Deep Eutectic Mixtures as Reaction Media for the Enantioselective Organocatalyzed α-Amination of 1,3-Dicarbonyl Compounds




Abstract
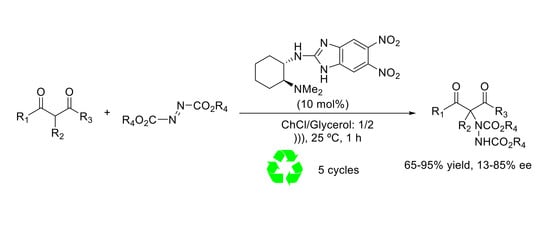
:
1. Introduction
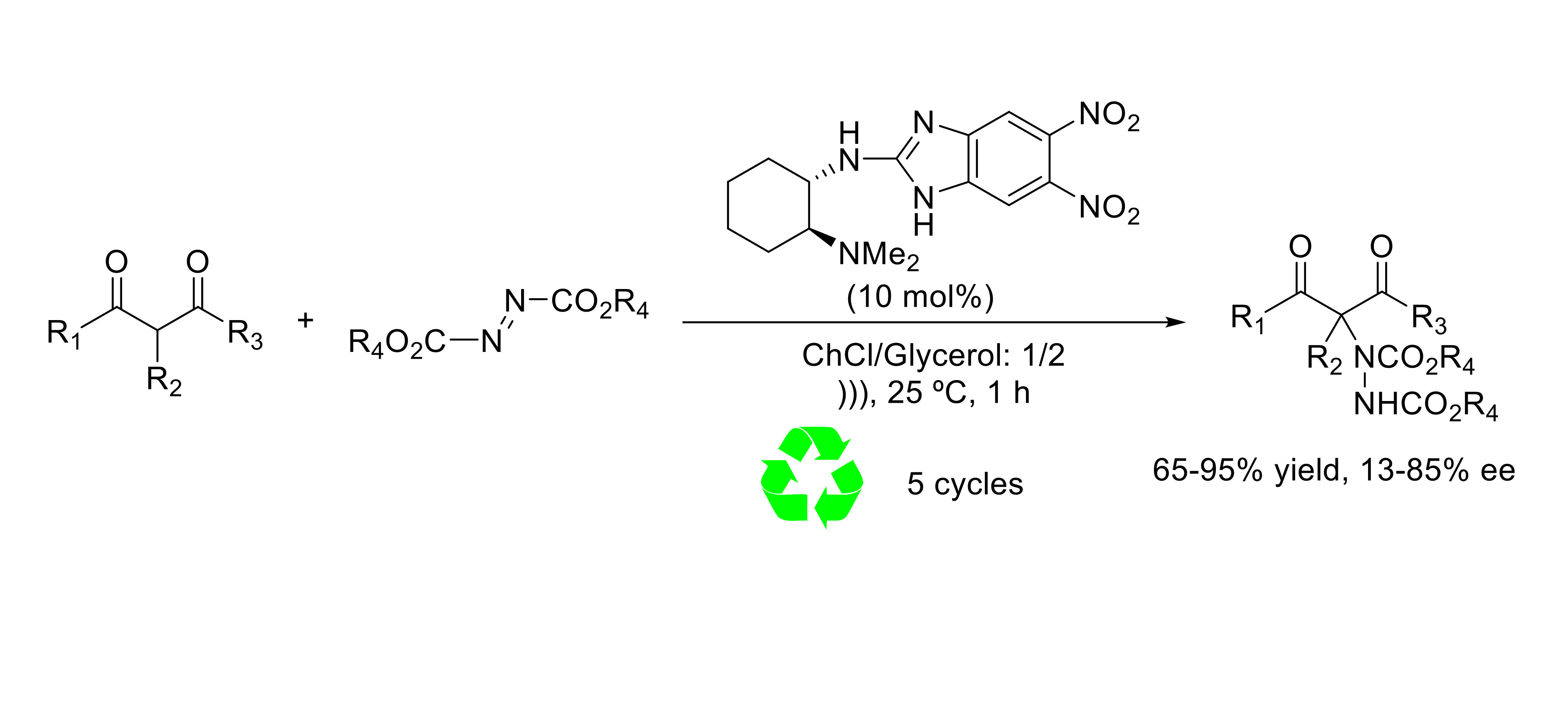
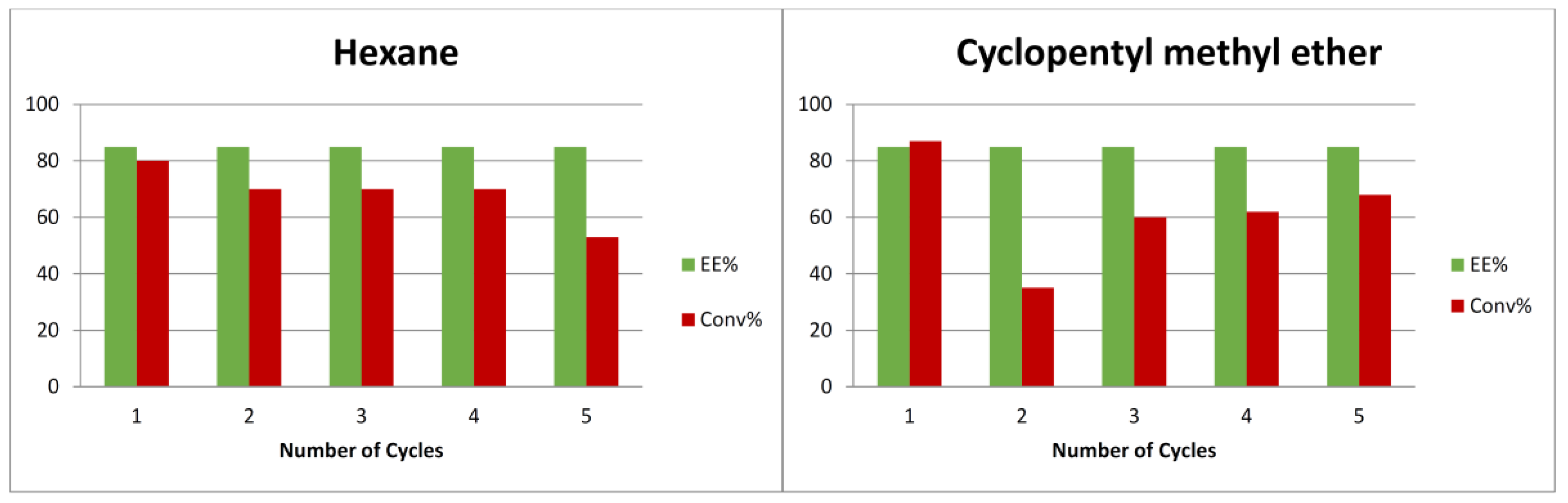
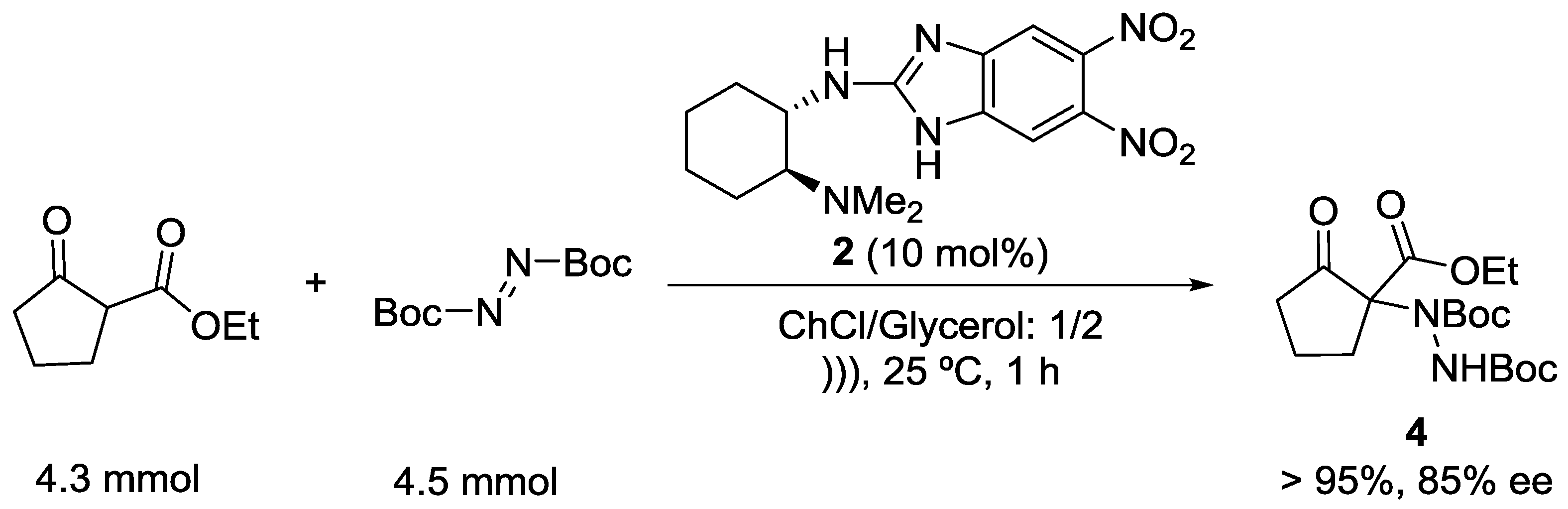
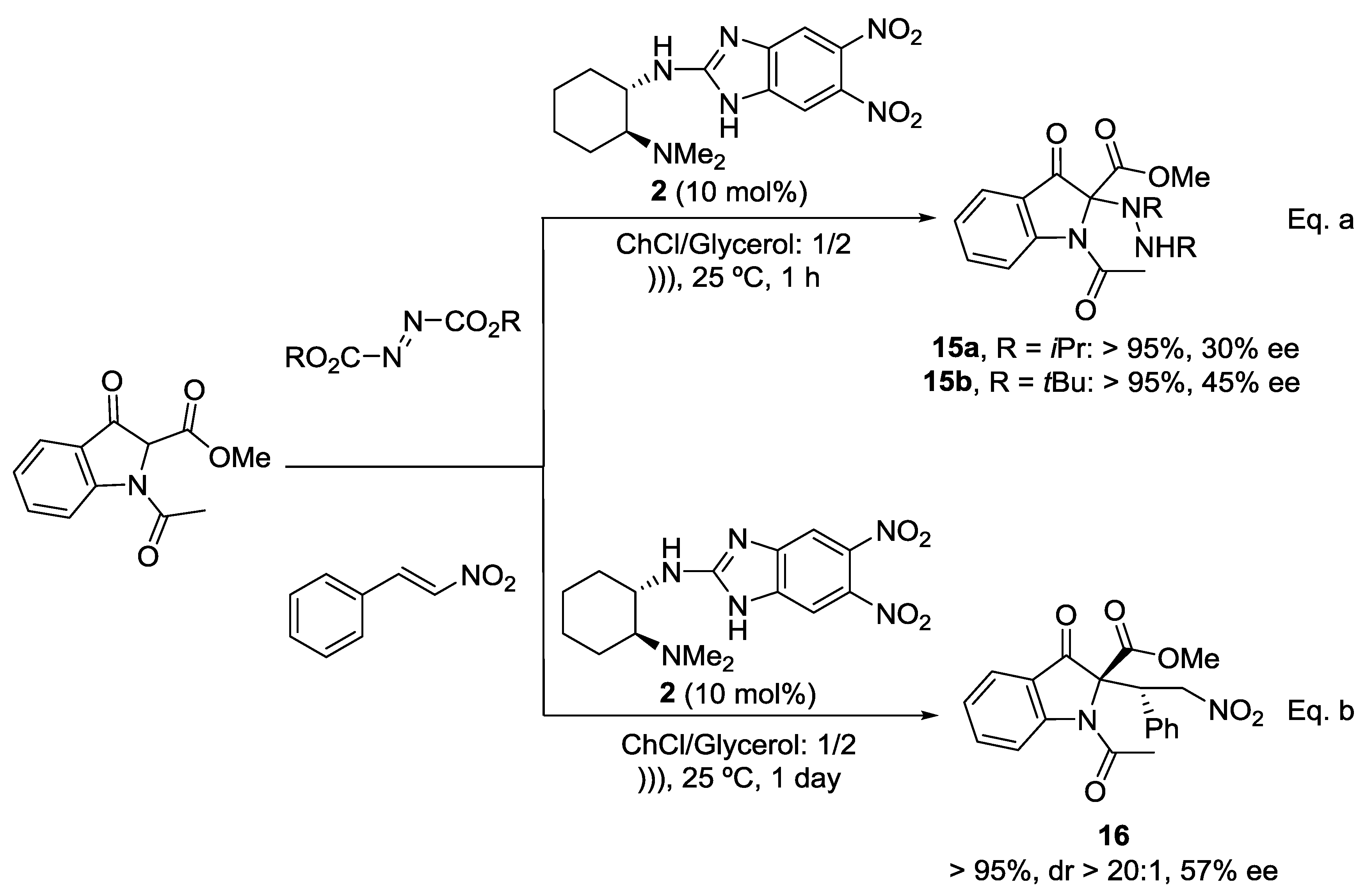
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Synthesis of Catalyst 2
3.3. Typical Procedure for the α-Amination Reaction
3.4. Typical Procedure for the Recovery of the Catalyst in the α-Amination Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References and Note
- Mahrwald, R. Enantioselective Organocatalyzed Reactions; Springer: Dordrecht, The Netherlands, 2011. [Google Scholar]
- Berkessel, A.; Gröger, H. Asymmetric Organocatalysis—From Biomimetic Concepts to Applications in Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Dalko, P. Enantioselective Organocatalysis; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Clarke, C.J.; Tu, W.C.; Levers, O.; Bröhl, A.; Hallett, J.P. Green and Sustainable Solvents in Chemical Processes. Chem. Rev. 2018, 118, 747–800. [Google Scholar] [CrossRef] [PubMed]
- Kerton, F.M. Solvent Systems for Sustainable Chemistry. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons: New York, NY, USA, 2016. [Google Scholar]
