Synthesis, Characterization and CO Tolerance Evaluation in PEMFCs of Pt2RuMo Electrocatalysts



Abstract
:
1. Introduction
2. Results and Discussion
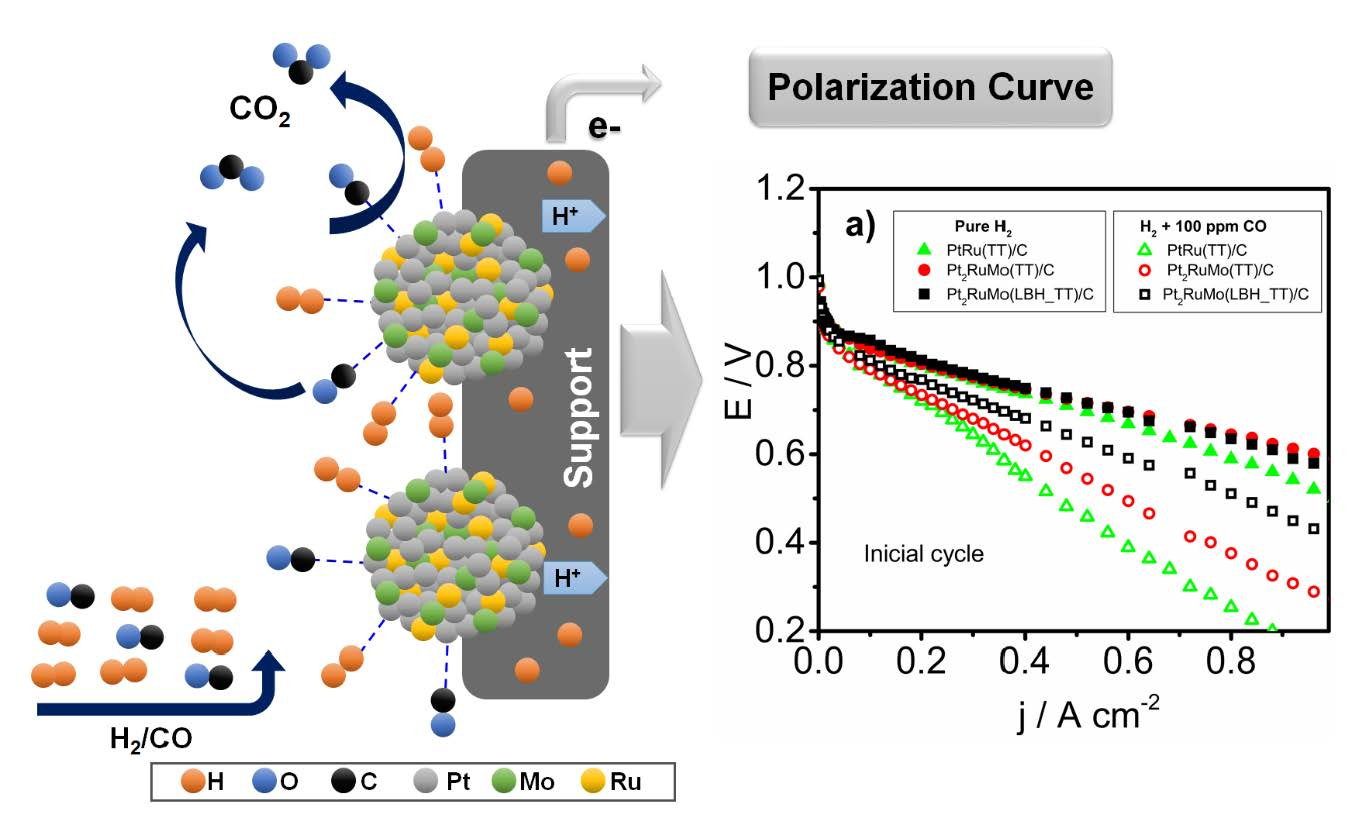
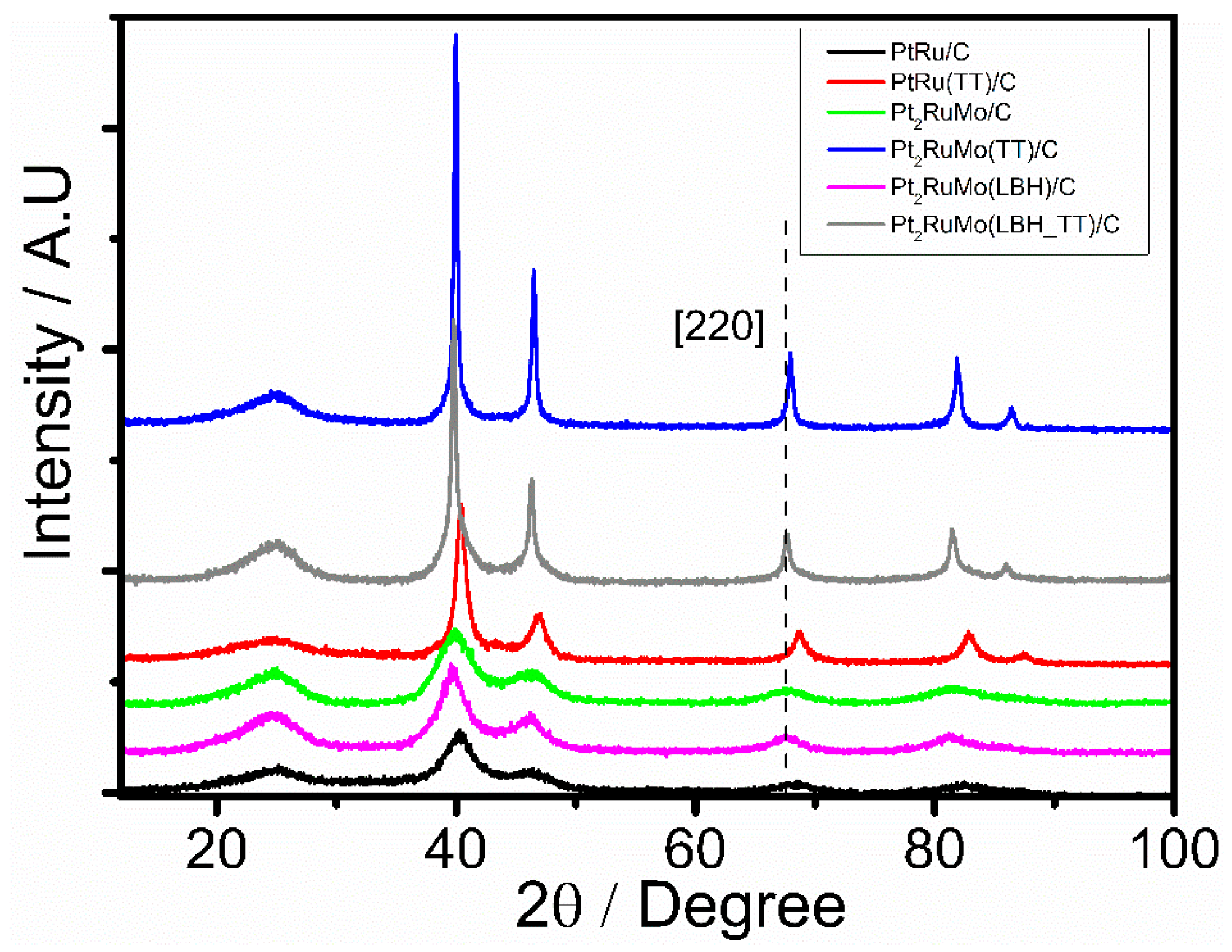
2.1. Physical Characterization
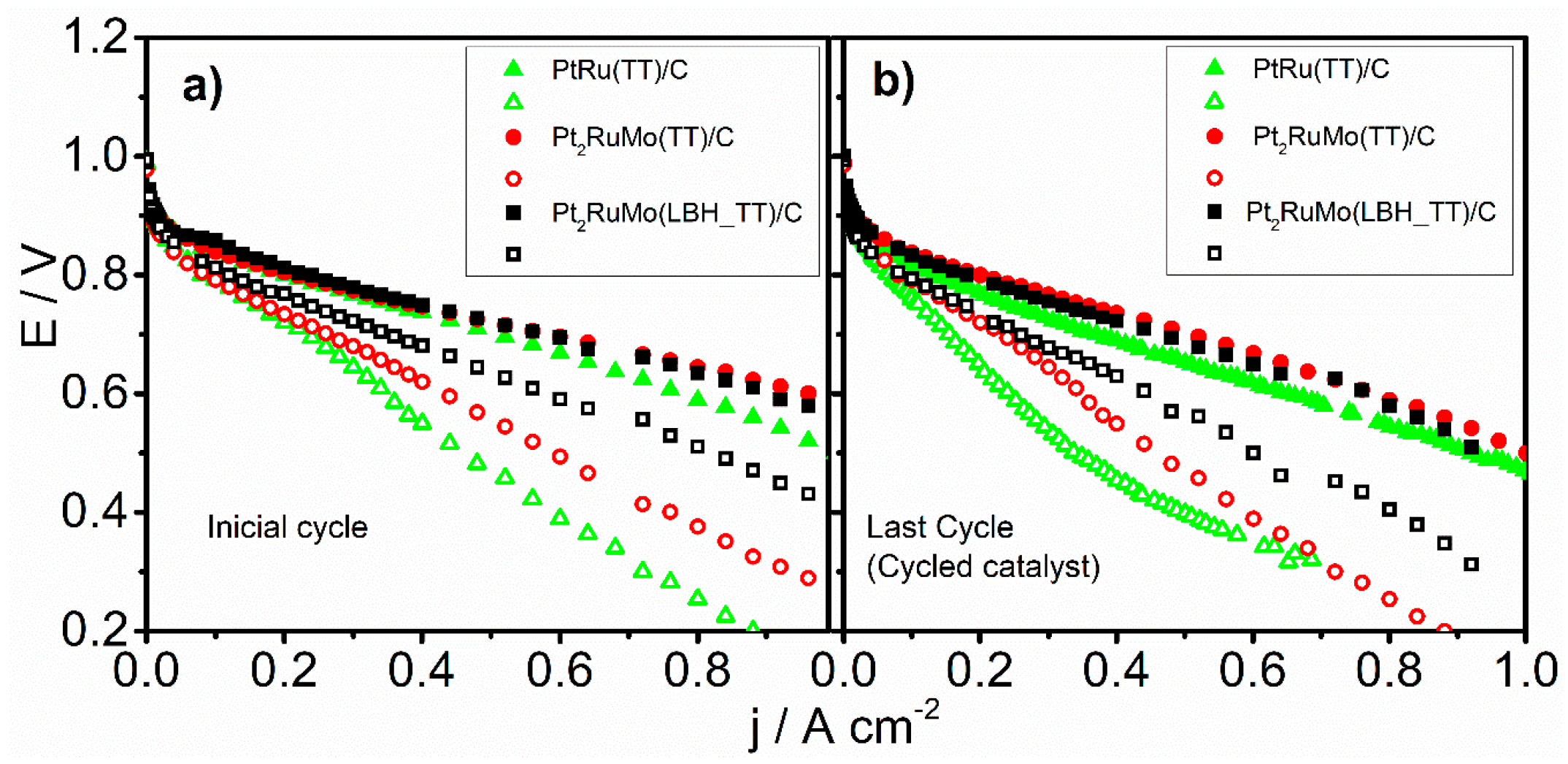
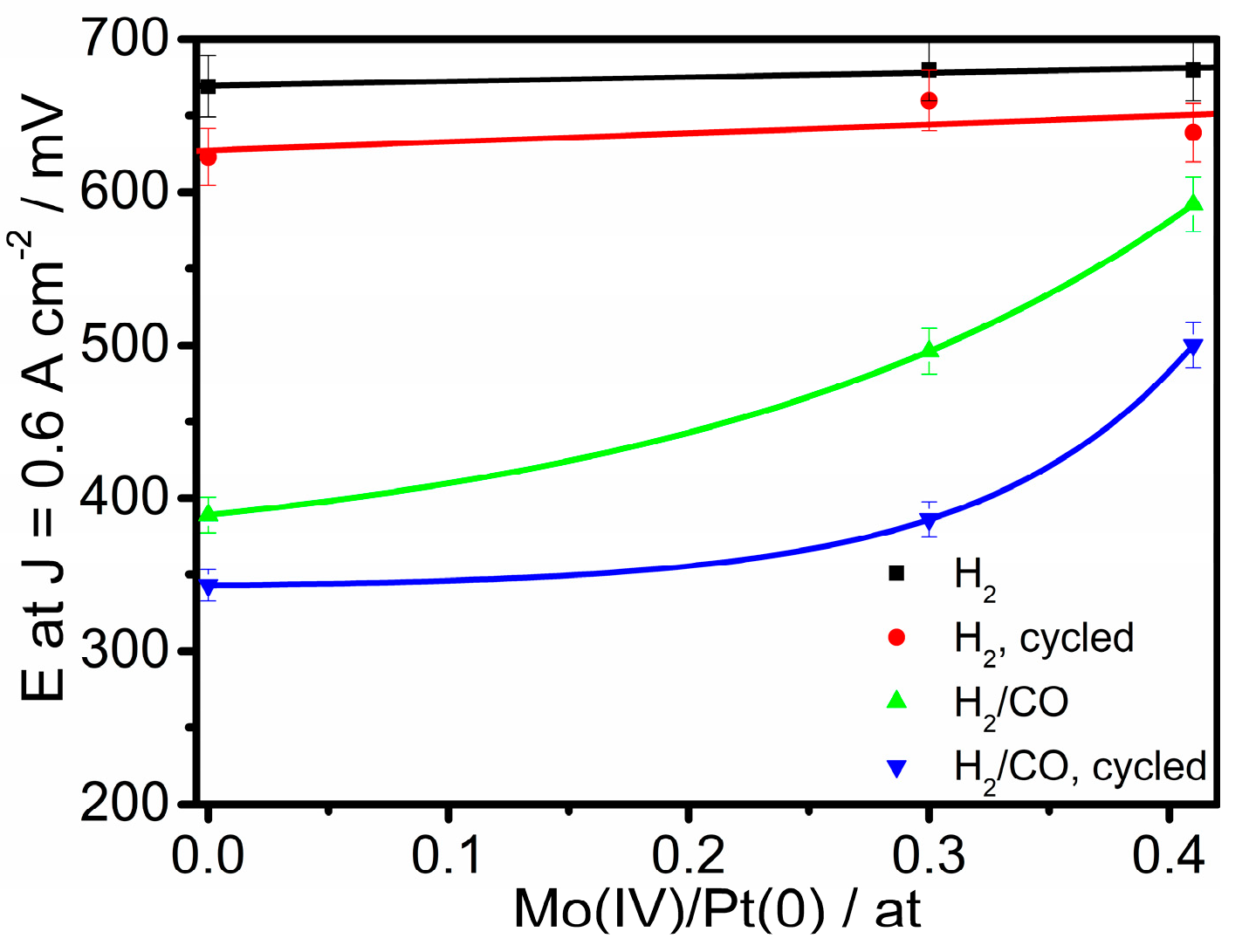
2.2. Electrochemical Characterization
3. Materials and Methods
3.1. Synthesis of the Catalysts
3.2. Physical Characterization
3.3. Electrochemical Measurements
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McBreen, J.; Mukerjee, S. In Situ X-ray Absorption Studies of a Pt-Ru Electrocatalyst. J. Electrochem. Soc. 1995, 142, 3399–3404. [Google Scholar] [CrossRef]
- Wagner, N.; Schulze, M. Change of electrochemical impedance spectra during CO poisoning of the Pt and Pt–Ru anodes in a membrane fuel cell (PEFC). Electrochim. Acta 2003, 48, 3899–3907. [Google Scholar] [CrossRef]
- Dimakis, N.; Cowan, M.; Hanson, G.; Smotkin, E.S. Attraction−Repulsion Mechanism for Carbon Monoxide Adsorption on Platinum and Platinum−Ruthenium Alloys. J. Phys. Chem. C 2009, 113, 18730–18739. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, H.; Fujino, T.; Watanabe, M. Hydrogen electro-oxidation on platinum catalysts in the presence of trace carbon monoxide. J. Electroanal. Chem. 1995, 391, 119–123. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Abild-Pedersen, F.; Studt, F.; Bligaard, T. Density functional theory in surface chemistry and catalysis. Proc. Natl. Acad. Sci. 2011, 108, 937–943. [Google Scholar] [CrossRef] [Green Version]
