Synthesis, X-ray Single Crystal, Conformational Analysis and Cholinesterase Inhibitory Activity of a New Spiropyrrolidine Scaffold Tethered Benzo[b]Thiophene Analogue

 ,
,  ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. General Methods
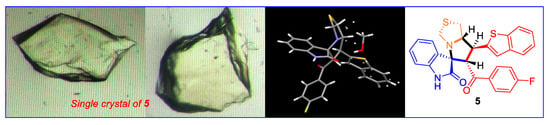
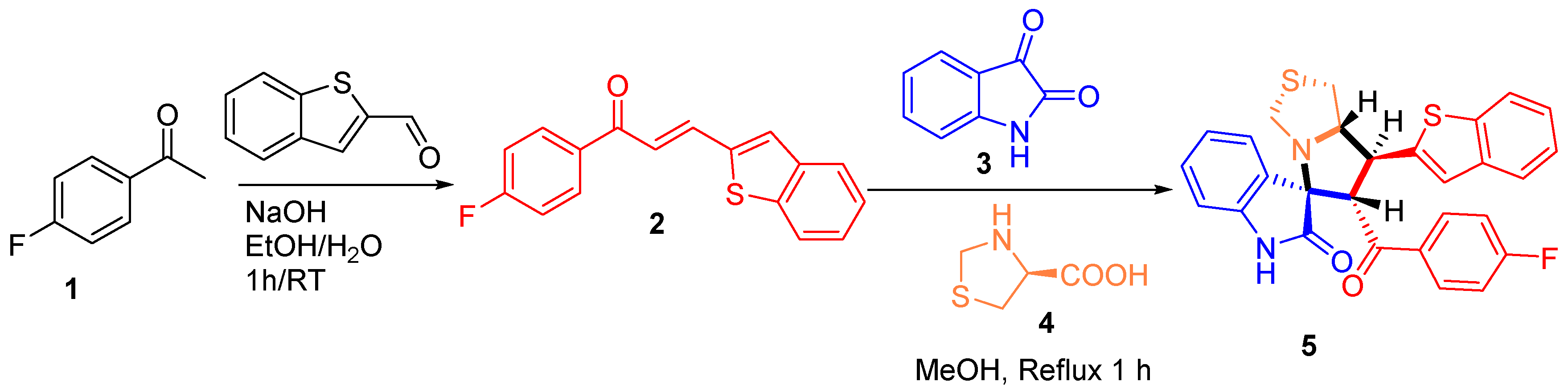
2.2. Synthesis of 7'-(Benzo[b]thiophen-2-yl)-6'-(4-fluorobenzoyl)-1',6',7',7a'-tetrahydro-3'H-spiro[indoline- 3,5'-pyrrolo[1,2-c]thiazol]-2-one (5)
2.3. Single-Crystal X-ray Diffraction Analysis
2.4. Hirshfeld Surface Analysis
2.5. Computational Methods
3. Results and Discussion
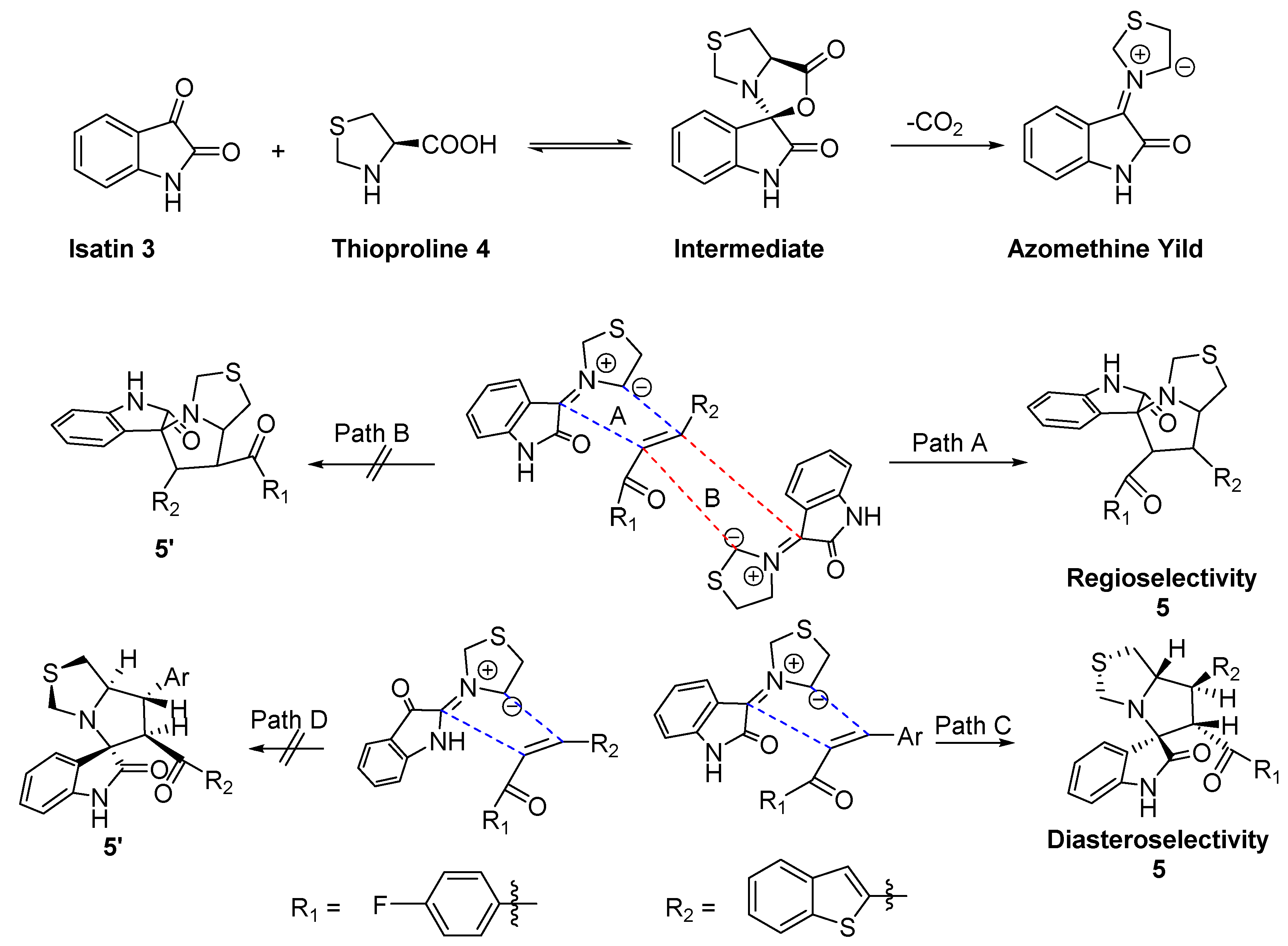
3.1. Chemistry
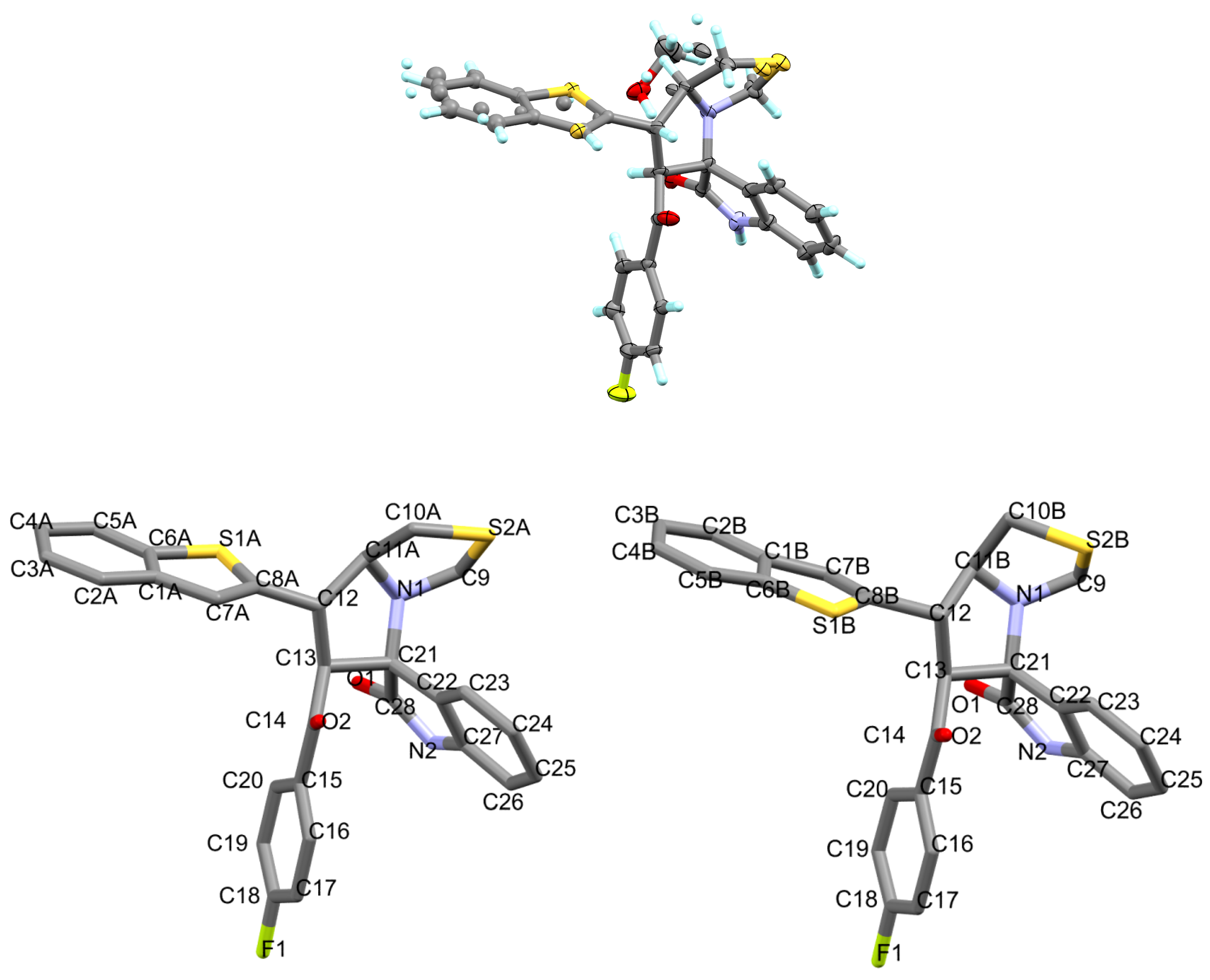
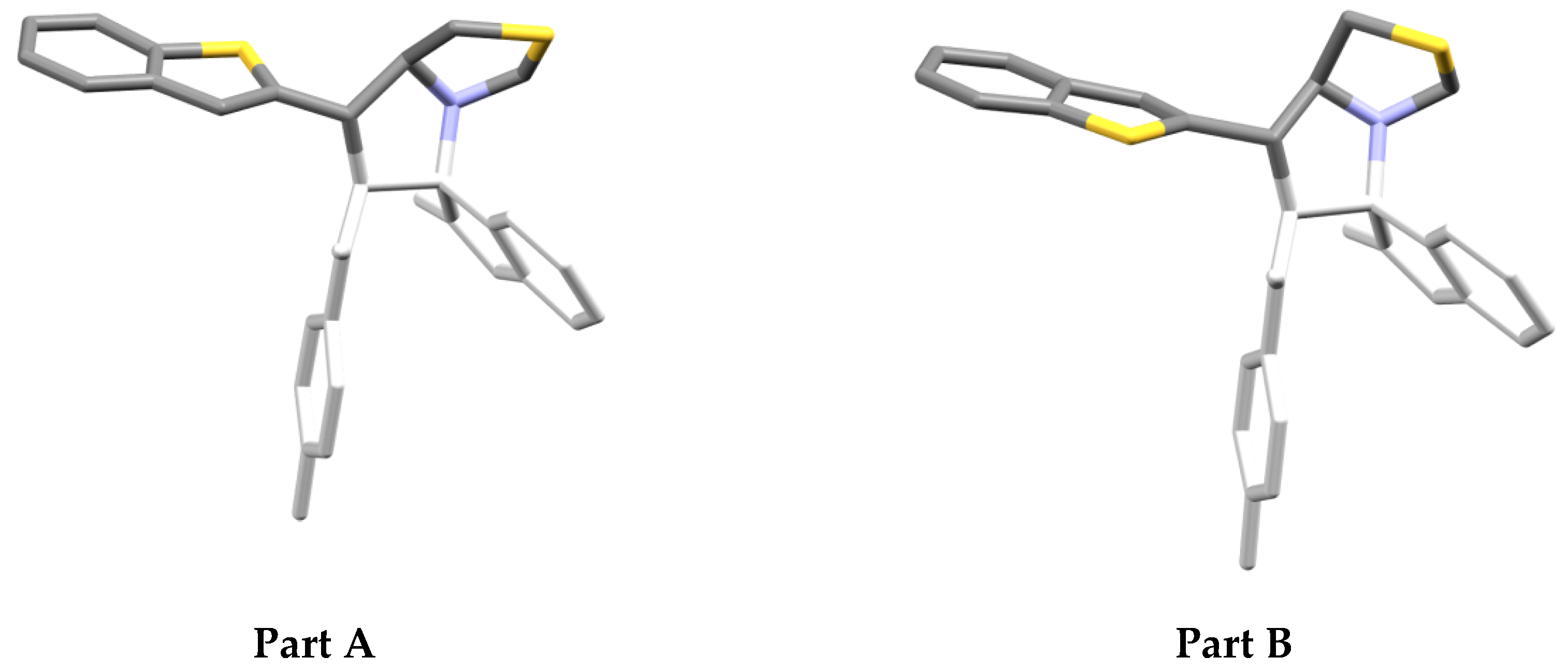
3.2. Crystal Structure Discription
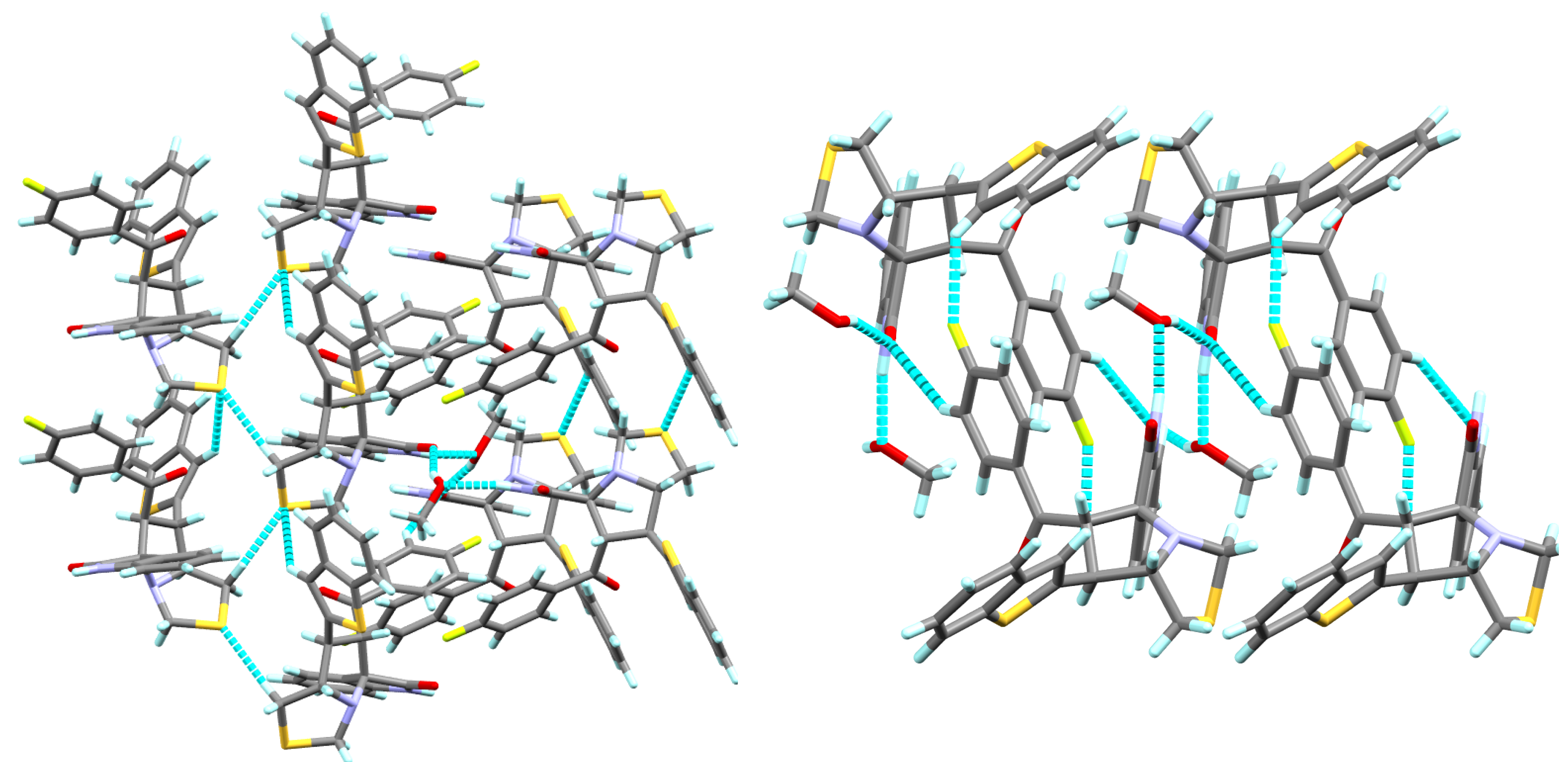
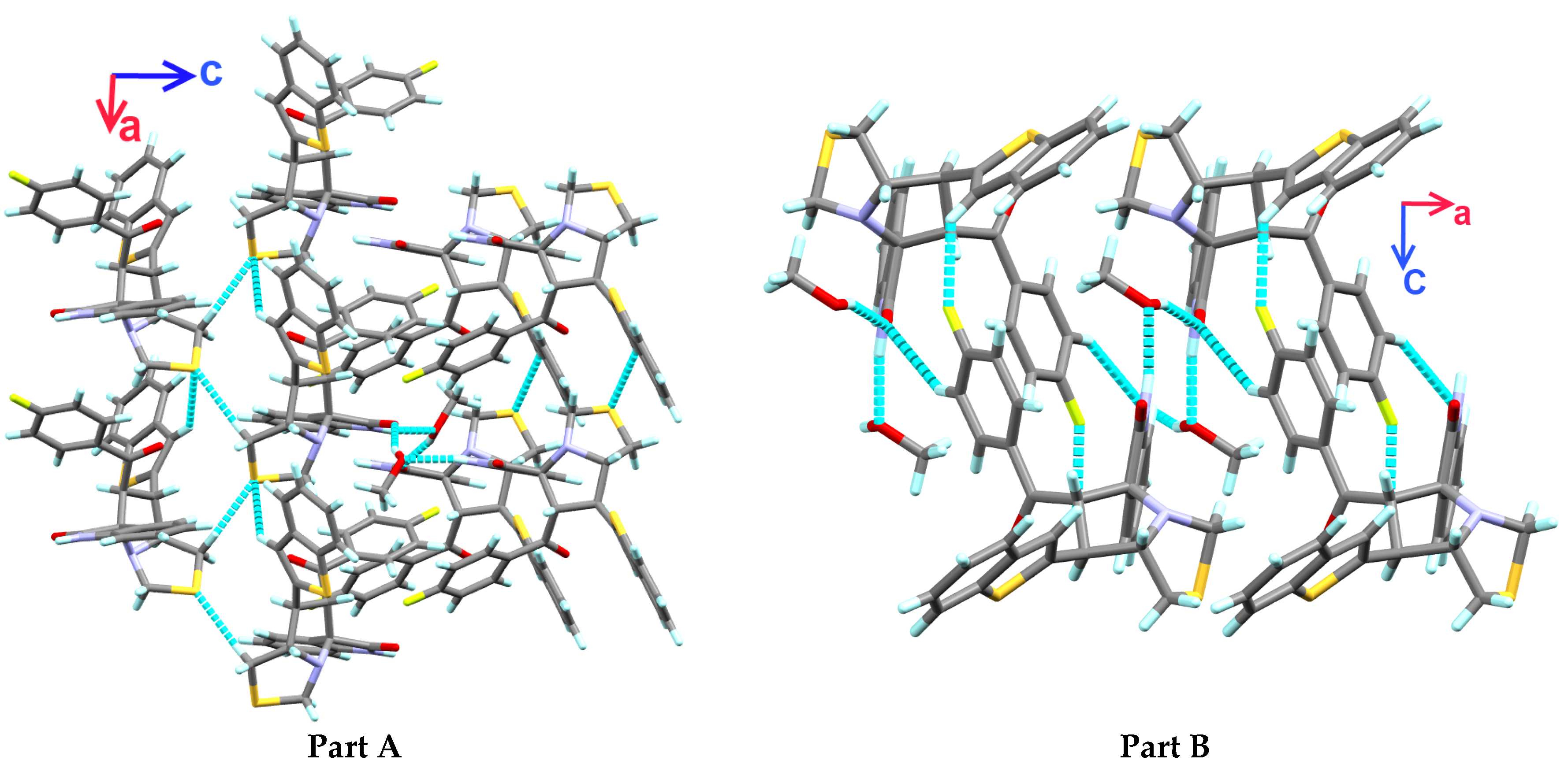
3.3. Crystal Packing
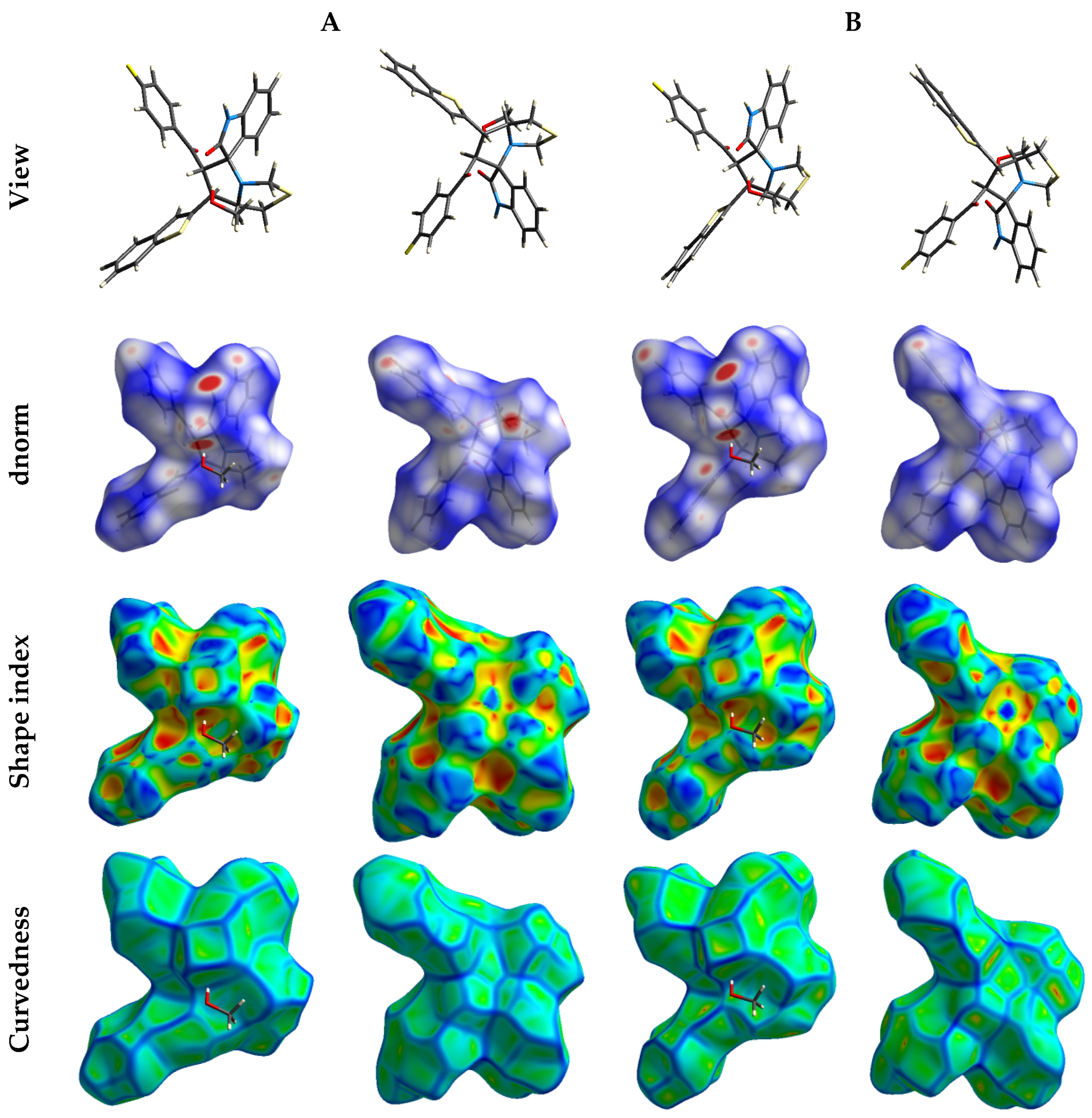
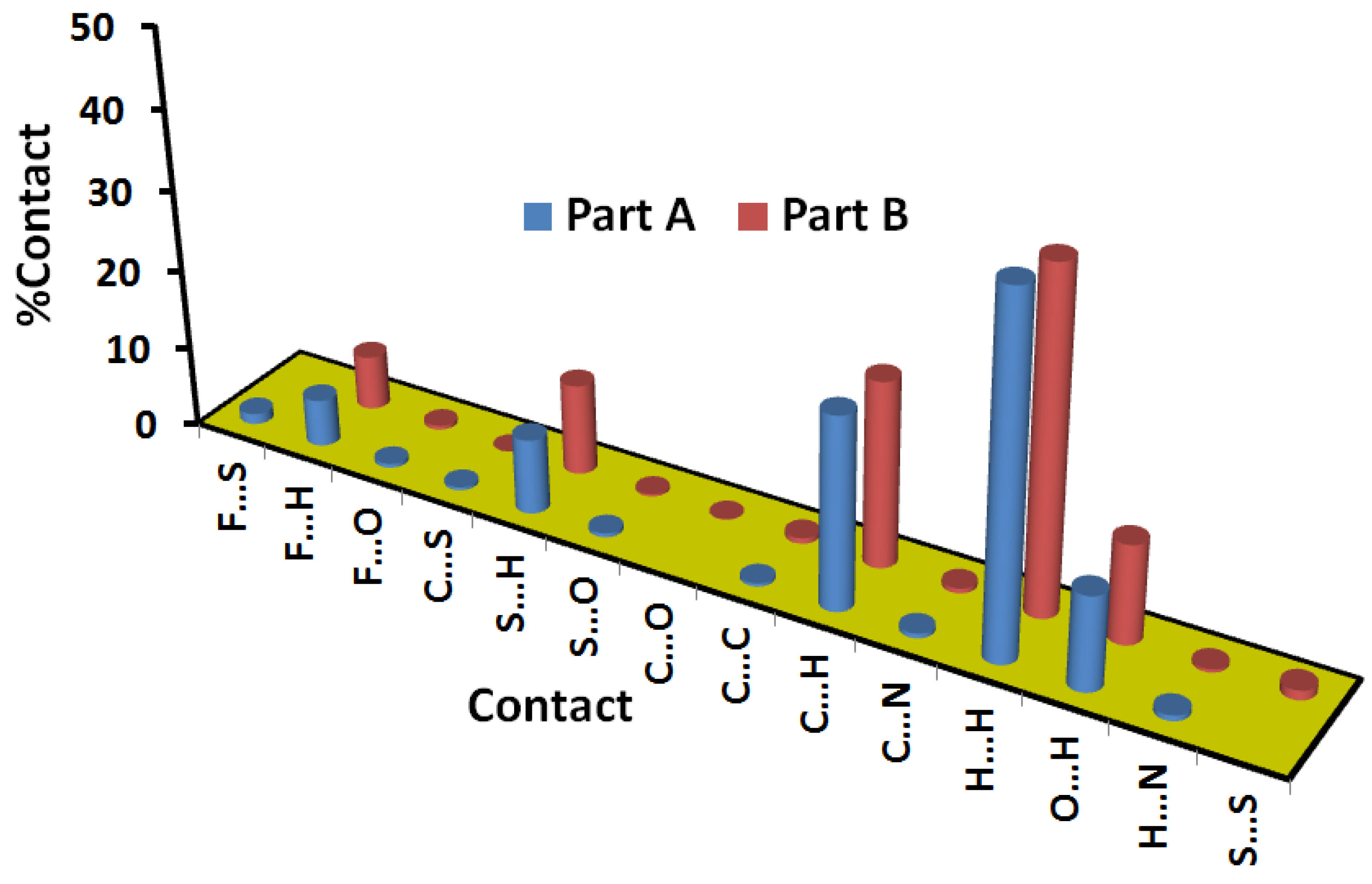
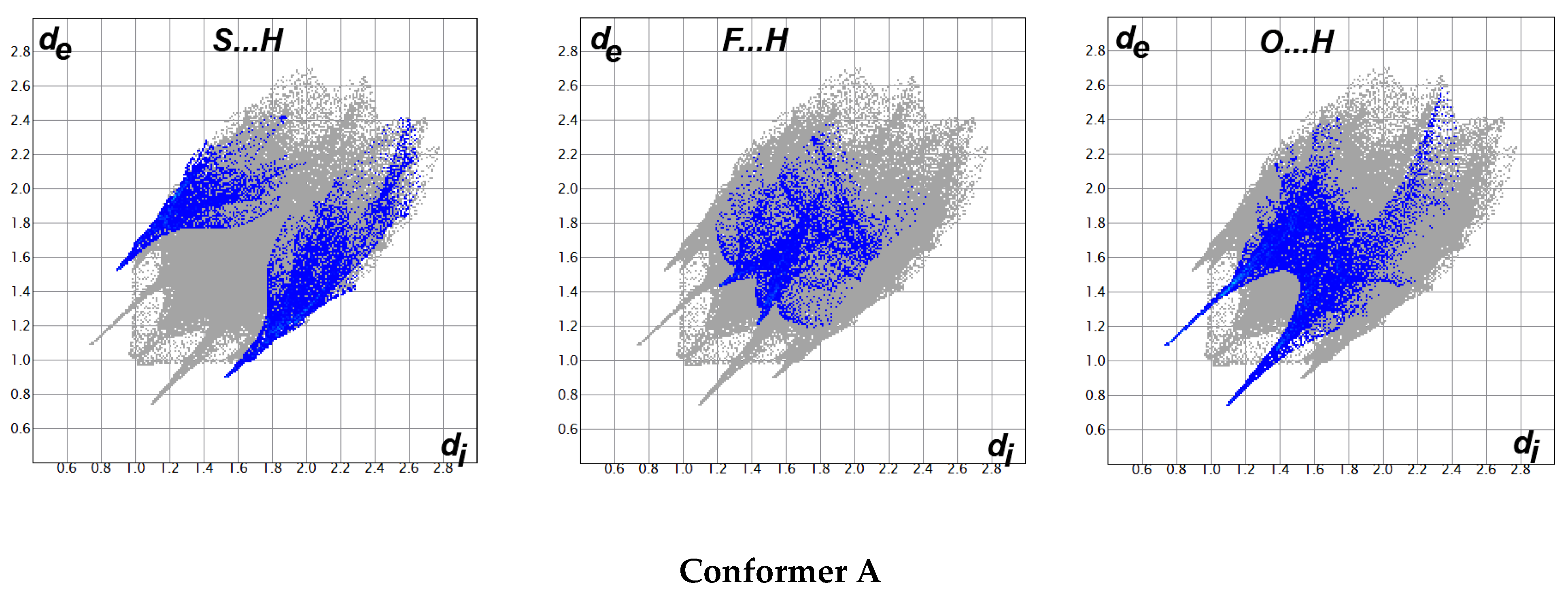
3.4. Hirshfeld Analysis of the Molecular Packing
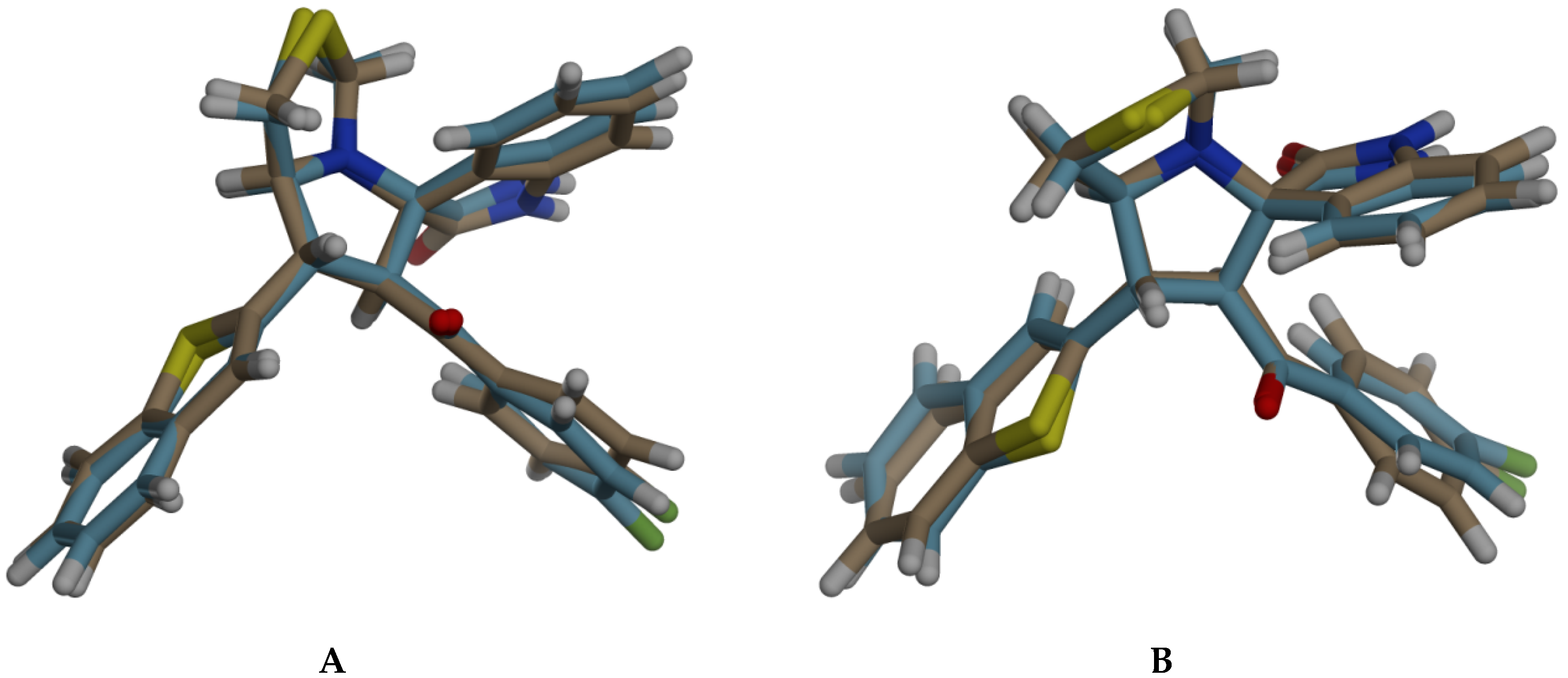
3.5. DFT Studies
3.6. Biological Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar]
