
A Bibliometric Analysis on Pulsed Electrolysis: Electronic Effect, Double Layer Effect, and Mass Transport



Abstract
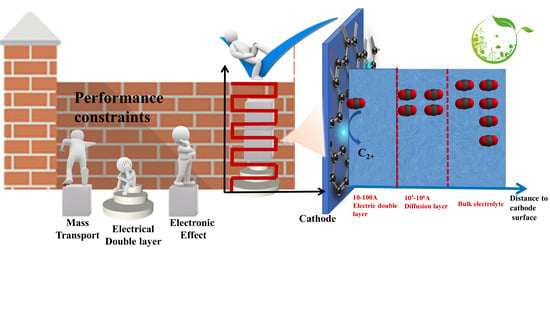
:
1. Introduction
2. Historical Analysis of Pulsed Electrolysis via a Bibliometric Method
- (1)
- Early stage of development: 1985–1995
- (2)
- Exploration and development period: 1996–2015
- (3)
- Diversification period from 2016 to 2023
- (a)
- A shorter pulse to tune product selectivity is used, especially for electrochemical CO2 reduction to covert CO2 into the high-value C2+ product [74]. To our knowledge, the first study on sub-second pulses was reported by Kumar et al. in 2016, who confirmed millisecond (square-wave) pulses boosted the selectivity of CO2R on Cu electrodes [75]. The influence of adsorbed species on the electrode could be one reason to improve selectivity when pulsed electrolysis was used [75]. Additionally, HER inhibition could be another factor in boosting selectivity during pulsed CO2RR electrolysis. Ding et al. [76] provided a new perspective on the mechanism of pulsed electrocatalysis to promote H2O2 production via 2e−-ORR;
- (b)
- In the meantime, with the advancement of various technologies as the mature of various technologies, researchers used DFT calculations, XPS, and ATR-SEIRAS to study the changes in the electrode surface and electrolyte during pulsed electrolysis. For example, Zhang et al. [77] proved that the low-frequency asymmetric pulse strategy could modulate the Cu oxidation state using ex situ spectroscopy (XPS, AES and XANES) and, in situ XANES coupled with in situ ATR-FTIR/Raman and DFT calculations.
- (c)
- Various visualization methods have been developed to study mass transfer processes in pulsed electrolysis, such as differential electrochemical mass spectroscopy (DEMS) [78], a micromachined electrochemical cell (MEC) combined with a laser scanning confocal microscope (LSCM)-fluorescence coupled detection system (MEC-LSCM) [79].
3. Definition of Pulsed Electrolysis and Its Key Parameters
4. How Pulsed Electrolysis Regulates Electrochemical Performance?
4.1. EDL Effect
4.1.1. Effect of Pulse Parameters on the EDL
- (1)
- toff < td: The bilayer is not fully discharged, leading to residual power, and EDL takes less time to fully recharge in the next pulse. After several cycles, bilayer capacitance saturates, causing alternating cycles of complete charging and incomplete discharging. It’s important to note that the faradaic current becomes zero when the faradaic potential difference falls below the reaction potential.
- (2)
- toff > td: In this case, the double layer is fully discharged, but it has not achieved complete charge, then the faradaic current with time will repeat the upward and downward trend. Consequently, the high-efficiency current from the double layer capacitor is not fully utilized, preventing the faradaic current from reaching its peak.
4.1.2. Analysis of the Effect of Applied Pulses from the EDL Perspective
4.2. Electronic Effect

4.3. Mass Transport Effect

5. In Situ Characterization Methods Used in the Pulsed Electrolysis
- (1)
- In situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS)
- (2)
- In situ X-ray absorption near-edge spectroscopy (XANES) measurements
- (3)
- X-ray absorption spectroscopy (XAS), high-energy X-ray diffraction (XRD), and quasi-in situ X-ray photoelectron spectroscopy (XPS).
- (4)
- Selected-Ion Flow Tube Mass Spectrometry (SIFT-MS)

6. Application of the Pulsed Electrolysis
7. Conclusions and Perspectives
- (1)
- Temporal and Spatial Substance Concentration: It becomes clear that obtaining precise temporal and spatial distribution data of substance concentrations in close vicinity to the electrode surface during both ’ton’ and ’toff’ phases is significant. At present, the primary focus is on simulation techniques. Bridging the gap between simulation and real-world electrolysis conditions is now a top priority. In addition, further exploration of mass transfer phenomena within the microenvironment of the electrode is necessary.
- (2)
- Adsorption Species, Reaction Path, and Product Selectivity: There is a noticeable gap in our knowledge concerning adsorption species coverage, reaction pathways, and product selectivity on the electrode surface during pulsed electrolysis. Creating micro-dynamic models is crucial in clarifying these processes, as it plays a pivotal role in unraveling the mechanisms involved in pulsed electrolysis.
- (3)
- Double Layer: At present, there is a scarcity of studies that examining the microenvironment and theoretical characterization of the double layer. Understanding the factors that influence changes in the double electric layer under pulsed conditions remains an unexplored frontier.
- (4)
- In Situ Detection Techniques: To achieve a comprehensive understanding of the changes occurring at the electrode-solution interface during pulsed electrolysis, the development of more in situ detection techniques is essential. The utilization of non-in situ characterization methods in many current studies hinders our ability to gather real-time information during pulse electrolysis.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Deng, F.; Jiang, J.; Sirés, I. State-of-the-art review and bibliometric analysis on electro-Fenton process. Carbon Lett. 2023, 33, 17–34. [Google Scholar] [CrossRef]
- Qu, J.; Li, Z.; Bi, F.; Zhang, X.; Zhang, B.; Li, K.; Wang, S.; Sun, M.; Ma, J.; Zhang, Y. A multiple Kirkendall strategy for converting nanosized zero-valent iron to highly active Fenton-like catalyst for organics degradation. Proc. Natl. Acad. Sci. USA 2023, 120, e2304552120. [Google Scholar] [CrossRef]
- Bai, S.; Chen, J.; Guo, M.; Ren, N.; Zhao, X. Vertical-scale spatial influence of radial oxygen loss on rhizosphere microbial community in constructed wetland. Environ. Int. 2023, 171, 107690. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Yang, G.; Meng, X.; Zhang, T.; Li, C.; Bai, S.; Zhao, X. Illuminating plant–microbe interaction: How photoperiod affects rhizosphere and pollutant removal in constructed wetland? Environ. Int. 2023, 179, 108144. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tian, H.; Ma, L.; Wang, Y.; Liu, X.; Gao, X. Low-temperature water electrolysis: Fundamentals, progress, and new strategies. Mater. Adv. 2022, 3, 5598–5644. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Sequeira, C.A.C.; Figueiredo, J.L. Hydrogen production by alkaline water electrolysis. Quim. Nova 2013, 36, 1176–1193. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, B. Recent advances in electrochemical hydrogen production from water assisted by alternative oxidation reactions. Chemelectrochem 2019, 6, 3214–3226. [Google Scholar] [CrossRef]