- Liu, Y.; Friesen, J.B.; McAlpine, J.B.; Lankin, D.C.; Chen, S.-N.; Pauli, G.F. Natural Deep Eutectic Solvents: Properties, Applications, and Perspectives. J. Nat. Prod. 2018, 81, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; De Oliveira Vigier, K.; Royera, S.; Jerome, F. Deep eutectic solvents: Syntheses, properties and applications. Chem. Soc. Rev. 2012, 41, 7108–7146. [Google Scholar] [CrossRef] [PubMed]
- García-Álvarez, J. Deep eutectic solvents and their applications as new green and biorenewable reaction media. In Handbook of Solvents; Wypych, G., Ed.; ChemTec Publishing: Toronto, ON, Canada, 2014; Volume 2, pp. 813–844. [Google Scholar]
- Guajardo, N.; Müller, C.R.; Schrebler, R.; Carlesi, C.; Domínguez de María, P. Deep eutectic solvents for organocatalysis, biotransformations, and multistep organocatalyst/enzyme combinations. ChemCatChem 2016, 8, 1020–1027. [Google Scholar] [CrossRef]
- Alonso, D.A.; Baeza, A.; Chinchilla, R.; Guillena, G.; Pastor, I.P.; Ramón, D.J. Deep Eutectic Solvents: The Organic Reaction Medium of the Century. Eur. J. Org. Chem. 2016, 2016, 612–632. [Google Scholar] [CrossRef]
- García-Álvarez, J. Deep Eutectic Mixtures: Promising Sustainable Solvents for Metal-Catalysed and Metal-Mediated Organic Reactions. Eur. J. Inorg. Chem. 2015, 2015, 5147–5157. [Google Scholar] [CrossRef]
- García-Álvarez, J.; Hevia, E.; Capriati, V. Reactivity of Polar Organometallic Compounds in Unconventional Reaction Media: Challenges and Opportunities. Eur. J. Org. Chem. 2015, 2015, 6779–6799. [Google Scholar] [CrossRef] [Green Version]
