Diastereoselective Synthesis of 7,8-Carvone Epoxides

 ,
,  , ,
, ,  , , and
, , and 
Abstract
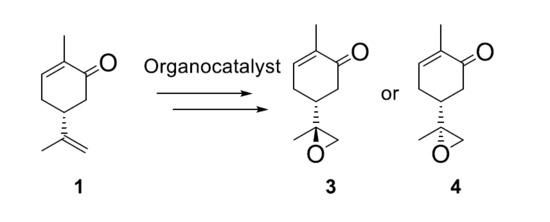
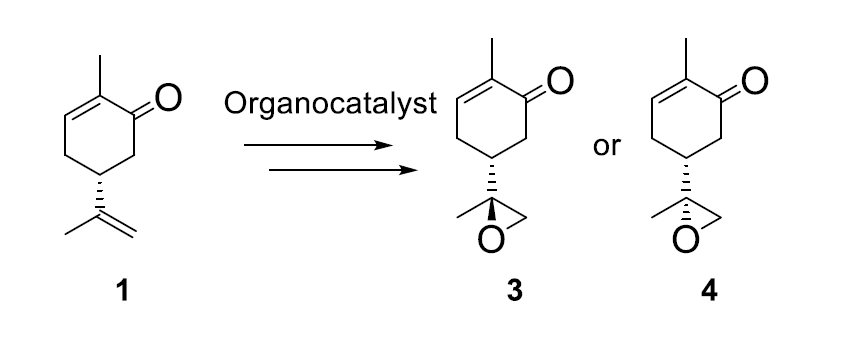
:
1. Introduction
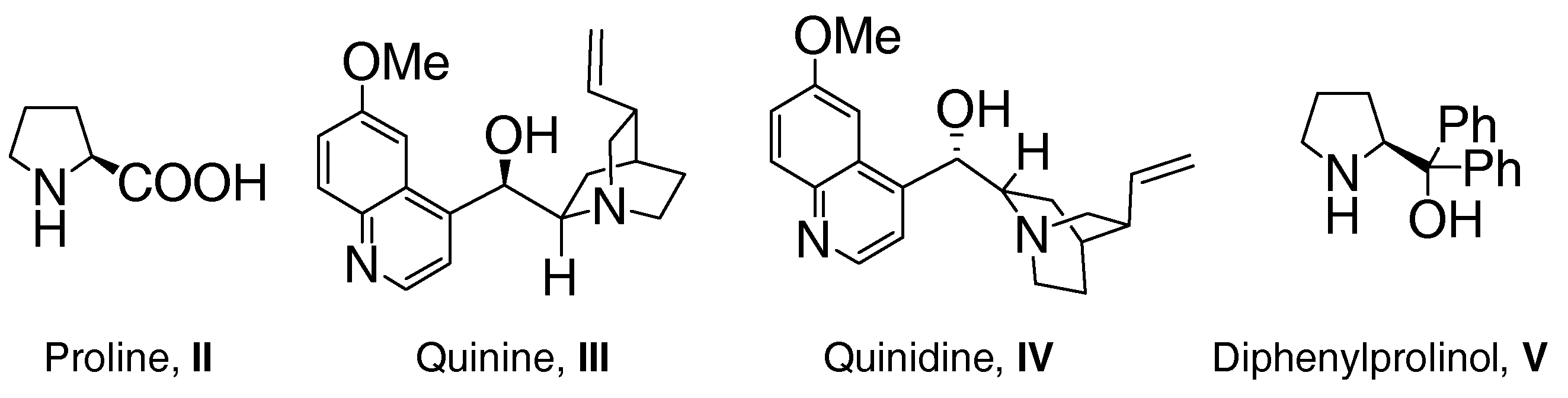
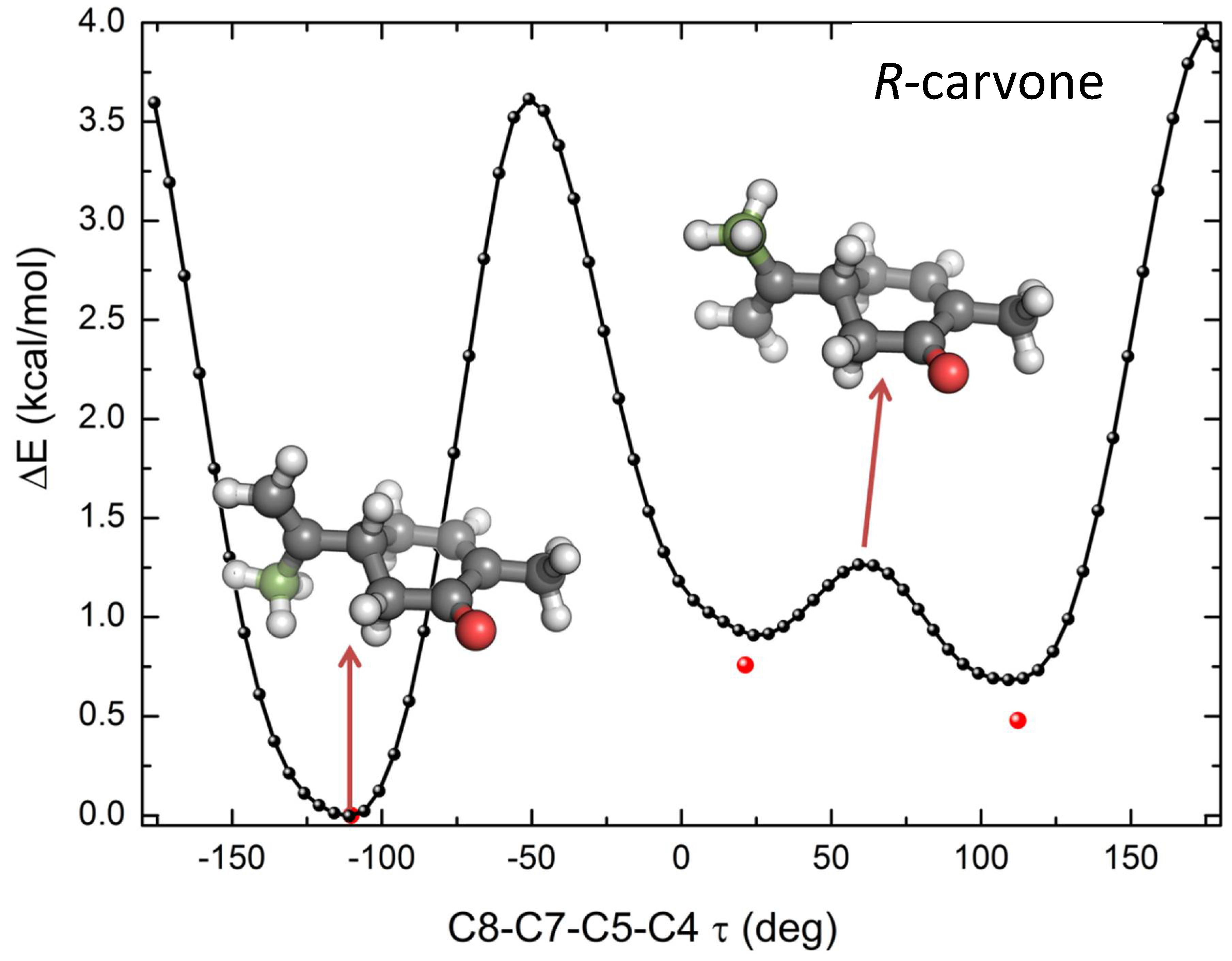
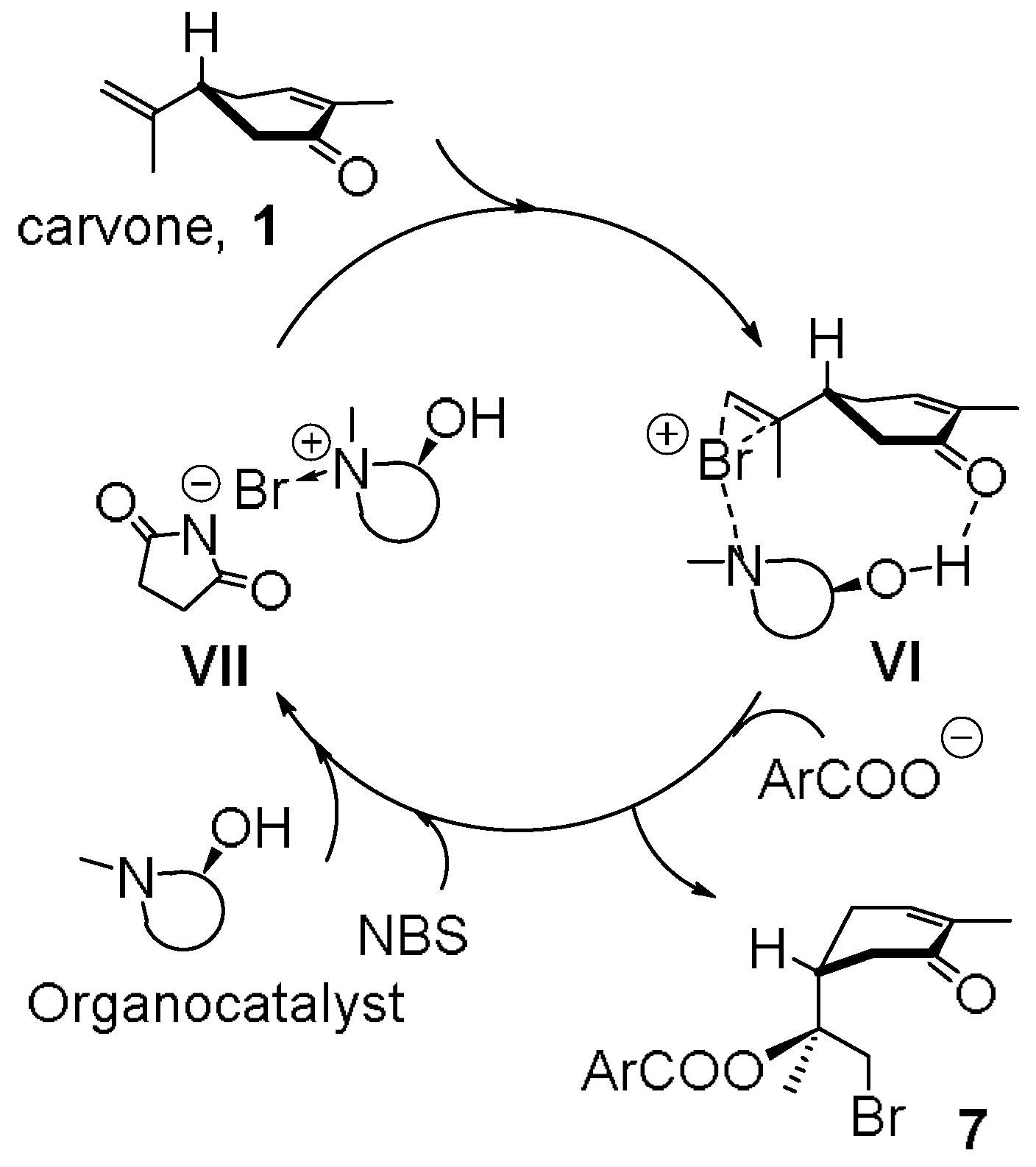
2. Results and Discussion
3. Materials and Methods
3.1. Reagents
3.2. Characterization
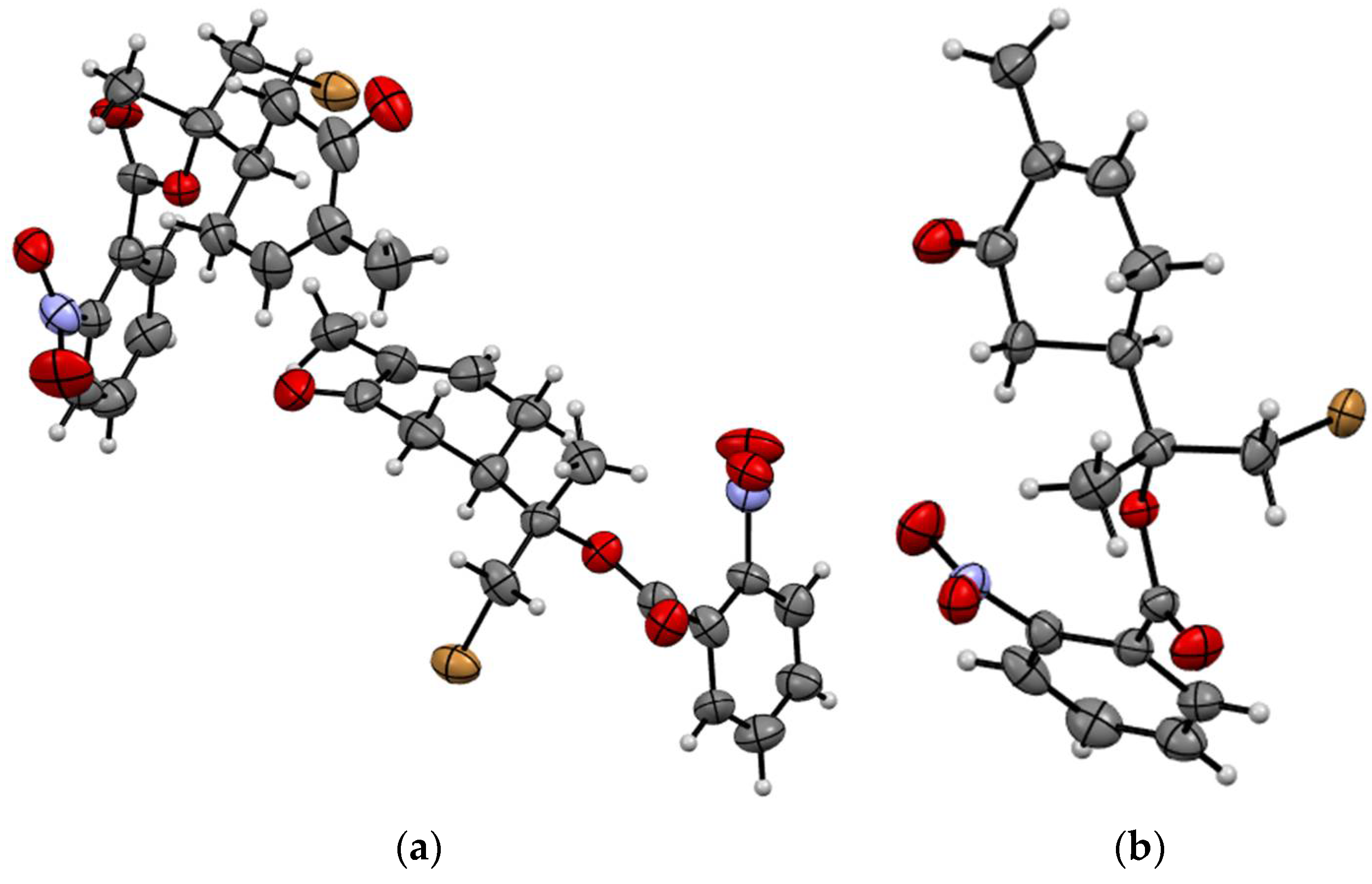
X-Ray Crystallography
3.3. Computational Methods
4. Experimental Section
4.1. General Procedure for the Synthesis of the Bromoesters 7 and 8
4.1.1. (5R)-9-Bromocarvone (5)
4.1.2. (5R,7RS)-7,8-Dibromocarvone (6)
4.1.3. (5R,7R)-8-Bromo-7-(2-nitrobenzoate)carvone (7)
4.1.4. (5R,7S)-8-Bromo-7-(2-nitrobenzoate)carvone (8)
4.2. Hydrolysis Reaction of Bromoesters 7 and 8
4.2.1. (5R,7S)-7,8-Epoxycarvone (3)
4.2.2. (5R,7R)-7,8-Epoxycarvone (4)
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Finkbeiner, P.; Murai, K.; Röpke, M.; Sarpong, R. Total synthesis of terpenoids employing a “benzannulation of carvone” strategy: Synthesis of (−)-crotogoudin. J. Am. Chem. Soc. 2017, 139, 11349–11352. [Google Scholar] [CrossRef] [PubMed]
- Nannini, L.J.; Nemat, S.J.; Carreira, E.M. Total synthesis of (+)-sarcophytin. Angew. Chem. Int. Ed. 2018, 57, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Kunimura, R.; Takagi, H.; Hirai, M.; Kogen, H.; Hirota, H.; Kuroda, C. Total synthesis of highly oxygenated bisabolane sesquiterpene isolated from ligularia lankongensis: Relative and absolute configurations of the natural product. J Org Chem 2018, 83, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Senthil