- Ursua, A.; Gandia, L.M.; Sanchis, P. Hydrogen production from water electrolysis: Current status and future trends. Proc. IEEE 2012, 100, 410–426. [Google Scholar] [CrossRef]
- Zhang, Q.; Tong, Y.; Wang, Z.; Jing, B.; Zhu, Y.; Qiu, S.; Cui, C.; Deng, F. Improved alkaline water electrolysis system for green energy: Sulfonamide antibiotic-assisted anodic oxidation integrated with hydrogen generation. J. Mater. Chem. A 2023, 11, 6129–6143. [Google Scholar] [CrossRef]
- Küngas, R. Review—Electrochemical CO2 reduction for CO Production: Comparison of low-and high-temperature electrolysis technologies. J. Electrochem. Soc. 2020, 167, 44508. [Google Scholar] [CrossRef]
- Jones, J.; Prakash, G.K.S.; Olah, G.A. Electrochemical CO2 reduction: Recent advances and current trends. Isr. J. Chem. 2014, 54, 1451–1466. [Google Scholar] [CrossRef]
- Hiragond, C.B.; Kim, H.; Lee, J.; Sorcar, S.; Erkey, C.; In, S. Electrochemical CO2 Reduction to CO catalyzed by 2D nanostructures. Catalysts 2020, 10, 98. [Google Scholar] [CrossRef]
- Song, J.; Cho, S. Catalytic materials for efficient electrochemical production of hydrogen peroxide. APL Mater. 2020, 8, 050701. [Google Scholar] [CrossRef]
- Qu, C.; Liang, D. Novel electrochemical advanced oxidation processes with H2O2 generation cathode for water treatment: A review. J. Environ. Chem. Eng. 2022, 10, 107896. [Google Scholar] [CrossRef]
- Crispim, A.C.; Da Silva Mendonça De Paiva, S.; de Araújo, D.M.; Souza, F.L.; Dos Santos, E.V. Ultrasound and UV technologies for wastewater treatment using boron-doped diamond anodes. Curr. Opin. Electrochem. 2022, 33, 100935. [Google Scholar] [CrossRef]
- Fan, R.; Li, C.; Liu, H.; Wang, C. Advanced electro-catalysis oxidation process with bdd film electrode for nonbiodegradable organic wastewater treatment. In Proceedings of the International Conference on Energy and Environment Technology (ICEET 2009), Guilin, China, 16–18 October 2009. [Google Scholar]
- Deng, F.; Brillas, E. Advances in the decontamination of wastewaters with synthetic organic dyes by electrochemical Fenton-based processes. Sep. Purif. Technol. 2023, 316, 123764. [Google Scholar] [CrossRef]
- Su, F.; Yao, K. Facile Fabrication of superhydrophobic surface with excellent mechanical abrasion and corrosion resistance on copper substrate by a novel method. ACS Appl. Mater. Interfaces 2014, 6, 8762–8770. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xue, J.; Luo, D.; Wang, H.; Gong, X.; Han, Z.; Ren, L. One-step fabrication of biomimetic superhydrophobic surface by electrodeposition on magnesium alloy and its corrosion inhibition. J. Colloid Interface Sci. 2017, 491, 313–320. [Google Scholar] [CrossRef]
- Tan, J.; Hao, J.; An, Z.; Liu, C. Simple fabrication of superhydrophobic nickel surface on steel substrate via electrodeposition. Int. J. Electrochem. Sci. 2017, 12, 40–49. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, H.; Zhang, F.; Yuan, J.; Dong, D.; Zhang, L.; Du, L. Electrochemical treatment of malachite green dye wastewater by pulse three-dimensional electrode method. Environ. Technol. 2022. [Google Scholar] [CrossRef]
- Ni, Q.; Kirk, D.W.; Thorpe, S.J. Pulse electrolysis in the electro-oxidation on the Ti/SnO2-Sb2O5 anode for wastewater treatment. Ecs Trans. 2010, 28, 33. [Google Scholar] [CrossRef]
- Son, M.; Cho, K.H.; Jeong, K.; Park, J. Membrane and electrochemical processes for water desalination: A short perspective and the role of nanotechnology. Membranes 2020, 10, 280. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Jing, B.; Zhang, Q.; Xie, J.; Qiu, S.; Deng, F. Carbon spheres modified titanium air diffusion cathode for boosting H2O2 and application in disinfection. J. Environ. Chem. Eng. 2023, 11, 110012. [Google Scholar] [CrossRef]
- An, J.; Li, N.; Zhao, Q.; Qiao, Y.; Wang, S.; Liao, C.; Zhou, L.; Li, T.; Wang, X.; Feng, Y. Highly efficient electro-generation of H2O2 by adjusting liquid-gas-solid three phase interfaces of porous carbonaceous cathode during oxygen reduction reaction. Water Res. 2019, 164, 114933. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Li, S.; Cao, Y.; Fang, M.A.; Qu, J.; Chen, Z.; Qiu, S. A dual-cathode pulsed current electro-Fenton system: Improvement for H2O2 accumulation and Fe3+ reduction. J. Power Sources 2020, 466, 228342. [Google Scholar] [CrossRef]
- Deng, F.; Olvera-Vargas, H.; Zhou, M.; Qiu, S.; Sirés, I.; Brillas, E. Critical review on the mechanisms of Fe2+ regeneration in the electro-Fenton process: Fundamentals and boosting strategies. Chem. Rev. 2023, 123, 4635–4662. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Jiang, Y.; Hu, Y.; Men, Y.; Liu, Y.; Cai, W.; Chen, S. Hydrogen bond network connectivity in the electric double layer dominates the kinetic pH effect in hydrogen electrocatalysis on Pt. Nat. Catal. 2022, 5, 900–911. [Google Scholar] [CrossRef]
- Ding, Y.; Zhou, W.; Li, J.; Wang, J.; Xie, L.; Meng, X.; Gao, J.; Sun, F.; Zhao, G.; Qin, Y. Revealing the in situ dynamic regulation of the interfacial microenvironment induced by pulsed electrocatalysis in the oxygen reduction reaction. ACS Energy Lett. 2023, 8, 3122–3130. [Google Scholar] [CrossRef]
- Herradon, C.; Le, L.; Meisel, C.; Huang, J.; Chmura, C.; Kim, Y.D.; Cadigan, C.O.; Hayre, R.; Sullivan, N.P. Proton-conducting ceramics for water electrolysis and hydrogen production at elevated pressure. Front. Energy Res. 2022, 10, 1020960. [Google Scholar] [CrossRef]
- Dufek, E.J.; Lister, T.E.; Stone, S.G.; Mcilwain, M.E. Operation of a pressurized system for continuous reduction of CO2. J. Electrochem. Soc. 2012, 159, F514–F517. [Google Scholar] [CrossRef]
- Chak, S.K.; Venkateswara Rao, P. The drilling of Al2O3 using a pulsed DC supply with a rotary abrasive electrode by the electrochemical discharge process. Int. J. Adv. Manuf. Technol. 2008, 39, 633–641. [Google Scholar] [CrossRef]
- Qiu, S.; Tang, W.; Yang, S.; Xie, J.; Yu, D.; Garcia-Rodriguez, O.; Qu, J.; Bai, S.; Deng, F. A microbubble-assisted rotary tubular titanium cathode for boosting Fenton’s reagents in the electro-Fenton process. J. Hazard. Mater. 2022, 424, 127403. [Google Scholar] [CrossRef]