- Massolo, E.; Palmieri, S.; Benaglia, M.; Capriati, V.; Perna, F.M. Stereoselective organocatalysed reactions in deep eutectic solvents: Highly tunable and biorenewable reaction media for sustainable organic synthesis. Green Chem. 2016, 18, 792–797. [Google Scholar] [CrossRef]
- Branco, L.C.; Faisca Phillips, A.M.; Marques, M.M.; Gago, S.; Branco, P.S. Recent advances in sustainable organocatalysis. In Recent Advances in Organocatalysis; Karame, I., Srour, H., Eds.; InTech: Rijeka, Croatia, 2016; pp. 141–182. ISBN 978-953-51-2673-7. [Google Scholar]
- Genet, J.-P.; Creck, C.; Lavergne, D. Modern Amination Methods; Ricci, A., Ed.; Wiley-VCH: Weinheim, Germany, 2000; Chapter 3. [Google Scholar]
- Amino Group Chemistry: From Synthesis to the Life Sciences; Ricci, A. (Ed.) Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Janey, J.M. Recent Advances in Catalytic, Enantioselective α-Aminations and α-Oxygenations of Carbonyl Compounds. Angew. Chem. Int. Ed. 2005, 44, 4292–4300. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.R.; Hii, K.K. Transition Metal Catalyzed Enantioselective α-Heterofunctionalization of Carbonyl Compounds. Chem. Rev. 2011, 111, 1637–1656. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Liao, F.-M.; Yu, J.-S.; Zhou, J. Catalytic Asymmetric Electrophilic Amination Reactions to Form Nitrogen-Bearing Tetrasubstituted Carbon Stereocenters. Synthesis 2014, 46, 2983–3003. [Google Scholar] [CrossRef]