Kumaran, R.; Mehta, G. A versatile, rcm based approach to eudesmane and dihydroagarofuran sesquiterpenoids from (−)-carvone: A formal synthesis of (−)-isocelorbicol. Tetrahedron 2015, 71, 1718–1731. [Google Scholar] [CrossRef]
- Abad, A.; Agulló, C.; Cuñat, A.C.; Llosá, M.C. Stereoselective construction of the tetracyclic scalarane skeleton from carvone. Chem. Commun. 1999, 427–428. [Google Scholar] [CrossRef] [Green Version]
- Abad, A.; Agulló, C.; Cuñat, A.; De Alfonso, I.; Navarro, I.; Vera, N. Synthesis of highly functionalised enantiopure bicyclo[3.2.1]-octane systems from carvone. Molecules 2004, 9, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.K.W.; Lai, Y.M.; Siu, Y.-H. Regiospecific epoxidation of carvone: A discovery-oriented experiment for understanding the selectivity and mechanism of epoxidation reactions. J. Chem. Ed. 2006, 83, 1058. [Google Scholar] [CrossRef]
- Masarwa, A.; Weber, M.; Sarpong, R. Selective c–c and c–h bond activation/cleavage of pinene derivatives: Synthesis of enantiopure cyclohexenone scaffolds and mechanistic insights. J. Am. Chem. Soc. 2015, 137, 6327–6334. [Google Scholar] [CrossRef] [PubMed]
- Valeev, R.F.; Bikzhanov, R.F.; Selezneva, N.K.; Gimalova, F.A.; Miftakhov, M.S. Synthesis of 6-hydroxycarvone derivatives and their oxidative decyclization with lead tetraacetate. Russ. J. Org. Chem. 2011, 47, 1287. [Google Scholar] [CrossRef]
- Sepúlveda-Arias, J.C.; Veloza, L.A.; Escobar, L.M.; Orozco, L.M.; Lopera, I.A. Anti-inflammatory effects of the main constituents and epoxides derived from the essential oils obtained from tagetes lucida, cymbopogon citratus, lippia alba and eucalyptus citriodora. J. Essent. Oil Res. 2013, 25, 186–193. [Google Scholar] [CrossRef]