- Birdja, Y.Y.; Pérez-Gallent, E.; Figueiredo, M.C.; Göttle, A.J.; Calle-Vallejo, F.; Koper, M.T.M. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 2019, 4, 732–745. [Google Scholar] [CrossRef]
- Butler, J.A.V.; Armstrong, G.; Kendall, J.P. The kinetics of electrode processes. Part II.―Reversible reduction and oxidation processes. In Proceedings of the Royal Society of London. Series A, Containing Papers of a Mathematical and Physical Character; Royal Society: London, UK, 1997; Volume 139, pp. 406–416. [Google Scholar]
- Kimura, K.W.; Fritz, K.E.; Kim, J.; Suntivich, J.; Abruna, H.D.; Hanrath, T. Controlled selectivity of CO2 reduction on copper by pulsing the electrochemical potential. Chemsuschem 2018, 11, 1781–1786. [Google Scholar] [CrossRef]
- Rocha, F.; de Radiguès, Q.; Thunis, G.; Proost, J. Pulsed water electrolysis: A review. Electrochim. Acta 2021, 377, 138052. [Google Scholar] [CrossRef]
- Bui, J.C.; Kim, C.; Weber, A.Z.; Bell, A.T. Dynamic boundary layer simulation of pulsed CO2 electrolysis on a copper catalyst. Acs Energy Lett. 2021, 6, 1181–1188. [Google Scholar] [CrossRef]
- Liu, T.; Wang, J.; Yang, X.; Gong, M. A review of pulse electrolysis for efficient energy conversion and chemical production. J. Energy Chem. 2021, 59, 69–82. [Google Scholar] [CrossRef]
- Ghoroghchian, J.; Bockris, J.O. Use of a homopolar generator in hydrogen production from water. Int. J. Hydrog. Energy 1985, 10, 101–112. [Google Scholar] [CrossRef]
- Martins, V.; Janis, K.; Gunars, B. Water electrolysis with inductive voltage pulses. In Electrolysis; Vladimir, L., Janis, K., Eds.; IntechOpen: Rijeka, Croatia, 2012; p. 2. [Google Scholar]
- Arán-Ais, R.M.; Scholten, F.; Kunze, S.; Rizo, R.; Roldan Cuenya, B. The role of in situ generated morphological motifs and Cu(i) species in C2+ product selectivity during CO2 pulsed electroreduction. Nat. Energy 2020, 5, 317–325. [Google Scholar] [CrossRef]
- Shiratsuchi, R.; Aikoh, Y.; Nogami, G. Pulsed electroreduction of CO2 on copper electrodes. J. Electrochem. Soc. 1993, 140, 3479. [Google Scholar] [CrossRef]
- Nouri-Nigjeh, E.; Permentier, H.P.; Bischoff, R.; Bruins, A.P. Electrochemical oxidation by square-wave potential pulses in the imitation of oxidative drug metabolism. Anal. Chem. 2011, 83, 5519–5525. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Gao, J.; Ding, Y.; Zhao, H.; Meng, X.; Wang, Y.; Kou, K.; Xu, Y.; Wu, S.; Qin, Y. Drastic enhancement of H2O2 electro-generation by pulsed current for ibuprofen degradation: Strategy based on decoupling study on H2O2 decomposition pathways. Chem. Eng. J. 2018, 338, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Vanags, M.; Kleperis, J.; Bajars, G.; Nemcevs, V. Electrodeposition of nanoporous nickel layers using inductive voltage pulses. In Proceedings of the IOP Conference Series: Materials Science and Engineering; Iop Publishing Ltd.: Briatol, UK, 2013; Volume 49, p. 12008. [Google Scholar]
- Tseung, A.C.C.; Vassie, P.R. A study of gas evolution in teflon bonded porous electrodes—III. Performance of teflon bonded Pt black electrodes for H2 evolution. Electrochim. Acta 1976, 21, 315–318. [Google Scholar] [CrossRef]
- Viswanathan, K.; Cheh, H.Y.; Standart, G.L. Electrolysis by intermittent potential. J. Appl. Electrochem. 1980, 10, 37–41. [Google Scholar] [CrossRef]
- Ibl, N. Some theoretical aspects of pulse electrolysis. Surf. Technol. 1980, 10, 81–104. [Google Scholar] [CrossRef]
- Andricacos, P.C.; Cheh, H.Y. The application of linear sweep voltammetry to a rotating disk electrode for a reversible reaction with soluble product. J. Electrochem. Soc. 1980, 127, 2385. [Google Scholar] [CrossRef]
- Viswanathan, K.; Cheh, H.Y. The application of pulsed potential and pulsed current to a rotating disc electrode system. J. Appl. Electrochem. 1979, 9, 537–543. [Google Scholar] [CrossRef]
- Ibl, N.; Puippe, J.C.; Angerer, H. Electrocrystallization in pulse electrolysis. Surf. Technol. 1978, 6, 287–300. [Google Scholar] [CrossRef]
- Bockris, J.O.; Dandapani, B.; Cocke, D.; Ghoroghchian, J. On the splitting of water. Int. J. Hydrog. Energy 1985, 10, 179–201. [Google Scholar] [CrossRef]
- Dewulf, D.W.; Jin, T.; Bard, A.J. Electrochemical and surface studies of carbon dioxide reduction to methane and ethylene at copper electrodes in aqueous solutions. J. Electrochem. Soc. 1989, 136, 1686. [Google Scholar] [CrossRef]
- Wasmus, S.; Cattaneo, E.; Vielstich, W. Reduction of carbon dioxide to methane and ethene—An on-line MS study with rotating electrodes. Electrochim. Acta 1990, 35, 771–775. [Google Scholar] [CrossRef]
- Shiratsuchi, R.; Nogami, G. Pulsed electroreduction of CO2 on silver electrodes. J. Electrochem. Soc. 1996, 143, 582. [Google Scholar] [CrossRef]
- David, M.; Ocampo-Martínez, C.; Sánchez-Peña, R. Advances in alkaline water electrolyzers: A review. J. Energy Storage 2019, 23, 392–403. [Google Scholar] [CrossRef]
- Brandon, N.P.; Kelsall, G.H. Growth kinetics of bubbles electrogenerated at microelectrodes. J. Appl. Electrochem. 1985, 15, 475–484. [Google Scholar] [CrossRef]
- Khosla, N.K.; Venkatachalam, S.; Somasundaran, P. Pulsed electrogeneration of bubbles for electroflotation. J. Appl. Electrochem. 1991, 21, 986–990. [Google Scholar] [CrossRef]
- Polatides, C.; Dortsiou, M.; Kyriacou, G. Electrochemical removal of nitrate ion from aqueous solution by pulsing potential electrolysis. Electrochim. Acta 2005, 50, 5237–5241. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Grover, A.K.; Dey, G.K.; Totlani, M.K. Nanocrystalline Ni–Cu alloy plating by pulse electrolysis. Surf. Coat. Technol. 2000, 126, 48–63. [Google Scholar] [CrossRef]
- Maeda, H.; Fukumoto, H.; Mitsuda, N. Reduction of CO concentration through CO electrooxidation for modified gas fuel PEFC. Electrochemistry 2002, 70, 615–621. [Google Scholar] [CrossRef]
- Ishimaru, S.; Shiratsuchi, R.; Nogami, G. Pulsed electroreduction of CO2 on Cu-Ag alloy electrodes. J. Electrochem. Soc. 2000, 147, 1864. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Liu, Q.; Li, X.; Jiang, F. The effects of electro-deposition current parameters on performance of tungsten coating. Int. J. Refract. Met. Hard Mater. 2012, 35, 241–245. [Google Scholar] [CrossRef]
- Mao, X.; Hong, S.; Zhu, H.; Lin, H.; Wei, L.; Gan, F. Alternating pulse current in electrocoagulation for wastewater treatment to prevent the passivation of al electrode. J. Wuhan Univ. Technol. Mater. Sci. Ed. 2008, 23, 239–241. [Google Scholar] [CrossRef]