- Genç, H.; Kalin, R.; Köksal, Z.; Sadeghian, N.; Kocyigit, U.M.; Zengin, M.; Gülçin, İ.; Özdemir, H. Discovery of Potent Carbonic Anhydrase and Acetylcholinesterase Inhibitors: 2-Aminoindan β-Lactam Derivatives. Int. J. Mol. Sci. 2016, 17, 1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topal, F.; Gulcin, I.; Dastan, A.; Guney, M. Novel eugenol derivatives: Potent acetylcholinesterase and carbonic anhydrase inhibitors. Int. J. Biol. Macromol. 2017, 94, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Kia, Y.; Osman, H.; Kumar, R.S.; Murugaiyah, V.; Basiri, A.; Perumal, S.; Wahab, H.A.; Bing, C.S. Synthesis and discovery of novel piperidone-grafted mono-and bis-spirooxindole. Bioorg. Med. Chem. 2013, 21, 1696–1707. [Google Scholar] [CrossRef]
- Kia, Y.; Osman, H.; Kumar, R.S.; Murugaiyah, V.; Basiri, A.; Perumal, S.; Razak, I.A. A facile chemo-, regio-and stereoselective synthesis and cholinesterase inhibitory activity of spirooxindole–pyrrolizine–piperidine hybrids. Bioorg. Med. Chem. Lett. 2013, 23, 2979–2983. [Google Scholar] [CrossRef]
- Keri, R.S.; Chand, K.; Budagumpi, S.; Somappa, S.B.; Patil, S.A.; Nagaraja, B.M. An overview of benzo [b] thiophene-based medicinal chemistry. Eur. J. Med. Chem. 2017, 138, 1002–1033. [Google Scholar] [CrossRef]
- Meixner, C.N.; Aref, M.W.; Gupta, A.; McNerny, E.M.B.; Brown, D.; Wallace, J.M.; Allen, M.R. Raloxifene mproves Bone Mechanical Properties in Mice Previously Treated with Zoledronate. Calcif. Tissue Int. 2017, 101, 75. [Google Scholar] [CrossRef]
