; Boukhachem, A.; Amlouk, M.; Manoubi, T. Physical properties of La-doped NiO sprayed thin films for optoelectronic and sensor applications. Ceram. Int. 2016, 42, 5963–5978. [Google Scholar] [CrossRef]
- Wang, W.; Liu, Y.; Xu, C.; Zheng, C.; Wang, G. Synthesis of NiO nanorods by a novel simple precursor thermal decomposition approach. Chem. Phys. Lett. 2002, 362, 119–122. [Google Scholar]
- Choi, H.C.; Jung, Y.M.; Kim, S.B. Size effects in the Raman spectra of TiO2 nanoparticles. Vib. Spectrosc. 2005, 37, 33–38. [Google Scholar] [CrossRef]
- Laguna-Bercero, M.A.; Sanjuán, M.L.; Merino, R.I. Raman spectroscopic study of cation disorder in poly- and single crystals of the nickel aluminate spinel. J. Physics. Condens. Matter 2007, 19, 186217. [Google Scholar] [CrossRef]
- Lubas, M.; Jasinski, J.J.; Sitarz, M.; Kurpaska, L.; Podsiad, P.; Jasinski, J. Raman spectroscopy of TiO2 thin films formed by hybrid treatment for biomedical applications. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 133, 867–871. [Google Scholar] [CrossRef]
- Jiménez-González, C.; Boukha, Z.; de Rivas, B.; Delgado, J.J.; Cauqui, M.Á.; González-Velasco, J.R.; Gutiérrez-Ortiz, J.I.; López-Fonseca, R. Structural characterisation of Ni/alumina reforming catalysts activated at high temperatures. Appl. Catal. A Gen. 2013, 466, 9–20. [Google Scholar] [CrossRef]
- Kitakatsu, N.; Maurice, V.; Hinnen, C.; Marcus, P. Surface hydroxylation and local structure of NiO thin films formed on Ni(111). Surf. Sci. 1998, 407, 36–58. [Google Scholar] [CrossRef]
- Natile, M.M.; Glisenti, A. Surface Reactivity of NiO: Interaction with Methanol. Chem. Mater. 2002, 14, 4895–4903. [Google Scholar] [CrossRef]
- Riman, D.; Spyrou, K.; Karantzalis, A.E.; Hrbac, J.; Prodromidis, M.I. Glucose sensing on graphite screen-printed electrode modified by sparking of copper nickel alloys. Talanta 2017, 165, 466–473. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, N.S.; Cook, M.G. X-ray photoelectron studies on some oxides and hydroxides of cobalt, nickel, and copper. Anal. Chem. 1975, 47, 2208–2213. [Google Scholar] [CrossRef]
- Kugler, E.L.; Leta, D.P. Nickel and vanadium on equilibrium cracking catalysts by imaging secondary ion mass spectrometry. J. Catal. 1988, 109, 387–395. [Google Scholar] [CrossRef]
- Lappas, A.A.; Nalbandian, L.; Iatridis, D.K.; Voutetakis, S.S.; Vasalos, I.A. Effect of metals poisoning on FCC products yields: Studies in an FCC short contact time pilot plant unit. Catal. Today 2001, 65, 233–240. [Google Scholar] [CrossRef]
- Pan, S.S.; Lin, L.T.X.; Komvokis, V.; Spann, A.; Clough, M.; Yilmaz, B. Nanomaterials Fueling the World. In Nanomaterials for Sustainable Energy; American Chemical Society: Washington, DC, USA, 2015; pp. 3–18. [Google Scholar]
- Cheng, W.C.; Juskelis, M.V.; Sua´rez, W. Reducibility of metals on fluid cracking catalyst. Appl. Catal. A Gen. 1993, 103, 87–103. [Google Scholar] [CrossRef]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Ni (e) (ppm) | V (ppm) | Fe (wt.%) | Na (wt.%) | REO (wt.%) | Al2O3 (wt.%) |
|---|---|---|---|---|---|---|