- Gao, J.; Jin, H.; Zhang, J.; Shi, J.; Li, L. Influence of depositing parameters on microstructure of Ni-Co pulse plating. In Proceedings of the International Conference on Green Power, Materials and Manufacturing Technology and Applications (GPMMTA 2011), Chongqing, China, 15–18 July 2011; Volume 84–85, pp. 86–90. [Google Scholar]
- Yano, J.; Morita, T.; Shimano, K.; Nagami, Y.; Yamasaki, S. Selective ethylene formation by pulse-mode electrochemical reduction of carbon dioxide using copper and copper-oxide electrodes. J. Solid State Electrochem. 2007, 11, 554–557. [Google Scholar] [CrossRef]
- Huang, C. Solar hydrogen production via pulse electrolysis of aqueous ammonium sulfite solution. Sol. Energy 2013, 91, 394–401. [Google Scholar] [CrossRef]
- Karastoyanov, V.I.; Tzvetkoff, T.B. Pulse electrolysis of alkaline solutions as highly efficient method of production of hydrogen/oxygen gas mixtures. Bulg. Chem. Commun. 2013, 45, 99–102. [Google Scholar]
- Yu, M.; Li, H.; Wang, Y. Effects of process parameters on the morphologies and composition of pulse electrodeposition of nickel-rich Co-Ni alloys. In Progress in Materials and Processes, PTS 1–3; Shi, Z., Dong, J.H., Ma, W., Eds.; Trans Tech Publications Ltd.: Zurich, Switzerland, 2013; Volume 602–604, pp. 565–569. [Google Scholar]
- Dikusar, A.I.; Globa, P.G.; Belevskii, S.S.; Sidel Nikova, S.P. On limiting rate of dimensional electrodeposition at meso- and nanomaterial manufacturing by template synthesis. Surf. Eng. Appl. Electrochem. 2009, 45, 171–179. [Google Scholar] [CrossRef]
- Jadhav, H.S.; Kalubarme, R.S.; Ahn, S.; Yun, J.H.; Park, C. Effects of duty cycle on properties of CIGS thin films fabricated by pulse-reverse electrodeposition technique. Appl. Surf. Sci. 2013, 268, 391–396. [Google Scholar] [CrossRef]
- Tamagawa, Y.; Yatsuo, Y.; Horikawa, H.; Iwasaki, M. Fine patterning of titanium oxide film loaded with hydroxyapatite using photopatterning and anodic oxidation. Mater. Trans. 2010, 51, 2225–2229. [Google Scholar] [CrossRef]
- Didomenico, R.C.; Hanrath, T. Pulse symmetry impacts the C2 product selectivity in pulsed electrochemical CO2 reduction. ACS Energy Lett. 2022, 7, 292–299. [Google Scholar] [CrossRef]
- Kumar, B.; Brian, J.P.; Atla, V.; Kumari, S.; Bertram, K.A.; White, R.T.; Spurgeon, J.M. Controlling the product syngas H2:CO ratio through pulsed-bias electrochemical reduction of CO2 on copper. ACS Catal. 2016, 6, 4739–4745. [Google Scholar] [CrossRef]
- Ding, Y.; Zhou, W.; Xie, L.; Chen, S.; Gao, J.; Sun, F.; Zhao, G.; Qin, Y. Pulsed electrocatalysis enables an efficient 2-electron oxygen reduction reaction for H2O2 production. J. Mater. Chem. A 2021, 9, 15948–15954. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, T.; Liu, C.; Zheng, D.; Huang, J.; Liu, Q.; Yuan, W.; Yin, Y.; Huang, L.; Xu, M.; et al. Asymmetric low-frequency pulsed strategy enables ultralong CO2 reduction stability and controllable product selectivity. J. Am. Chem. Soc. 2023, 145, 2195–2206. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Weng, L.; Bell, A.T. Impact of pulsed electrochemical reduction of CO2 on the formation of C2+ products over Cu. Acs Catal. 2020, 10, 12403–12413. [Google Scholar] [CrossRef]
- Xin, H.; Wang, H.; Zhang, W.; Chen, Y.; Ji, Q.; Zhang, G.; Liu, H.; Taylor, A.D.; Qu, J. In operando visualization and dynamic manipulation of electrochemical processes at the electrode–solution interface. Angew. Chem. Int. Ed. 2022, 61, e202206236. [Google Scholar] [CrossRef] [PubMed]
- Rocha, F.; Proost, J. Discriminating between the effect of pulse width and duty cycle on the hydrogen generation performance of 3-D electrodes during pulsed water electrolysis. Int. J. Hydrogen Energy 2021, 46, 28925–28935. [Google Scholar] [CrossRef]
- Nomura, K.; Shibata, N.; Maeda, M. Orientation control of zinc oxide films by pulsed current electrolysis. J. Cryst. Growth 2002, 235, 224–228. [Google Scholar] [CrossRef]
- Li, H.; Yu, M.; Wang, Y.; Shi, M. Influence of process parameters on pulse electroforming of nickel-rich nickel-cobalt alloys from sulfamate electrolyte. In Proceedings of the 2nd International Conference on Biotechnology, Chemical and Materials Engineering (CBCME 2012), Xiamen, China, 28–29 December 2013; Volume 641–642, pp. 440–443. [Google Scholar]
- Miličić, T.; Sivasankaran, M.; Blümner, C.; Sorrentino, A.; Vidaković-Koch, T. Pulsed electrolysis- explained. Faraday Discuss 2023, 246, 179–197. [Google Scholar] [CrossRef]
- Kireev, S.Y.; Frolov, A.V. Electrodeposition of nickel coatings from acetate-chloride electrolyte using galvanostatic pulse electrolysis. Prot. Met. Phys. Chem. Surf. 2021, 57, 1375–1379. [Google Scholar] [CrossRef]
- Puippe, J.C.; Ibl, N. Influence of charge and discharge of electrical double layer in pulse plating. J. Appl. Electrochem. 1980, 10, 775–784. [Google Scholar] [CrossRef]
- Chamelot, P.; Taxil, P.; Oquab, D.; Serp, J.; Lafage, B. Niobium electrodeposition in molten fluorides using pulsed electrolysis. J. Electrochem. Soc. 2000, 147, 4131. [Google Scholar] [CrossRef]
- Vincent, I.; Choi, B.; Nakoji, M.; Ishizuka, M.; Tsutsumi, K.; Tsutsumi, A. Pulsed current water splitting electrochemical cycle for hydrogen production. Int. J. Hydrogen Energy 2018, 43, 10240–10248. [Google Scholar] [CrossRef]
- Ostanina, T.N.; Rudoi, V.M.; Nikitin, V.S.; Darintseva, A.B.; Ostanin, N.I. Effect of parameters of pulse electrolysis on concentration changes in the loose zinc deposit and deposit properties. Russ. Chem. Bull. 2017, 66, 1433–1438. [Google Scholar] [CrossRef]
- Strain, J.M.; Gulati, S.; Pishgar, S.; Spurgeon, J.M. Pulsed electrochemical carbon monoxide reduction on oxide-derived copper catalyst. Chemsuschem 2020, 13, 3028–3033. [Google Scholar] [CrossRef] [PubMed]
- Blom, M.J.W.; Smulders, V.; van Swaaij, W.P.M.; Kersten, S.R.A.; Mul, G. Pulsed electrochemical synthesis of formate using Pb electrodes. Appl. Catal. B Environ. 2020, 268, 118420. [Google Scholar] [CrossRef]
- Engelbrecht, A.; Uhlig, C.; Stark, O.; Hämmerle, M.; Schmid, G.; Magori, E.; Wiesner-Fleischer, K.; Fleischer, M.; Moos, R. On the electrochemical CO2 Reduction at copper sheet electrodes with enhanced long-term stability by pulsed electrolysis. J. Electrochem. Soc. 2018, 165, J3059. [Google Scholar] [CrossRef]
- Murata, A.; Hori, Y. Product selectivity affected by cationic species in electrochemical reduction of CO2 and CO at a Cu Electrode. Bull. Chem. Soc. Jpn. 1991, 64, 123–127. [Google Scholar] [CrossRef]