- Khose, V.N.; John, M.E.; Pandey, A.D.; Karnik, A.V. Chiral benzimidazoles and their applications in stereodiscrimination processes. Tetrahedron Asymmetry 2017, 28, 1233–1289. [Google Scholar] [CrossRef]
- Nájera, C.; Yus, M. Chiral benzimidazoles as hydrogen bonding organocatalysts. Tetrahedron Lett. 2015, 56, 2623–2633. [Google Scholar] [CrossRef] [Green Version]
- Almaşi, D.; Alonso, D.A.; Gómez-Bengoa, E.; Nájera, C. Chiral 2-Aminobenzimidazoles as Recoverable Organocatalysts for the Addition of 1,3-Dicarbonyl Compounds to Nitroalkenes. J. Org. Chem. 2009, 74, 6163–6168. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Torres, E.; Alonso, D.A.; Gómez-Bengoa, E.; Nájera, C. Conjugate Addition of 1,3-Dicarbonyl Compounds to Maleimides Using a Chiral C2-Symmetric Bis(2-aminobenzimidazole) as Recyclable Organocatalyst. Org. Lett. 2011, 13, 6106–6109. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Torres, E.; Alonso, D.A.; Gómez-Bengoa, E.; Nájera, C. Enantioselective Synthesis of Succinimides by Michael Addition of 1,3-Dicarbonyl Compounds to Maleimides Catalyzed by a Chiral Bis(2-aminobenzimidazole) Organocatalyst. Eur. J. Org. Chem. 2013, 2013, 1434–1440. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Martínez, M.; Alonso, D.A.; Pastor, I.P.; Guillena, G.; Baeza, A. Organocatalyzed Assembly of Chlorinated Quaternary Stereogenic Centers. Asian J. Org. Chem. 2016, 5, 1428–1437. [Google Scholar] [CrossRef]