- Bligaard, T.; Nørskov, J.K. Ligand effects in heterogeneous catalysis and electrochemistry. Electrochim. Acta 2007, 52, 5512–5516. [Google Scholar] [CrossRef]
- Mavrikakis, M.; Hammer, B.; Nørskov, J.K. Effect of Strain on the Reactivity of Metal Surfaces. Phys. Rev. Lett. 1998, 81, 2819–2822. [Google Scholar] [CrossRef] [Green Version]
- Dimakis, N.; Flor, F.A.; Navarro, N.E.; Salgado, A.; Smotkin, E.S. Adsorption of Carbon Monoxide on Platinum–Ruthenium, Platinum–Osmium, Platinum–Ruthenium–Osmium, and Platinum–Ruthenium–Osmium–Iridium Alloys. J. Phys. Chem. C 2016, 120, 10427–10441. [Google Scholar] [CrossRef]
- Ianniello, R.; Schmidt, V.M.; Stimming, U.; Stumper, J.; Wallau, A. CO adsorption and oxidation on Pt and Pt Ru alloys: dependence on substrate composition. Electrochim. Acta 1994, 39, 1863–1869. [Google Scholar] [CrossRef]
- Obradović, M.; Gojkovic, S. Electrochemical Instability of Pt Nanoparticles Probed by Formic Acid Oxidation. J. Electroanal. Chem. 2011, 664. [Google Scholar] [CrossRef]
- Igarashi, H.; Fujino, T.; Zhu, Y.; Uchida, H.; Watanabe, M. CO Tolerance of Pt alloy electrocatalysts for polymer electrolyte fuel cells and the detoxification mechanism. Phys. Chem. Chem. Phys. 2001, 3, 306–314. [Google Scholar] [CrossRef]
- Dimakis, N.; Iddir, H.; Díaz-Morales, R.R.; Liu, R.; Bunker, G.; Chung, E.-H.; Smotkin, E.S. A Band Dispersion Mechanism for Pt Alloy Compositional Tuning of Linear Bound CO Stretching Frequencies. J. Phys. Chem. B 2005, 109, 1839–1848. [Google Scholar] [CrossRef]