- Croxtall, J.D.; Plosker, G.L. Sertaconazole: A review of its use in the management of superficial mycoses in dermatology and gynaecology. Drugs 2009, 69, 339–359. [Google Scholar] [CrossRef]
- Sarret, C.; Pichard, S.; Afenjar, A.; Boespflug-Tanguy, O. Lack of long-term neurologic efficacy of zileuton in Sjogren-Larsson’s syndrome. Neuropediatrics 2017, 48, 205–206. [Google Scholar]
- Vogel, V.G.; Costantino, J.P.; Wickerham, D.L.; Cronin, W.M.; Cecchini, R.S.; Atkins, J.N.; Bevers, T.B.; Fehrenbacher, L.; Pajon, E.R.; Wade, J.L.; et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: The NSABP study of tamoxifen and raloxifene (STAR) P-2 trial. JAMA 2006, 295, 2727–2741. [Google Scholar] [CrossRef] [Green Version]
- Chonan, T.; Wakasugi, D.; Yamamoto, D.; Yashiro, M.; Oi, T.; Tanaka, H.; Ohoka-Sugita, A.; Io, F.; Koretsune, H.; Hiratate, A. Discovery of novel (4-piperidinyl)-piperazines as potent and orally active acetyl-CoA carboxylase 1/2 non-selective inhibitors: F-Boc and triF-Boc groups are acid-stable bioisosteres for the Boc group. Bioorg. Med. Chem. 2011, 19, 1580–1593. [Google Scholar] [CrossRef] [PubMed]
- Pinney, K.G.; Bounds, A.D.; Dingeman, K.M.; Mocharla, V.P.; Pettit, G.R.; Bai, R.; Hamel, E. A new anti-tubulin agent containing the benzo [b] thiophene ring system. Bioorg. Med. Chem. Lett. 1999, 9, 1081–1086. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cara, C.L.; Preti, D.; Fruttarolo, F.; Pavani, M.G.; Tabrizi, M.A.; Tolomeo, M.; Grimaudo, S.; et al. Synthesis and biological evaluation of 2-and 3-aminobenzo [b] thiophene derivatives as antimitotic agents and inhibitors of tubulin polymerization. J. Med. Chem. 2007, 50, 2273–2277. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.C.; Moon, B.S.; Lee, J.H.; Chung, K.H.; Katzenellenbogen, J.A.; Chi, D.Y. Synthesis and binding affinities of fluoroalkylated raloxifenes. Bioorg. Med. Chem. 2003, 11, 3649–3658. [Google Scholar] [CrossRef]
- Zaher, A.F.; Khalil, N.A.; Ahmed, E.M. Synthesis and anticonvulsant activity of new 3′-aryl-7-bromo-spiro[[1]benzothiophene-3,2′-[1,3] thiazolidine]-2,4′-dione derivatives. Orient. J. Chem. 2010, 26, 1241–1248. [Google Scholar]
- Moinet, G.; Leriche, C.; Kergoat, M. Antidiabetic Compounds Comprising Benzofuran and Benzothiophene Derivatives. EP1685121 A1. PCT/EP2004/012075, 11 June 2008. [Google Scholar]
- Rao, G.K.; Subramaniam, R. Synthesis, antitubercular and antibacterial activities of some quinazolinone analogs substituted with benzothiophene. Chem. Sci. J. 2015, 6, 92–96. [Google Scholar] [CrossRef]
- Jagtap, V.A.; Agasimundin, Y.S. Synthesis and preliminary evaluation of some 2-amino-n′-[substituted]-4,5,6,7-tetrahydro-1-benzothiophene-3-carbohydrazide as antimicrobial agents. J. Pharm. Res. 2015, 9, 10–14. [Google Scholar]