- Sánchez, D.; Baeza, A.; Alonso, D. Organocatalytic Asymmetric α-Chlorination of 1,3-Dicarbonyl Compounds Catalyzed by 2-Aminobenzimidazole Derivatives. Symmetry 2016, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Trillo, P.; Gómez-Martínez, M.; Alonso, D.A.; Baeza, A. 2-Aminobenzimidazole Organocatalyzed Asymmetric Amination of Cyclic 1,3-Dicarbonyl Compounds. Synlett 2015, 26, 95–100. [Google Scholar]
- Benavent, L.; Puccetti, F.; Baeza, A.; Gómez-Martínez, M. Readily Available Chiral Benzimidazoles-Derived Guanidines as Organocatalysts in the Asymmetric α-Amination of 1,3-Dicarbonyl Compounds. Molecules 2017, 22, 1333. [Google Scholar] [CrossRef] [PubMed]
- Ñíguez, D.R.; Guillena, G.; Alonso, D.A. Chiral 2-Aminobenzimidazoles in Deep Eutectic Mixtures: Recyclable Organocatalysts for the Enantioselective Michael Addition of 1,3-Dicarbonyl Compounds to β-Nitroalkenes. ACS Sustain. Chem. Eng. 2017, 5, 10649–10656. [Google Scholar] [CrossRef]
- Muller, C.R.; Meiners, I.; Domínguez de María, P. Highly enantioselective tandem enzyme–organocatalyst crossed aldol reactions with acetaldehyde in deep-eutectic-solvents. RSC Adv. 2014, 4, 46097–46101. [Google Scholar] [CrossRef]
- Muller, C.R.; Rosen, A.; Domínguez de María, P. Multi-step enzyme-organocatalyst C–C bond forming reactions in deep-eutectic-solvents: Towards improved performances by organocatalyst design. Sustain. Chem. Process 2015, 3, 12–20. [Google Scholar] [CrossRef]