- Watanabe, M.; Motoo, S. Electrocatalysis by ad-atoms: Part II. Enhancement of the oxidation of methanol on platinum by ruthenium ad-atoms. J. Electroanal. Chem. Interfacial Electrochem. 1975, 60, 267–273. [Google Scholar] [CrossRef]
- Urian, R.C.; Gullá, A.F.; Mukerjee, S. Electrocatalysis of reformate tolerance in proton exchange membranes fuel cells: Part I. J. Electroanal. Chem. 2003, 554–555, 307–324. [Google Scholar] [CrossRef]
- Santiago, E.I.; Camara, G.A.; Ticianelli, E.A. CO tolerance on PtMo/C electrocatalysts prepared by the formic acid method. Electrochim. Acta 2003, 48, 3527–3534. [Google Scholar] [CrossRef]
- Maillard, F.; Lu, G.Q.; Wieckowski, A.; Stimming, U. Ru-Decorated Pt Surfaces as Model Fuel Cell Electrocatalysts for CO Electrooxidation. J. Phys. Chem. B 2005, 109, 16230–16243. [Google Scholar] [CrossRef]
- Ioroi, T.; Akita, T.; Yamazaki, S.-I.; Siroma, Z.; Fujiwara, N.; Yasuda, K. Comparative study of carbon-supported Pt/Mo-oxide and PtRu for use as CO-tolerant anode catalysts. Electrochim. Acta 2006, 52, 491–498. [Google Scholar] [CrossRef]
- Alcaide, F.; Álvarez, G.; Tsiouvaras, N.; Peña, M.A.; Fierro, J.L.G.; Martínez-Huerta, M.V. Electrooxidation of H2/CO on carbon-supported PtRu-MoOx nanoparticles for polymer electrolyte fuel cells. Int. J. Hydrogen Energy 2011, 36, 14590–14598. [Google Scholar] [CrossRef]
- Liang, Y.M.; Zhang, H.M.; Zhong, H.X.; Zhu, X.B.; Tian, Z.Q.; Xu, D.Y.; Yi, B.L. Preparation and characterization of carbon-supported PtRuIr catalyst with excellent CO-tolerant performance for proton-exchange membrane fuel cells. J. Catal. 2006, 238, 468–476. [Google Scholar] [CrossRef]