- Le Duff, C.S.; Lawrence, M.J.; Rodriguez, P. Role of the adsorbed oxygen species in the selective electrochemical reduction of CO2 to alcohols and carbonyls on copper electrodes. Angew. Chem. Int. Ed. 2017, 56, 12919–12924. [Google Scholar] [CrossRef]
- Dinh, C.T.; Burdyny, T.; Kibria, M.G.; Seifitokaldani, A.; Gabardo, C.M.; De Arquer, F.P.G.; Kiani, A.; Edwards, J.P.; De Luna, P.; Bushuyev, O.S.; et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef]
- Kimura, K.W.; Casebolt, R.; Cimada Dasilva, J.; Kauffman, E.; Kim, J.; Dunbar, T.A.; Pollock, C.J.; Suntivich, J.; Hanrath, T. Selective electrochemical CO2 reduction during pulsed potential stems from dynamic interface. ACS Catal. 2020, 10, 8632–8639. [Google Scholar] [CrossRef]
- Timoshenko, J.; Bergmann, A.; Rettenmaier, C.; Herzog, A.; Arán-Ais, R.M.; Jeon, H.S.; Haase, F.T.; Hejral, U.; Grosse, P.; Kühl, S.; et al. Steering the structure and selectivity of CO2 electroreduction catalysts by potential pulses. Nat. Catal. 2022, 5, 259–267. [Google Scholar] [CrossRef]
- De Luna, P.; Quintero-Bermudez, R.; Dinh, C.; Ross, M.B.; Bushuyev, O.S.; Todorović, P.; Regier, T.; Kelley, S.O.; Yang, P.; Sargent, E.H. Catalyst electro-redeposition controls morphology and oxidation state for selective carbon dioxide reduction. Nat. Catal. 2018, 1, 103–110. [Google Scholar] [CrossRef]
- Chou, T.; Chang, C.; Yu, H.; Yu, W.; Dong, C.; Velasco-Vélez, J.; Chuang, C.; Chen, L.; Lee, J.; Chen, J.; et al. Controlling the oxidation state of the cu electrode and reaction intermediates for electrochemical CO2 reduction to ethylene. J. Am. Chem. Soc. 2020, 142, 2857–2867. [Google Scholar] [CrossRef]
- Lin, S.; Chang, C.; Chiu, S.; Pai, H.; Liao, T.; Hsu, C.; Chiang, W.; Tsai, M.; Chen, H.M. Operando time-resolved X-ray absorption spectroscopy reveals the chemical nature enabling highly selective CO2 reduction. Nat. Commun. 2020, 11, 3525. [Google Scholar] [CrossRef]
- Chang, C.; Hung, S.; Hsu, C.; Chen, H.; Lin, S.; Liao, Y.; Chen, H.M. Quantitatively unraveling the redox shuttle of spontaneous oxidation/electroreduction of CuOx on silver nanowires using in situ x-ray absorption spectroscopy. ACS Central Sci. 2019, 5, 1998–2009. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Sullivan, I.; Larson, D.M.; Liu, G.; Toma, F.M.; Xiang, C.; Drisdell, W.S. Correlating oxidation state and surface area to activity from operando studies of copper co electroreduction catalysts in a gas-fed device. Acs Catal. 2020, 10, 8000–8011. [Google Scholar] [CrossRef]
- Lum, Y.; Ager, J.W. Stability of residual oxides in oxide-derived copper catalysts for electrochemical CO2 reduction investigated with 18O labeling. Angew. Chem. Int. Ed. 2018, 57, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chang, X.; Malkani, A.S.; Yang, X.; Thompson, L.; Jiao, F.; Xu, B. Speciation of Cu surfaces during the electrochemical CO reduction reaction. J. Am. Chem. Soc. 2020, 142, 9735–9743. [Google Scholar] [CrossRef]
- Iijima, G.; Inomata, T.; Yamaguchi, H.; Ito, M.; Masuda, H. Role of a hydroxide layer on cu electrodes in electrochemical CO2 reduction. ACS Catal. 2019, 9, 6305. [Google Scholar] [CrossRef]
- Chernyshev, A.A.; Apisarov, A.P.; Isakov, A.V.; Shmygalev, A.S.; Arkhipov, S.P.; Zaikov, Y.P. Molybdenum electrodeposition in NaCl–KCl–MoCl3 melt using pulse electrolysis. Mater. Chem. Phys. 2023, 298, 127475. [Google Scholar] [CrossRef]
- Simon, G.H.; Kley, C.S.; Roldan Cuenya, B. Potential-Dependent morphology of copper catalysts during CO2 electroreduction revealed by in situ atomic force microscopy. Angew. Chem. Int. Ed. 2021, 60, 2561–2568. [Google Scholar] [CrossRef] [PubMed]
- Burdyny, T.; Graham, P.J.; Pang, Y.; Dinh, C.; Liu, M.; Sargent, E.H.; Sinton, D. Nanomorphology-enhanced gas-evolution intensifies CO2 reduction electrochemistry. Acs Sustain. Chem. Eng. 2017, 5, 4031–4040. [Google Scholar] [CrossRef]
- Liu, M.; Pang, Y.; Zhang, B.; De Luna, P.; Voznyy, O.; Xu, J.; Zheng, X.; Dinh, C.T.; Fan, F.; Cao, C.; et al. Enhanced electrocatalytic CO2 reduction via field-induced reagent concentration. Nature 2016, 537, 382–386. [Google Scholar] [CrossRef]
- Klinkova, A.; Luna, P.; Dinh, C.T.; Voznyy, O.; Larin, E.; Kumacheva, E.; Sargent, E. Rational design of efficient palladium catalysts for electroreduction of carbon dioxide to formate. ACS Catal. 2016, 6, 8115–8120. [Google Scholar] [CrossRef]
- Saberi Safaei, T.; Mepham, A.; Zheng, X.; Pang, Y.; Dinh, C.; Liu, M.; Sinton, D.; Kelley, S.O.; Sargent, E.H. High-density nanosharp microstructures enable efficient CO2 electroreduction. Nano Lett. 2016, 16, 7224–7228. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.; Shen, J.; Schouten, K.J.P.; Calle-Vallejo, F.; Koper, M.T.M. Catalysts and reaction pathways for the electrochemical reduction of carbon dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. [Google Scholar] [CrossRef]
- Jiang, K.; Sandberg, R.B.; Akey, A.J.; Liu, X.; Bell, D.C.; Nørskov, J.K.; Chan, K.; Wang, H. Metal ion cycling of Cu foil for selective C–C coupling in electrochemical CO2 reduction. Nat. Catal. 2018, 1, 111–119. [Google Scholar] [CrossRef]
- Jermann, B.; Augustynski, J. Long-term activation of the copper cathode in the course of CO2 reduction. Electrochim. Acta 1994, 39, 1891–1896. [Google Scholar] [CrossRef]
- Jeon, H.S.; Timoshenko, J.; Rettenmaier, C.; Herzog, A.; Yoon, A.; Chee, S.W.; Oener, S.; Hejral, U.; Haase, F.T.; Roldan Cuenya, B. Selectivity control of Cu nanocrystals in a gas-fed flow cell through CO2 pulsed electroreduction. J. Am. Chem. Soc. 2021, 143, 7578–7587. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.N.; Xie, L.; Zhou, W.; Sun, F.; Gao, J.H.; Yang, C.W.; Zhao, G.B.; Qin, Y.K.; Ma, J. Pulsed electrocatalysis enables the stabilization and activation of carbon-based catalysts towards H2O2 production. Appl. Catal. B Environ. 2022, 316, 121688. [Google Scholar] [CrossRef]
- Jiao, H.; Liu, M.; Gao, Y.; Song, J.; Jiao, S. Dynamic evolution of high-temperature molten salt electrolysis of titanium under different operational conditions. Inorg. Chem. Front. 2023, 10, 529–534. [Google Scholar] [CrossRef]
- Clark, E.L.; Resasco, J.; Landers, A.; Lin, J.; Chung, L.; Walton, A.; Hahn, C.; Jaramillo, T.F.; Bell, A.T. Standards and protocols for data acquisition and reporting for studies of the electrochemical reduction of carbon dioxide. Acs Catal. 2018, 8, 6560–6570. [Google Scholar] [CrossRef]