- Kimbaris, A.C.; González-Coloma, A.; Andrés, M.F.; Vidali, V.P.; Polissiou, M.G.; Santana-Méridas, O. Biocidal compounds from mentha sp. Essential oils and their structure–activity relationships. Chem. Biodivers. 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Mandelli, D.; Kozlov, Y.N.; da Silva, C.A.R.; Carvalho, W.A.; Pescarmona, P.P.; Cella, D.d.A.; de Paiva, P.T.; Shul’pin, G.B. Oxidation of olefins with H2O2 catalysed by gallium(III) nitrate and aluminum(III) nitrate in solution. J. Mol. Catal. A Chem. 2016, 422, 216–220. [Google Scholar] [CrossRef]
- Kamata, K.; Sugahara, K.; Yonehara, K.; Ishimoto, R.; Mizuno, N. Efficient epoxidation of electron-deficient alkenes with hydrogen peroxide catalysed by [γ-PW10O38V2(μ-OH)2]3−. Chemistry 2011, 17, 7549–7559. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Garcia-Bosch, I.; Company, A.; Sala, X.; Fontrodona, X.; Ribas, X.; Costas, M. Chiral manganese complexes with pinene appended tetradentate ligands as stereoselective epoxidation catalysts. Dalton Trans. 2007, 5539–5545. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Miao, C.-X.; Wang, D.; Wang, S.; Xia, C.; Sun, W. Mnii complexes with tetradentate n4 ligands: Highly efficient catalysts for the epoxidation of olefins with H2O2. J. Mol. Catal. A Chem. 2012, 353–354, 185–191. [Google Scholar] [CrossRef]
- Garcia-Bosch, I.; Ribas, X.; Costas, M. A broad substrate-scope method for fast, efficient and selective hydrogen peroxide-epoxidation. Adv. Syn. Catal. 2009, 351, 348–352. [Google Scholar] [CrossRef]