- Martínez, R.; Berbegal, L.; Guillena, G.; Ramón, D.J. Bio-renewable enantioselective aldol reaction in natural deep eutectic solvents. Green Chem. 2016, 18, 1724–1730. [Google Scholar] [CrossRef] [Green Version]
- Fanjul-Mosteirín, N.; Concellon, C.; del Amo, V. l-Isoleucine in a choline chloride/ethylene glycol deep eutectic solvent: A reusable reaction kit for the asymmetric cross-aldol carboligation. Org. Lett. 2016, 18, 4266–4269. [Google Scholar] [CrossRef] [PubMed]
- Brenna, D.; Massolo, E.; Puglisi, A.; Rossi, S.; Celentano, G.; Benaglia, M.; Capriati, V. Towards the development of continuous, organocatalytic, and stereoselective reactions in deep eutectic solvents. Beilstein J. Org. Chem. 2016, 12, 2620–2626. [Google Scholar] [CrossRef] [PubMed]
- Flores-Ferrándiz, J.; Chinchilla, R. Organocatalytic enantioselective conjugate addition of aldehydes to maleimides in deep eutectic solvents. Tetrahedron Asymmetry 2017, 28, 302–306. [Google Scholar] [CrossRef]
- Mason, T.J. Ultrasound in synthetic organic chemistry. Chem. Soc. Rev. 1997, 26, 443–451. [Google Scholar] [CrossRef]
- Chatel, G.; MacFarlane, D.R. Ionic liquids and ultrasound in combination: Synergies and challenges. Chem. Soc. Rev. 2014, 43, 8132–8149. [Google Scholar] [CrossRef] [PubMed]
- For the full catalyst optimization study, see the SI.