- Clark, E.L.; Bell, A.T. Direct observation of the local reaction environment during the electrochemical reduction of CO2. J. Am. Chem. Soc. 2018, 140, 7012–7020. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Oh, C.; Kim, K.; Lee, J.; Kim, T. Electrical double layer mechanism analysis of PEM water electrolysis for frequency limitation of pulsed currents. Energies 2021, 14, 7822. [Google Scholar] [CrossRef]
- Datta, M.; Landolt, D. Experimental investigation of mass transport in pulse plating. Surf. Technol. 1985, 25, 97–110. [Google Scholar] [CrossRef]
- Gupta, N.; Gattrell, M.; Macdougall, B. Calculation for the cathode surface concentrations in the electrochemical reduction of CO2 in KHCO3 solutions. J. Appl. Electrochem. 2006, 36, 161–172. [Google Scholar] [CrossRef]
- Cui, Y.; He, B.; Liu, X.; Sun, J. Ionic liquids-promoted electrocatalytic reduction of carbon dioxide. Ind. Eng. Chem. Res. 2020, 59, 20235–20252. [Google Scholar] [CrossRef]
- Wang, L.; Nitopi, S.A.; Bertheussen, E.; Orazov, M.; Morales-Guio, C.G.; Liu, X.; Higgins, D.C.; Chan, K.; Nørskov, J.K.; Hahn, C.; et al. Electrochemical carbon monoxide reduction on polycrystalline copper: Effects of potential, pressure, and pH on selectivity toward multicarbon and oxygenated products. ACS Catal. 2018, 8, 7445–7454. [Google Scholar] [CrossRef]
- Hori, Y.; Takahashi, R.; Yoshinami, Y.; Murata, A. Electrochemical reduction of CO at a copper Electrode. J. Phys. Chem. B 1997, 101, 7075–7081. [Google Scholar] [CrossRef]
- Liu, X.; Schlexer, P.; Xiao, J.; Ji, Y.; Wang, L.; Sandberg, R.B.; Tang, M.; Brown, K.S.; Peng, H.; Ringe, S.; et al. pH effects on the electrochemical reduction of CO(2) towards C2 products on stepped copper. Nat. Commun. 2019, 10, 32. [Google Scholar] [CrossRef]
- Mandal, L.; Yang, K.R.; Motapothula, M.R.; Ren, D.; Lobaccaro, P.; Patra, A.; Sherburne, M.; Batista, V.S.; Yeo, B.S.; Ager, J.W.; et al. Investigating the role of copper oxide in electrochemical CO2 reduction in real time. ACS Appl. Mater. Interfaces 2018, 10, 8574–8584. [Google Scholar] [CrossRef]
- Lobaccaro, P.; Mandal, L.; Motapothula, M.R.; Sherburne, M.; Martin, J.; Venkatesan, T.; Ager, J.W. Initial application of selected-ion flow-tube mass spectrometry to real-time product detection in electrochemical CO2 Reduction. Energy Technol. 2018, 6, 110–121. [Google Scholar] [CrossRef]
- Ma, X.; He, C.; Yan, Y.; Chen, J.; Feng, H.; Hu, J.; Zhu, H.; Xia, Y. Energy-efficient electrochemical degradation of ciprofloxacin by a Ti-foam/PbO2-GN composite electrode: Electrode characteristics, parameter optimization, and reaction mechanism. Chemosphere 2023, 315, 137739. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; You, S.; Zhang, J. Application of pulsed electrochemistry to enhanced water decontamination. Acs Es&T Eng. 2021, 1, 1502–1508. [Google Scholar]
- Obasanjo, C.A.; Gao, G.; Khiarak, B.N.; Pham, T.H.; Crane, J.; Dinh, C. Progress and perspectives of pulse electrolysis for stable electrochemical carbon dioxide reduction. Energy Fuels 2023, 37, 13601–13623. [Google Scholar] [CrossRef]
- Greenwell, F.; Siritanaratkul, B.; Sharma, P.K.; Yu, E.H.; Cowan, A.J. Pulsed electrolysis with a nickel molecular catalyst improves selectivity for carbon dioxide reduction. J. Am. Chem. Soc. 2023, 145, 15078–15083. [Google Scholar] [CrossRef]
- Wu, A.; Li, C.; Han, B.; Liu, W.; Zhang, Y.; Hanson, S.; Guan, W.; Singhal, S.C. Pulsed electrolysis of carbon dioxide by large-scale solid oxide electrolytic cells for intermittent renewable energy storage. Carbon Energy 2023, 5, e262. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Hatsukade, T.; Cave, E.R.; Abram, D.N.; Kibsgaard, J.; Jaramillo, T.F. Electrocatalytic conversion of carbon dioxide to methane and methanol on transition metal surfaces. J. Am. Chem. Soc. 2014, 136, 14107–14113. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Savéant, J. Catalysis of the electrochemical reduction of carbon dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, W.; Li, J.; Yang, C.; Meng, X.; Gao, J. Insights into the effects of pulsed parameters on H2O2 synthesis by two-electron oxygen reduction under pulsed electrocatalysis. Electrochem. Commun. 2023, 146, 107414. [Google Scholar] [CrossRef]
- Jännsch, Y.; Leung, J.J.; Hämmerle, M.; Magori, E.; Wiesner-Fleischer, K.; Simon, E.; Fleischer, M.; Moos, R. Pulsed potential electrochemical CO2 reduction for enhanced stability and catalyst reactivation of copper electrodes. Electrochem. Commun. 2020, 121, 106861. [Google Scholar] [CrossRef]
- Lu, Z.; Tang, J.; de Lourdes Mendoza, M.; Chang, D.; Cai, L.; Zhang, L. Electrochemical decrease of sulfide in sewage by pulsed power supply. J. Electroanal. Chem. 2015, 745, 37–43. [Google Scholar] [CrossRef]
- Besra, L.; Uchikoshi, T.; Suzuki, T.S.; Sakka, Y. Application of constant current pulse to suppress bubble incorporation and control deposit morphology during aqueous electrophoretic deposition (EPD). J. Eur. Ceram. Soc. 2009, 29, 1837–1845. [Google Scholar] [CrossRef]
- Shimizu, N.; Hotta, S.; Sekiya, T.; Oda, O. A novel method of hydrogen generation by water electrolysis using an ultra-short-pulse power supply. J. Appl. Electrochem. 2006, 36, 419–423. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Year | Details for Configurations | Working Electrode | Pulse Parameters | Applications Area | Enhancement Compared to Steady State Electrolysis | References |
|---|---|---|---|---|---|---|---|
| 1 | 2023 | A three-electrode setup was used for electrolysis, consisting of counter electrode (a platinum electrode, 1.5 cm × 1.5 cm × 1 mm), working electrode (graphite felt, 2 cm × 2 cm × 5 mm), and reference electrode (a silver chloride electrode, Ag/AgCl, saturated potassium chloride). The electrolyte, stirred at 150 rmp, was a 0.5 M Na2SO4 solution saturated with O2. | Graphite felt | The pulse waveform was sine wave and frequency was 0.2 Hz along with the amplitude 10 mV. | H2O2 production by 2-e−ORR | When subjected to pulsed potential, H2O2 production rate and FE of 2-e−ORR increased by 1.4 times and 3.62 times compared to constant potential conditions. In addition, energy consumption reduced by 75%. | [135] |
| 2 | 2023 | A 10 mL-H-type cell equipped with three electrodes, using Pt foil, Ag/AgCl, Cu3(DMPz)3, and 0.1 M KCl as the counter electrode, reference electrode, working electrode, and electrolyte. A nafion 117 proton exchange membrane separated the cathode and anode compartments. | Cu-dimethylpyrazole complex Cu3(DMPz)3 | There were two different asymmetric low-frequency pulsed strategy profiles (ALPS) to improve e selectivity of CH4 and C2H4 products.