- Spannring, P.; Yazerski, V.A.; Chen, J.; Otte, M.; Weckhuysen, B.M.; Bruijnincx, P.C.A.; Klein Gebbink, R.J.M. Regioselective cleavage of electron-rich double bonds in dienes to carbonyl compounds with [Fe(OTf)2(mix-BPBP)] and a combination of H2O2 and NaiO4. Eur. J. Inorg. Chem. 2015, 2015, 3462–3466. [Google Scholar] [CrossRef]
- Oddon, F.; Girgenti, E.; Lebrun, C.; Marchi-Delapierre, C.; Pécaut, J.; Ménage, S. Iron coordination chemistry of N2Py2 ligands substituted by carboxylic moieties and their impact on alkene oxidation catalysis. Eur. J. Inorg. Chem. 2012, 2012, 85–96. [Google Scholar] [CrossRef]
- Clemente-Tejeda, D.; López-Moreno, A.; Bermejo, F.A. Non-heme iron catalysis in C=C, C–H, and CH2 oxidation reactions. Oxidative transformations on terpenoids catalysed by Fe(bpmen)(OTf)2. Tetrahedron 2013, 69, 2977–2986. [Google Scholar] [CrossRef]
- Yazerski, V.A.; Spannring, P.; Gatineau, D.; Woerde, C.H.M.; Wieclawska, S.M.; Lutz, M.; Kleijn, H.; Klein Gebbink, R.J.M. Making Fe(BPBP)-catalysed C-H and C=C oxidations more affordable. Org. Biomol. Chem. 2014, 12, 2062–2070. [Google Scholar] [CrossRef] [PubMed]
- Thatte, C.S.; Rathnam, M.V.; Kumar, M.S.S. Synthesis, characterization and application of chitosan based schiff base-transition metal complexes (Mn, Cu, Co, Ni). JOAC 2013, 2, 1192. [Google Scholar]
- Stok, J.E.; Yamada, S.; Farlow, A.J.; Slessor, K.E.; De Voss, J.J. Cytochrome P450cin (CYP176A1) D241N: Investigating the role of the conserved acid in the active site of cytochrome P450s. Biochim. Biophys. Acta 2013, 