- Mourey, R.J.; Burnette, B.L.; Brustkern, S.J.; Daniels, J.S.; Hirsch, J.L.; Hood Meyers, M.J.; Mnich, S.J.; Pierce, B.S.; Saabye, M.J.; Schindler, J.F.; et al. A benzothiophene inhibitor of mitogen-activated protein kinase-activated protein kinase 2 inhibits tumor necrosis factor α production and has oral anti-inflammatory efficacy in acute and chronic models of inflammation. J. Pharmacol. Exp. Ther. 2010, 333, 797–807. [Google Scholar] [CrossRef] [Green Version]
- Rackham, M.D.; Brannigan, J.A.; Moss, D.K.; Yu, Z.; Wilkinson, A.J.; Holder, A.A.; Tate, E.W.; Leatherbarrow, R.J. Discovery of novel and ligand-efficient inhibitors of plasmodium falciparum and plasmodium vivax N-myristoyltransferase. J. Med. Chem. 2013, 56, 371–375. [Google Scholar] [CrossRef]
- Bruker, A. SAINT Software Reference Manual; Bruker-AXS: Madison, WI, USA, 1998. [Google Scholar]
- Spek, L.A. Single-crystal structure validation with the program PLATON. J. Appl. Chem. 2003, 36, 7–13. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer17, University of Western Australia. 2017. Available online: http://hirshfeldsurface.net (accessed on 15 February 2020).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. GAUSSIAN 09. Revision A02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Keith, T.; Millam, J. GaussView, Version 4.1, R. Dennington II; Semichem Inc.: Shawnee Mission, KS, USA, 2007. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar]