- Stevens, D.A.; Rouleau, J.M.; Mar, R.E.; Atanasoski, R.T.; Schmoeckel, A.K.; Debe, M.K.; Dahn, J.R. Enhanced CO-Tolerance of Pt–Ru–Mo Hydrogen Oxidation Catalysts. J. Electrochem. Soc. 2007, 154, B1211–B1219. [Google Scholar] [CrossRef]
- Martínez-Huerta, M.V.; Tsiouvaras, N.; Peña, M.A.; Fierro, J.L.G.; Rodríguez, J.L.; Pastor, E. Electrochemical activation of nanostructured carbon-supported PtRuMo electrocatalyst for methanol oxidation. Electrochim. Acta 2010, 55, 7634–7642. [Google Scholar] [CrossRef]
- Suo, C.; Zhang, W.; Shi, X.; Ma, C. Investigation of nano Pt and Pt-based alloys electrocatalysts for direct methanol fuel cells and their properties. AIP Adv. 2014, 4, 031340. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, F.; Chan, K.-Y. Synthesis of Pt–Ru–Mo ternary metal nanoparticles by microemulsions, their characterization and electrocatalytic properties. J. Mater. Sci. 2004, 39, 5845–5848. [Google Scholar] [CrossRef]
- Zhou, W.J.; Li, W.Z.; Song, S.Q.; Zhou, Z.H.; Jiang, L.H.; Sun, G.Q.; Xin, Q.; Poulianitis, K.; Kontou, S.; Tsiakaras, P. Bi- and tri-metallic Pt-based anode catalysts for direct ethanol fuel cells. J. Power Sources 2004, 131, 217–223. [Google Scholar] [CrossRef]
- Chen, S.; Ye, F.; Lin, W. Effect of operating conditions on the performance of a direct methanol fuel cell with PtRuMo/CNTs as anode catalyst. Int. J. Hydrogen Energy 2010, 35, 8225–8233. [Google Scholar] [CrossRef]