1834, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Sánchez, D.; Ríos-Lombardía, N.; Gotor, V.; Gotor-Fernández, V. Chemoenzymatic epoxidation of alkenes based on peracid formation by a rhizomucor miehei lipase-catalysed perhydrolysis reaction. Tetrahedron 2014, 70, 1144–1148. [Google Scholar] [CrossRef]
- Rodilla, J.M.; Neves, P.P.; Pombal, S.; Rives, V.; Trujillano, R.; Díez, D. Hydrotalcite catalysis for the synthesis of new chiral building blocks. Nat. Prod. Res. 2016, 30, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Majetich, G.; Shimkus, J.; Li, Y. Epoxidation of olefins by β-bromoalkoxydimethylsulfonium ylides. Tetrahedron Lett. 2010, 51, 6830–6834. [Google Scholar] [CrossRef]
- Dalko, P.I.; Moisan, L. Enantioselective organocatalysis. Angew. Chem. Int. Ed. 2001, 40, 3726–3748. [Google Scholar] [CrossRef]
- List, B. Asymmetric aminocatalysis. Synlett 2001, 2001, 1675–1686. [Google Scholar] [CrossRef]
- Brown, S.P.; Brochu, M.P.; Sinz, C.J.; MacMillan, D.W.C. The direct and enantioselective organocatalytic α-oxidation of aldehydes. J. Am. Chem. Soc. 2003, 125, 10808–10809. [Google Scholar] [CrossRef] [PubMed]
- Pidathala, C.; Hoang, L.; Vignola, N.; List, B. Direct catalytic asymmetric enolexo aldolizations. Angew. Chem. Int. Ed. 2003, 42, 2785–2788. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, D. Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2005; p. 454. [Google Scholar]
- Jiang, X.; Liu, H. 4.07 electrophilic cyclization a2-knochel, paul. In Comprehensive Organic Synthesis II, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 412–494. [Google Scholar]