| CO2RR |
| [77] |
| 3 | 2023 | A 250 mL reactor containing a 0.1 M Na2SO4 electrolyte was used for electrolysis. | Ti sheet cathode and PbO2 composite anode | Current density = 25.00 mA cm−2, pulse duty cycle = 50.0%, and pulse frequency = 5000 Hz, pulse electrolysis time = 120 min. | Organic wastewater treatment | Pulse mode (21.08 kWh m−3) can reduce energy consumption by 70.7% compared to DC mode (6.17 kWh m−3), although the CIP removal by pulse mode (89.7%) was reduced by 2.2% compared with DC mode (91.9%). | [128] |
| 4 | 2023 | The flat-tube solid oxide electrolytic cells (SOEC) include a supporting layer (NiO-3YSZ), fuel electrode (NiO-8YSZ), electrolyte (8YSZ), barrier layer (GDC(Gd0.1Ce0.9O2−δ)), and oxygen electrode (LSCF). | Fuel electrode was made of NiO-8YSZ(8 mol. % yttria-stabilized zirconia), and the oxygen electrode was made of LSCF (La0.6Sr0.4Co0.2Fe0.8O3−δ) | Pulse current = −300 mA/cm2. | CO2 conversion in solid oxide electrolytic cells | After 100 cycles of pulsed current, the voltage attenuated by 0.041% per cycle. The calculated efficiency approached 98.2% Additionally, the total conversion rate of CO2 was 52%, while it was about 20% with open circuit voltage. The theoretical lifespan of SOEC can exceed 500 cycles at −300 mA/cm2. | [132] |
| 5 | 2023 | Mo electroreduction is carried out in a cell with a c-axial cathode position in an argon atmosphere. The cell has a graphite container for the melt (MPG grade 8 structural graphite). The interior of the graphite container is lined with a Mo plate. Molybdenum rods are used as anode current leads for graphite containers. The electrolyte was NaCl-KCl-MoCl3 melt. | SY-200 grade glass carbon cathode with an area of 3 cm2 | Cathode current ranged from 2 up to 6 A/cm2, average cathode current density ranged from 0.286 up to 0.875 A/cm2, and ratio of the relaxation time to the pulse time (Tr/Tc) ranged from 6 up to 18. | Electrodeposition | Pulsed electrolysis for electrodeposition of Mo coatings from chloride molten salts shorted time to form dense deposits compared to constant current electrolysis. Optimized current density and Tr/Tc parameters increased the electrodeposition rate by a factor of 5 compared to static current electrolysis. | [105] |
| 6 | 2022 | CO2 reduction reaction was carried out in a custom-made glass two-compartment hydrogen cell, with the cathode chamber and anode chamber separated by a Nafion anion exchange membrane. The electrolyte was 0.5 M KCl. Ag/AgCl electrode was the reference electrode, and platinum mesh was the counter electrode. | A polycrystalline copper foil electrode | The anode pulse potential was 0.2 V, the cathode pulse potential is −1.2 V, the duty cycle was 50%, and the pulse period was 500 ms. | CO2RR | A high pulse potential with positive anode current improved reaction stability and increased C2 selectivity (FE = 76%). | [74] |
| 7 | 2021 | The electrolysis cell, constructed from plexiglass, was rectangular in shape with dimensions of 10 cm × 10 cm × 5 cm. Positioned at opposite ends of the cell, a Ti4O7 anode (with a geometric area of 10 cm2) and a stainless steel sheet cathode (10 cm2) were utilized without the need for a separator. | Ti4O7 anode, stainless steel sheet cathode | Pulsed-current mode adopted square current wave forms. Current density = 20 mA cm−2, reaction time = 120 min, anodic time/resting time = 100 ms. | Organic wastewater treatment | Pulsed-current mode: The rate constant for phenol oxidation was 1.48 h−1 and the energy consumption was 57.1%, which was lower than the direct-current mode (0.97 h−1). | [129] |
| 8 | 2021 | The apparatus consisted of a counter electrode (Pt), a reference electrode (Ag/AgCl) and a working electrode (graphite felt). The electrolyte was 0.05 M Na2SO4, stirred at 350 rpm, and the volume was 150 mL. | Graphite felt | Pulsed width = 1 s, duty ratio =30%, pulsed potential= −1.0 V vs. Ag/AgCl, and reaction time = 50 min. | H2O2 production via 2-e- ORR | H2O2 production increased by 138.12% using pulse potential compared to constant potential. | [76] |
| 9 | 2021 | A flow cell is composed of CO2 gas, cathode electrolyte, and anode electrolyte. The cathode and anode chambers were separated by an anion exchange membrane and equipped with Ag/AgCl reference electrodes, platinum-mesh counter electrodes, and the working electrode. The electrolyte solution was 1 M KOH. | A gas diffusion electrode sprayed with Cu NCs catalyst | The anode pulse potential = 0.9 V vs. RHE or 1.2 V vs. RHE, the cathode CO2 reduction pulse potential = −0.7 V vs. RHE, the duty cycle = 50%, and the pulse period = 2 s. | CO2RR | Compared with a potentiostatic of CO2RR at −0.7 V vs. RHE, the faraday efficiency of C2H4 and C2H5OH products at an anode potential of 0.9V vs. RHE increased from 40.9% to 43.6% and from 11% to 19.8%, respectively. Additionally, the FECH4− was 48.3% at an anode potential of 1.2 V vs. RHE compared with the FECH4 = 48.3% at the conventional condition. | [114] |
| 10 | 2020 | A standard three-electrode setup with two compartments separated by a membrane was used for the electrolysis. The OD-Cu foil, Pt mesh, and Ag/AgCl were used as the working electrode, the counter electrode, and the reference electrode. The electrolyte was 0.1 M KOH saturated with CO. | Oxide-derived copper (OD-Cu) | The cathodic potential = −0.35 V vs. RHE, a resting potential = 0 V vs. RHE, duty cycle = 50%, pulsing time = 10 ms, and electrolysis time = 60 min. | CO reduction | Pulsed CORR electrolysis: a product distribution by charge of approximately 70% for CORR and 29.3% for HER. The charge fraction of C1 products was 97% of CORR products (C1 and C2 products), while the charge fraction of C1 products was less than 20% for non-pulsed CORR electrolysis. Non-pulsed CORR electrolysis at −0.35 V vs. RHE: a product distribution by charge of 70.3% for HER and 29.7% for CORR. | [89] |
| 11 | 2020 | The setup included an Ir-MMO anode, Cu-DHP working electrode (Cu-DHP), and reference electrode (Ag/AgCl), which were separated by a Nafion membrane. The cathode electrolyte (pH = 8.5) was composed of a 0.1 M KHCO3 solution saturated with CO2, while the anode electrolyte (pH = 8.3) was 1 M KHCO3. | Deoxygenized high phosphorous copper sheets (Cu-DHP) | Working potential of −1.38 V remained 25 s, followed by the potential of −1.0 V remained for 5 s. | CO2RR | Compared with constant potential electrolysis, pulse electrolysis improved stability of the catalyst. After 8 h of constant potential electrolysis, the ethylene selectivity decreased from 15% to 2%, followed by pulse electrolysis of 8 h, it recovered back to 15%. | [136] |