- Rooksby, H.P.; Lewis, B. Relations between the structures of phases in the system platinum-molybdenum. J. Less Common Met. 1964, 6, 451–460. [Google Scholar] [CrossRef]
- Martinez-Huerta, M.V.; Tsiouvaras, N.; García, G.; Pena, M.A.; Pastor, E.; Rodríguez, J.L.; Fierro, J.L.G. Carbon-Supported PtRuMo Electrocatalysts for Direct Alcohol Fuel Cells. Catalysts 2013, 3, 811–838. [Google Scholar] [CrossRef] [Green Version]
- Antolini, E.; Cardellini, F.; Giorgi, L.; Passalacqua, E. Effect of Me (Pt+Ru) content in Me/C catalysts on PtRu alloy formation: An XRD analysis. J. Mater. Sci. Lett. 2000, 19, 2099–2103. [Google Scholar] [CrossRef]
- Antolini, E.; Cardellini, F. Formation of carbon supported PtRu alloys: An XRD analysis. J. Alloys Compd. 2001, 315, 118–122. [Google Scholar] [CrossRef]
- El-Hinnawi, M.A.; El-Qaseer, A.K. Formation of lithium cyclopentadienide by the reaction of Li(C2H5)3BH with cyclopentadiene, and a convenient preparation of [M(CO)3(C5H5)] − (M = Cr, Mo and W). J. Organomet. Chem. 1985, 281, 119–122. [Google Scholar] [CrossRef]
- Kaydashev, V.; Janssens, E.; Lievens, P. Tolerance of platinum clusters to CO poisoning induced by molybdenum doping. Int. J. Mass Spectrom. 2015, 379, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Samjeské, G.; Wang, H.; Löffler, T.; Baltruschat, H. CO and methanol oxidation at Pt-electrodes modified by Mo. Electrochim. Acta 2002, 47, 3681–3692. [Google Scholar] [CrossRef]
- Sun, S.; Murray, C.B.; Weller, D.; Folks, L.; Moser, A. Monodisperse FePt Nanoparticles and Ferromagnetic FePt Nanocrystal Superlattices. Science 2000, 287, 1989–1992. [Google Scholar] [CrossRef] [PubMed]