- Bar, S. Organocatalysis in the stereoselective bromohydrin reaction of alkenes. Can. J. Chem. 2010, 88, 605–612. [Google Scholar] [CrossRef]
- Castellanos, A.; Fletcher, S.P. Current methods for asymmetric halogenation of olefins. Chem. Eur. J. 2011, 17, 5766–5776. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.K.; Yeung, Y.-Y. Recent advances in stereoselective bromofunctionalization of alkenes using n-bromoamide reagents. Chem. Commun. 2013, 49, 7985–7996. [Google Scholar] [CrossRef] [PubMed]
- Valeev, R.F.; Bikzhanov, R.F.; Yagafarov, N.Z.; Miftakhov, M.S. Synthesis of the northern fragment of an epothilone d analogue from (−)-carvone. Tetrahedron 2012, 68, 6868–6872. [Google Scholar] [CrossRef]
- Antelo, J.M.; Arce, F.; Crugeiras, J. Kinetics of electrophilic bromine transfer from n-bromosuccinimide to amines and amino acids. J. Chem. Soc. Perk. Trans. 1995, 2, 2275–2279. [Google Scholar] [CrossRef]
- Chen, G.; Ma, S. Enantioselective halocyclization reactions for the synthesis of chiral cyclic compounds. Angew. Chem. Int. Ed. 2010, 49, 8306–8308. [Google Scholar] [CrossRef] [PubMed]
- Crystallographic data for the structures reported in this paper has been deposited at the Cambridge Crystallographic Data Centre as supplementary material No. CCDC 1821654–1821653 the data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/getstructures.
- Avilés Moreno, J.R.; Huet, T.R.; González, J.J.L. Conformational relaxation of s-(+)-carvone and r-(+)-limonene studied by microwave fourier transform spectroscopy and quantum chemical calculations. Struct. Chem. 2013, 24, 1163–1170. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09 rev. B.