| 12 | 2018 | A glass H-cell with two compartments contained a counter electrode (a platinum wire coil) and a working electrode (copper sheet), respectively. The reference electrode (Ag/AgCl, 3 M KCl) embedded in a Luggin capillary tube was placed in proximity to the working electrode. A Nafion N117 membrane separated the two compartments. The catholyte was 0.1 M KHCO3 (125 mL, 20 °C), and the anolyte was 1 M KHCO3 (125 mL, 20 °C). | copper sheet | Cathodic time (tc) = 25 s, anodic time (ta) = 5 s, cathodic bias (Uc) = −1.6 V, and anodic bias (Ua) = −0.18 V. | CO2RR | The long-term stability could extend 95 h under pulsed electrolysis. The faradaic efficiency of carbon containing products (CO, CH4, and C2H4) was about 40% and the efficiency of hydrogen about 20%. Potentiostatic electrolysis at −1.6 V vs. Ag/AgCl: it had very poor stability, and FEH2 gradually increased, and reached 70% after 16 h electrolysis. | [91] |
| 13 | 2018 | Ni(OH)2 and metal hydride MH electrodes were employed as the positive and negative electrodes. Additionally, a manganese dioxide MnO2 electrode served as an intermediate electrode. To ensure proper separation, a pair of polypropylene spacers with a thickness of 120 μm were placed between each electrode. | Ni(OH)2 positive electrode, MH negative electrode, and MnO2 the intermediate | Current density = 0.2 A cm−2 and pulse frequency = 500 Hz. | Water splitting for hydrogen production | The optimal performance was obtained at the current density of 0.2 A cm−2, and the battery voltage was 1.69 V when the pulse frequency was 500 Hz at 25 °C, which was higher than the electrochemical cycle of conventional electrolysis. | [87] |
| 14 | 2016 | Two-compartment polycarbonate electrochemical cell, with the cathode chamber and anode chamber separated by an anion exchange membrane (Selemion AMV). The Ag/AgCl reference electrode was located in the cathode chamber, and the platinum mesh counterpart electrode was in the anode chamber. The electrolyte was 1.0 M HCl, and the pH value was maintained at 6.8. | Polycrystalline copper foil | The cathode pulse time = 10–80 ms, a pulse potential= −1.0 V vs. RHE was applied, anode pulse time = 50 ms, and potential = 0.61 V vs. RHE. | CO2RR | A change in pulse time in the range of 10 to 80 ms produced syngas in the absence of by-products, with CO:H2 molar ratio ranging from ~32:1 to 9:16. No comparison with DC electrolysis. | [75] |
| 15 | 2015 | The reactor with two graphite plates with a size of 12 cm × 5 cm × 4 mm was used for organic wastewater treatment, containing 1 L of simulated wastewater (pH 7.0 ± 0.1). | Graphite plates | Duty cycle = 50%, pulse frequency = 1000 Hz, and cell voltage = 3 V. | Organic wastewater treatment | Compared to DC power supply (73.2%), pulse power supply (93.2%) had a higher removal rate of sulfide. | [137] |
| 16 | 2013 | The experimental setup comprised a stack of 9 electrolytic cells and a pulse generator. The electrolytes were 0.1 M and 0.4 M KOH (2 L). Electrodes with an area of 20 cm2 and a spacing of 10 mm. | Stainless steel electrodes | Pulse frequency = 1 kHz, applied current = 1.4 A, and a duty cycle of 3%. | Water splitting for hydrogen production | Pulse current in water electrolysis significantly enhanced the rate of electrolysis, producing more hydrogen and oxygen than that of direct current (DC). | [69] |
| 17 | 2013 | The reaction was carried out in a three-electrode system with saturated calomel as the reference electrode and platinum gauze used for the counter electrode. The electrolyte consisted of 3 mM CuCl2, 15 mM InCl3, 32 mM Ga(NO3)3, and 75 mM H2SeO3 at a pH of 1.5. | A Mo coated glass substrate | The cathode pulse potential = −700 mV vs. SCE, the anode pulse potential = 180 mV vs. SCE, the pulse period = 3 s, duty cycle = 67%, and the total degradation time = 50 min. | Electrodeposition | The films prepared by pulsed electrodeposition were smoother, denser, and more uniform compared to DC electrodeposition. | [72] |
| 18 | 2009 | The experimental setup includes deposition and counter electrodes composed of stainless steel (316 L) plates with dimensions of 2 cm × 5 cm × 0.4 mm. The reactor was filled with a 5 vol% aluminum trioxide suspension, and the pH was 4.5. The electrodes were spaced 20 mm apart, and the deposition range on the electrodes was defined as 2 cm × 2 cm. | Stainless steel (316 L) plates | Applied current = 0.001 A–0.1 A. There were bubbles in the deposits above the upper limit of the pulse width and incomplete deposition below the lowest value. | Electrodeposition | There were bubble-free deposits and lower deposition yield with suitable pulse parameters, and bubbly deposits and higher deposition yield with the corresponding DC current. | [138] |
| 19 | 2005 | The experiment was carried out in an electrolytic cell containing electrolyte (3.4 L, 1 M KOH solution, 293 ± 2 K), anode (platinum plates), and cathode (platinum plates). The electrodes were spaced 3 cm apart. The ultra-short power supply consisted of a static induction thyristor (SIThy) and an inductive energy storage (IES) circuit. | Platinum plates | Voltage pulse-width = 300 ns, the secondary peak voltage ranged from 7.9 to 140 V, the frequency = 2–25 kHz, and input power changed by increasing the pulse frequency. | Water splitting for hydrogen production | Ultra-short-pulse electrolysis alleviated the issue of decreasing efficiency while increasing power under DC electrolysis. | [139] |
| 20 | 2000 | Conventional H-type gas-tight glass cell filled with CO2-saturated 0.1 M KHCO3 buffer solution (pH 6.8) at 10 °C. The counter electrode (platinum) and the working electrode (CuAg alloy) were separated by an ion exchange membrane (Nafion 417). The reference electrode was an Ag/AgCl electrode. | CuAg alloy electrodes with the atomic ratios (Cu/Ag) of 28/72 | An anodic bias = −0.4 V, a cathodic bias = −2.0 V vs. Ag/AgCl. Cathodic period (Tc)/anodic period (Ta) = 5 s. | CO2RR | The total value of faradaic efficiencies for these C2 compounds was 54.2% under the pulsed CO2 electroreduction. No comparison with the DC case. | [63] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Liu, Y.; Liu, S.; Cao, Y.; Qiu, S.; Deng, F. A Bibliometric Analysis on Pulsed Electrolysis: Electronic Effect, Double Layer Effect, and Mass Transport. Catalysts 2023, 13, 1410. https://doi.org/10.3390/catal13111410
Wang Z, Liu Y, Liu S, Cao Y, Qiu S, Deng F. A Bibliometric Analysis on Pulsed Electrolysis: Electronic Effect, Double Layer Effect, and Mass Transport. Catalysts. 2023; 13(11):1410. https://doi.org/10.3390/catal13111410
Chicago/Turabian StyleWang, Zhuowen, Yijun Liu, Sibei Liu, Yuxuan Cao, Shan Qiu, and Fengxia Deng. 2023. "A Bibliometric Analysis on Pulsed Electrolysis: Electronic Effect, Double Layer Effect, and Mass Transport" Catalysts 13, no. 11: 1410. https://doi.org/10.3390/catal13111410
APA StyleWang, Z., Liu, Y., Liu, S., Cao, Y., Qiu, S., & Deng, F. (2023). A Bibliometric Analysis on Pulsed Electrolysis: Electronic Effect, Double Layer Effect, and Mass Transport. Catalysts, 13(11), 1410. https://doi.org/10.3390/catal13111410