- Fievet, F.; Lagier, J.P.; Figlarz, M. Preparing monodisperse metal powders in micrometer and submicrometer sizes by poliol process. MRS Bull. 1989, 14, 29–34. [Google Scholar] [CrossRef]
- Pinheiro, A.L.N.; Oliveira-Neto, A.; de Souza, E.C.; Perez, J.; Paganin, V.A.; Ticianelli, E.A.; Gonzalez, E.R. Electrocatalysis on noble metal and noble metal alloys dispersed on high surface area carbon. J. New Mater. Electrochem. Syst. 2003, 6, 1–8. [Google Scholar]
- Nepel, T.C.M.; Lopes, P.P.; Paganin, V.A.; Ticianelli, E.A. CO tolerance of proton exchange membrane fuel cells with Pt/C and PtMo/C anodes operating at high temperatures: A mass spectrometry investigation. Electrochim. Acta 2013, 88, 217–224. [Google Scholar] [CrossRef]
- Paganin, V.A.; Ticianelli, E.A.; Gonzalez, E.R. Development and electrochemical studies of gas diffusion electrodes for polymer electrolyte fuel cells. J. Appl. Electrochem. 1996, 26, 297–304. [Google Scholar] [CrossRef]
- Bonnet, C.; Franck-Lacaze, L.; Ronasi, S.; Besse, S.; Lapicque, F. PEM fuel cell Pt anode inhibition by carbon monoxide: Non-uniform behaviour of the cell caused by the finite hydrogen excess. Chem. Eng. Sci. 2010, 65, 3050–3058. [Google Scholar] [CrossRef]
- Zhang, S.; Yuan, X.-Z.; Hin, J.N.C.; Wang, H.; Friedrich, K.A.; Schulze, M. A review of platinum-based catalyst layer degradation in proton exchange membrane fuel cells. J. Power Sources 2009, 194, 588–600. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | EDX Composition | a (Å) | xRu (at%) | A.D. (%) | dc (nm) | dn (nm) | |