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Scalmani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry a | Catalyst, Load | T (°C) | Time (days) | Yield (%) b | d.r. (%) c | ||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 5 | 6 | 7 | 8 | |||||
| 1 | No catalyst | rt | 6 | 15 | 28 | - | 15 | 12 | 11 |
| 2 | No catalyst | 39 | 3 | 15 | 27 | - | 18 | 16 | 6 |
| 3 | Proline, II, 2% | rt | 6 | - | 12 | 4 | 34 | 20 | 26 |
| 4 | Proline, II, 20% | rt | 6 | - | 13 | 5 | 36 | 18 | 33 |
| 5 | Proline, II, 20% | 39 | 6 | - | 17 | - | 34 | 18 | 31 |
| 6 | Quinine, III, 2% | 39 | 6 | - | 14 | - | 36 | 20 | 29 |
| 7 | Quinine, III, 20% | rt | 6 | - | 16 | - | 35 | 13 | 46 |
| 8 | Quinine, III, 20% | 39 | 6 | - | 17 | - | 36 | 16 | 38 |
| 9 | Quinidine, IV, 2% | rt | 6 | - | 13 | - | 56 | 11 | 67 |
| 10 | Quinidine, IV, 2% | 39 | 6 | - | 16 | - | 41 | 14 | 49 |
| 11 | Quinidine, IV, 20% | rt | 6 | - | 15 | - | 57 | 10 | 70 |
| 12 | Quinidine, IV, 20% | 39 | 6 | - | 11 | - | 54 | 16 | 54 |
| 13 | Diphenylprolinol, V, 2% | rt | 6 | - | 20 | - | 27 | 10 | 46 |
| 14 | Diphenylprolinol, V, 2% | 39 | 6 | - | 7 | - | 35 | 28 | 11 |
| 15 | Diphenylprolinol, V, 20% | rt | 6 | - | 21 | - | 50 | 17 | 50 |
| 16 | Diphenylprolinol, V, 20% | 39 | 6 | - | 23 | - | 44 | 24 | 29 |
| C | 3.166 | −0.027 | 1.285 |
| C | 2.926 | 0.247 | −1.184 |
| C | −3.461 | −0.554 | 0.118 |
| C | −1.150 | −1.514 | −0.146 |
| C | 0.348 | −1.433 | −0.231 |
| C | 0.098 | 1.040 | −0.298 |
| C | 2.369 | 0.033 | 0.201 |
| C | −1.967 | −0.440 | −0.014 |
| C | −1.386 | 0.926 | −0.015 |
| C | 0.872 | −0.114 | 0.351 |
| O | −2.090 | 1.917 | 0.171 |