- Merad, J.; Lalli, C.; Bernadat, G.; Maury, J.; Masson, G. Enantioselective Brønsted Acid Catalysis as a Tool for the Synthesis of Natural Products and Pharmaceuticals. Chem. Eur. J. 2018, 24, 3925–3943. [Google Scholar] [CrossRef] [PubMed]
- Dibello, E.; Gamenara, D.; Seoane, G. Organocatalysis in the Synthesis of Natural Products: Recent Developments in Aldol and Mannich Reactions, and 1,4-Conjugated Additions. Curr. Organocatal. 2015, 2, 124–149. [Google Scholar] [CrossRef]
- Sun, B.-F. Total synthesis of natural and pharmaceutical products powered by organocatalytic reactions. Tetrahedron Lett. 2015, 2133–2140. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, Y.; Chen, D.F.; Gong, L.-Z. Organocatalytic asymmetric synthesis of chiral nitrogenous heterocycles and natural products. Pure Appl. Chem. 2014, 86, 1217–1226. [Google Scholar] [CrossRef]
- Abbasov, M.E.; Romo, D. The ever-expanding role of asymmetric covalent organocatalysis in scalable, natural product synthesis. Nat. Prod. Rep. 2014, 31, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Xing, H.; Yu, D.-Q.; Liu, H.-M. Catalytic asymmetric synthesis of biologically important 3-hydroxyoxindoles: An update. Beilstein J. Org. Chem. 2016, 12, 1000–1039. [Google Scholar] [CrossRef] [PubMed]
- Macaev, F.Z.; Sucman, N.S.; Boldescu, V.V. Selective transformations of isatins to substituted 2-oxindoles. Russ. Chem. Bull. 2014, 63, 15–25. [Google Scholar] [CrossRef]
- Dalpozzo, R.; Bartoli, G.; Bencivenni, G. Recent advances in organocatalytic methods for the synthesis of disubstituted 2- and 3-indolinones. Chem. Soc. Rev. 2012, 41, 7247–7290. [Google Scholar] [CrossRef] [PubMed]
- Badillo, J.J.; Hanhan, N.V.; Franz, A.K. Enantioselective synthesis of substituted oxindoles and spirooxindoles with applications in drug discovery. Curr. Opin. Drug. Discov. Dev. 2010, 13, 758–776. [Google Scholar]
- Chauhan, P.; Chimni, S.S. Organocatalytic asymmetric synthesis of 3-amino-2-oxindole derivatives bearing a tetra-substituted stereocenter. Tetrahedron Asymmetry 2013, 24, 343–356. [Google Scholar] [CrossRef]
- Yarlagadda, S.; Ramesh, B.; Reddy, C.R.; Srinivas, L.; Sridhar, B.; Subba Reddy, B.V. Organocatalytic Enantioselective Amination of 2-Substituted Indolin-3-ones: A Strategy for the Synthesis of Chiral α-Hydrazino Esters. Org. Lett. 2017, 19, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.-Y.; Wang, Y.; Liu, Y.-Z.; Shen, C.; Xu, P.-F. Organocatalytic Asymmetric Michael Addition of Oxindoles to Nitroolefins for the Synthesis of 2,2-Disubstituted Oxindoles Bearing Adjacent Quaternary and Tertiary Stereocenters. J. Org. Chem. 2012, 77, 11307–11312. [Google Scholar] [CrossRef] [PubMed]