- Islam, M.S.; Ghawas, H.M.; El-Senduny, F.F.; Al-Majid, A.M.; Elshaier, Y.A.; Badria, F.A.; Barakat, A. Synthesis of new thiazolo-pyrrolidine–(spirooxindole) tethered to 3-acylindole as anticancer agents. Bioorg. Chem. 2019, 82, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Islam, M.S.; Ghawas, H.M.; Al-Majid, A.M.; El-Senduny, F.F.; Badria, F.A.; Elshaier, Y.A.M.; Ghabbour, H.A. Substituted spirooxindole derivatives as potent anticancer agents through inhibition of phosphodiesterase 1. RSC Adv. 2018, 8, 14335–14346. [Google Scholar] [CrossRef] [Green Version]
- Lotfy, G.; El Sayed, H.; Said, M.M.; Aziz, Y.M.A.; Al-Dhfyan, A.; Al-Majid, A.M.; Barakat, A. Regio-and stereoselective synthesis of new spirooxindoles via 1,3-dipolar cycloaddition reaction: Anticancer and molecular docking studies. J. Photochem. Photobiol. B Biol. 2018, 180, 98–108. [Google Scholar] [CrossRef]
- Lotfy, G.; Said, M.M.; El Sayed, H.; El Sayed, H.; Al-Dhfyan, A.; Aziz, Y.M.A.; Barakat, A. Synthesis of new spirooxindole-pyrrolothiazole derivatives: Anti-cancer activity and molecular docking. Biorg. Med. Chem. 2017, 25, 1514–1523. [Google Scholar] [CrossRef]