| H | 0.447 | 2.017 | 0.051 |
| H | 0.223 | 1.001 | −1.390 |
| H | −3.961 | −0.011 | −0.690 |
| H | −3.797 | −0.111 | 1.060 |
| H | −3.771 | −1.601 | 0.089 |
| H | −1.600 | −2.507 | −0.186 |
| H | 0.791 | −2.283 | 0.303 |
| H | 0.654 | −1.530 | −1.284 |
| H | 0.638 | −0.116 | 1.425 |
| H | 2.587 | 1.203 | −1.599 |
| H | 4.019 | 0.254 | −1.163 |
| H | 2.600 | −0.538 | −1.875 |
| H | 2.751 | −0.168 | 2.280 |
| H | 4.246 | 0.062 | 1.197 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pombal, S.; E. Tobal, I.; M. Roncero, A.; M. Rodilla, J.; M. Garrido, N.; Sanz, F.; Esteban, A.; Tostado, J.; F. Moro, R.; Sexmero, M.J.; et al. Diastereoselective Synthesis of 7,8-Carvone Epoxides. Catalysts 2018, 8, 250. https://doi.org/10.3390/catal8060250
Pombal S, E. Tobal I, M. Roncero A, M. Rodilla J, M. Garrido N, Sanz F, Esteban A, Tostado J, F. Moro R, Sexmero MJ, et al. Diastereoselective Synthesis of 7,8-Carvone Epoxides. Catalysts. 2018; 8(6):250. https://doi.org/10.3390/catal8060250
Chicago/Turabian StylePombal, Sofia, Ignacio E. Tobal, Alejandro M. Roncero, Jesus M. Rodilla, Narciso M. Garrido, Francisca Sanz, Alberto Esteban, Jaime Tostado, Rosalina F. Moro, Maria Jose Sexmero, and et al. 2018. "Diastereoselective Synthesis of 7,8-Carvone Epoxides" Catalysts 8, no. 6: 250. https://doi.org/10.3390/catal8060250
APA StylePombal, S., E. Tobal, I., M. Roncero, A., M. Rodilla, J., M. Garrido, N., Sanz, F., Esteban, A., Tostado, J., F. Moro, R., Sexmero, M. J., G. Jambrina, P., & Diez, D. (2018). Diastereoselective Synthesis of 7,8-Carvone Epoxides. Catalysts, 8(6), 250. https://doi.org/10.3390/catal8060250