|---|---|---|---|---|---|---|---|
| Pt:Ru:Mo | Pt:Ru | ||||||
| PtRu(TT)/C | 48:52:0 | 48:52 | 3.863 | 49 | 89 | 8.4 | 4.1 |
| Pt2RuMo(TT)/C | 55:19:26 | 74:26 | 3.900 | 19 | 67 | 19.5 | 4.2 |
| Pt2RuMo(LBH_TT)/C | 47:26:27 | 64:36 | 3.914 | 8 | 15 | 14.9 | 3.7 |
| Catalysts | Pt 4f | Ru 3p | Mo 3d | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Pt0 | Pt2+ | Pt4+ | Ru0 | Ru2+ | Ru4+ | Mo0 | Mo4+ | Mo6+ | |
| Pt-Ru(TT)/C | 70.0 | 18.5 | 11.5 | 36.0 | 46.3 | 17.7 | |||
| Pt2RuMo(TT)/C | 60.7 | 27.6 | 11.7 | 44.6 | 30.6 | 24.8 | 55.6 | 31.7 | 12.7 |
| Pt2RuMo(LBH_TT)/C | 63.2 | 24.9 | 11.8 | 35.5 | 43.3 | 21.2 | 27.7 | 45.1 | 27.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Hernández, M.; Antolini, E.; Perez, J. Synthesis, Characterization and CO Tolerance Evaluation in PEMFCs of Pt2RuMo Electrocatalysts. Catalysts 2019, 9, 61. https://doi.org/10.3390/catal9010061
González-Hernández M, Antolini E, Perez J. Synthesis, Characterization and CO Tolerance Evaluation in PEMFCs of Pt2RuMo Electrocatalysts. Catalysts. 2019; 9(1):61. https://doi.org/10.3390/catal9010061
Chicago/Turabian StyleGonzález-Hernández, Martin, Ermete Antolini, and Joelma Perez. 2019. "Synthesis, Characterization and CO Tolerance Evaluation in PEMFCs of Pt2RuMo Electrocatalysts" Catalysts 9, no. 1: 61. https://doi.org/10.3390/catal9010061
APA StyleGonzález-Hernández, M., Antolini, E., & Perez, J. (2019). Synthesis, Characterization and CO Tolerance Evaluation in PEMFCs of Pt2RuMo Electrocatalysts. Catalysts, 9(1), 61. https://doi.org/10.3390/catal9010061