- For the synthesis of catalyst 1 see SI.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | DES | T (°C) | t (h) | Conversion (%) 1 | Ee (%) 2 |
|---|---|---|---|---|---|
| 1 | ChCl/Urea: 1/2 | 25 | 5 | 61 | 77 |
| 2 | ChCl/Urea: 1/2 | 0 | 5 | 94 | 78 |
| 3 | AcChCl/Urea: 1/2 | 25 | 5 | 64 | 72 |
| 4 | AcChCl/Urea: 1/2 | 0 | 5 | 55 | 75 |
| 5 | ChCl/Glycerol: 1/2 | 25 | 5 | 94 | 73 |
| 6 | ChCl/Glycerol: 1/2 | 0 | 5 | 94 | 80 |
| 7 | ChCl/Ethyleneglycol: 1/2 | 25 | 5 | 78 | 72 |
| 8 | ChCl/Ethyleneglycol: 1/2 | 0 | 5 | 84 | 70 |
| 9 | ChCl/Malic acid: 1/1 | 25 | 5 | <5 | nd |
| 10 | ChCl/Tartaric acid: 1/1 | 25 | 5 | 64 | 76 |
| 11 | ChCl/Tartaric acid: 1/1 | 0 | 5 | 58 | 77 |
| 12 | ChCl/Urea: 1/2 | 25 3 | 1 | 92 | 76 |
| 13 | ChCl/Glycerol: 1/2 | 25 3 | 1 | 80 | 80 |

| Entry | Catalyst | DES | Conversion (%) 1 | Ee (%) 2 |
|---|---|---|---|---|
| 1 | 1 | ChCl/Urea: 1/2 | 92 | 76 |
| 2 | 1 | ChCl/Glycerol: 1/2 | 80 | 80 |
| 3 | 2 | ChCl/Urea: 1/2 | 85 | 84 |
| 4 | 2 | ChCl/Glycerol: 1/2 | 90 | 82 |
| 5 | 5 | ChCl/Urea: 1/2 | 95 | 74 |
| 6 | 5 | ChCl/Glycerol: 1/2 | 70 | 78 |
| 7 | 3 | ChCl/Urea: 1/2 | 90 | 40 |
| 8 | 3 | ChCl/Glycerol: 1/2 | 95 | 44 |
| 9 | 6 | ChCl/Urea: 1/2 | 92 | 40 |
| 10 | 6 | ChCl/Glycerol: 1/2 | 91 | 33 |

| Entry | Dicarbonyl | Azodicarboxylate | Product | Yield (%) 1 | Ee (%) 2 |
|---|---|---|---|---|---|
| 1 |  | BocN=NBoc | 4 | 78 | 85 |
| 2 | iPrO2CN=NCO2iPr | 7 | 52 | 60 | |
| 3 | EtO2CN=NCO2Et | 8 | 76 | 65 | |
| 4 | BnO2CN=NCO2Bn | 9 | 0 | nd | |
| 5 |  | BocN=NBoc | 10 | 66 | 36 |
| 6 |  | BocN=NBoc | 11 | 65 | 35 |
| 7 |  | BocN=NBoc | 12 | 65 | 13 |
| 8 |  | BocN=NBoc | 13 | 68 | 25 |
| 9 |  | BocN=NBoc | 14 | 75 | 53 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ros Ñíguez, D.; Khazaeli, P.; Alonso, D.A.; Guillena, G. Deep Eutectic Mixtures as Reaction Media for the Enantioselective Organocatalyzed α-Amination of 1,3-Dicarbonyl Compounds. Catalysts 2018, 8, 217. https://doi.org/10.3390/catal8050217
Ros Ñíguez D, Khazaeli P, Alonso DA, Guillena G. Deep Eutectic Mixtures as Reaction Media for the Enantioselective Organocatalyzed α-Amination of 1,3-Dicarbonyl Compounds. Catalysts. 2018; 8(5):217. https://doi.org/10.3390/catal8050217
Chicago/Turabian StyleRos Ñíguez, Diego, Pegah Khazaeli, Diego A. Alonso, and Gabriela Guillena. 2018. "Deep Eutectic Mixtures as Reaction Media for the Enantioselective Organocatalyzed α-Amination of 1,3-Dicarbonyl Compounds" Catalysts 8, no. 5: 217. https://doi.org/10.3390/catal8050217
APA StyleRos Ñíguez, D., Khazaeli, P., Alonso, D. A., & Guillena, G. (2018). Deep Eutectic Mixtures as Reaction Media for the Enantioselective Organocatalyzed α-Amination of 1,3-Dicarbonyl Compounds. Catalysts, 8(5), 217. https://doi.org/10.3390/catal8050217