- Al-Majid, A.M.; Ghawas, H.M.; Islam, M.S.; Soliman, S.M.; El-Senduny, F.F.; Badria, F.A.; Ali, M.; Shaik, M.R.; Ghabbour, H.A.; Barakat, A. Synthesis of spiroindolone analogue via three components reaction of olefin with isatin and sarcosine: Anti-proliferative activity and computational studies. J. Mol. Struct. 2020, 1204, 127500. [Google Scholar] [CrossRef]
- Lotfy, G.; Aziz, Y.M.A.; Said, M.M.; El Ashry, E.S.H.; El Tamany, E.S.H.; Barakat, A.; Ghabbour, H.A.; Yousuf, S.; Ul-Haq, Z.; Choudhary, M.I. Synthesis of oxindole analogues, biological activity, and in silico studies. ChemistrySelect 2019, 4, 10510–10516. [Google Scholar] [CrossRef]
- Barakat, A.; Soliman, S.M.; Al-Majid, A.M.; Ali, M.; Islam, M.S.; Elshaier, Y.A.; Ghabbour, H.A. New spiro-oxindole constructed with pyrrolidine/thioxothiazolidin-4-one derivatives: Regioselective synthesis, x-ray crystal structures, hirshfeld surface analysis, dft, docking and antimicrobial studies. J. Mol. Struct. 2018, 1152, 101–114. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foresman, J.B.; Frisch, A. Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Chang, R. Chemistry, 7th ed.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Kosar, B.; Albayrak, C. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino) methyl] phenol. Spectrochim. Acta-Part A Mol. Biomol. Spectrosc. 2011, 78, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, T.A. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den einzelnen Elektronen eines Atoms. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Chamorro, E.; Pérez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar]