| SiO2-Ni-X (a) X = 1000, 2500, 5000, 10,000, 15,000, 20,000 | 934, 2129, 4485, 10,990, 17,030, 16,510 | - | - | - | - | - |
| SiO2-Ni-oc-X (b) X = 500, 1000, 2500, 5000, 10,000, 15,000, 20,000 | X = 887, 2190, 5561, 10,960, 17,100, 24,100 | - | - | - | - | - |
| SiO2-CH-Ni-oc-X (c) X = 1000, 2500, 5000, 10,000, 15,000, 20,000 | X = 893, 2437, 5626, 12,100 19,700, 24,830 | - | - | - | - | - |
| Al2O3-CH-Ni-oc-X (c) X = 1000, 2500, 5000, 10,000, 15,000, 20,000, 100,000, 200,000 | Nominal values | - | - | - | - | rest |
| USY-CH-Ni-oc-X (c) X = 1000, 2500, 5000, 10,000, 15,000, 20,000, 50,000 | Nominal values | - | - | 0.03 | - | 11.2 |
| BLF-CAT-X (d) X = 1000, 2500, 5000, 10,000 | Nominal values 10,000/9547 | traces (f) | 0.26 | 0.17 | 2.2 | 44 |
| BLB-CAT-X (d) X = 1000, 2500, 5000, 10,000 | Nominal values 10,000/10,510 | traces (f) | 0.26 | 0.17 | 2.2 | 44 |
| Ecat-2 (g) (B-CAT-2M) | 4327 | 6153 | 0.73 | 0.26 | 2.1 | 40.2 |
| Ecat-3 (B-CAT-3M) | 3553 | 4761 | 0.62 | 0.40 | 4.6 | 53.6 |
| Ecat-4 (B-CAT-4M) | 2546 | 5009 | 0.89 | 0.26 | 2.7 | 41.0 |
| Ecat-5 (B-CAT-5M) | 5141 | 1949 | 0.89 | 0.46 | 3.0 | 44.3 |
| Ecat-6 (B-CAT-6M) | 1297 | 2608 | 0.89 | 0.28 | 1.9 | 39.6 |
| Sample | Total Ni Content (ppm) | Ni * (ppm) | Ni as NiO (ppm) |
|---|---|---|---|
| BLF-CAT-1 | 1000 | - | - |
| BLF-CAT-2 | 2500 | 217 | 276 |
| BLF-CAT-3 | 5000 | 243 | 310 |
| BLF-CAT-4 | 10,000 | 1391 | 1770 |
| BLB-CAT-1 | 1000 | - | - |
| BLB-CAT-2 | 2500 | - | - |
| BLB-CAT-3 | 5000 | 230 | 293 |
| BLB-CAT-4 | 10,000 | 722 | 919 |
| B-CAT-2M | 4327 | 359 | 456 |
| B-CAT-3M | 3553 | - | - |
| B-CAT-4M | 2546 | 108 | 137 |
| B-CAT-5M | 5141 | 150 | 191 |
| B-CAT-6M | 1297 | 348 | 442 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charisteidis, I.D.; Trikalitis, P.N.; Triantafyllidis, K.S.; Komvokis, V.; Yilmaz, B. Characterization of Ni-Phases and Their Transformations in Fluid Catalytic Cracking (FCC) Catalysts: Comparison of Conventional Versus Boron-Based Ni-Passivation. Catalysts 2023, 13, 3. https://doi.org/10.3390/catal13010003
Charisteidis ID, Trikalitis PN, Triantafyllidis KS, Komvokis V, Yilmaz B. Characterization of Ni-Phases and Their Transformations in Fluid Catalytic Cracking (FCC) Catalysts: Comparison of Conventional Versus Boron-Based Ni-Passivation. Catalysts. 2023; 13(1):3. https://doi.org/10.3390/catal13010003
Chicago/Turabian StyleCharisteidis, Ioannis D., Pantelis N. Trikalitis, Konstantinos S. Triantafyllidis, Vasileios Komvokis, and Bilge Yilmaz. 2023. "Characterization of Ni-Phases and Their Transformations in Fluid Catalytic Cracking (FCC) Catalysts: Comparison of Conventional Versus Boron-Based Ni-Passivation" Catalysts 13, no. 1: 3. https://doi.org/10.3390/catal13010003
APA StyleCharisteidis, I. D., Trikalitis, P. N., Triantafyllidis, K. S., Komvokis, V., & Yilmaz, B. (2023). Characterization of Ni-Phases and Their Transformations in Fluid Catalytic Cracking (FCC) Catalysts: Comparison of Conventional Versus Boron-Based Ni-Passivation. Catalysts, 13(1), 3. https://doi.org/10.3390/catal13010003