- Kumar, R.S.; Almansour, A.I.; Arumugam, N.; Althomili, D.M.Q.; Altaf, M.; Basiri, A.; Kotresha, D.; Manohar, T.S.; Venketesh, S. Ionic liquid-enabled synthesis, cholinesterase inhibitory activity, and molecular docking study of highly functionalized tetrasubstituted pyrrolidines. Bioorg. Chem. 2018, 77, 263–268. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCDC | 1972061 |
| Empirical formula | C29H25N2O3FS2 |
| Formula weight | 532.63 |
| Temperature | 100(2) K |
| Wavelength | 1.54178 Å |
| Crystal system | Orthorhombic |
| Space group | P 21 21 21 |
| Unit cell dimensions | a = 8.2588(4) Å b = 10.6003(5) Å, α = β = γ = 90°. c = 28.8478(14) Å |
| Volume | 2525.5(2)Å3 |
| Z | 4 |
| Calculated density | 1.401 mg/m3 |
| Absorption coefficient | 2.268 mm−1 |
| F(000) | 1112 |
| Crystal size | 0.11 × 0.09 × 0.07 mm3 |
| Theta range for data collection | 4.444 to 68.231 |
| Limiting indices | −9 ≤ h ≤ 8, −11 ≤ k ≤12, −33 ≤ l ≤34 |
| Reflections collected/unique | 16366/4587 |
| R(int) | 0.0332 |
| Completeness to theta = 67.679 | 99.5% |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 4587/0/334 |
| Goodness-of-fit on F2 | 1.038 |
| Final R indices [I>2sigma(I)] | R1 = 0.0396, wR2 = 0.0970 |
| Final R indexes [all data] | R1 = 0.0418, wR2 = 0.0987 |
| Largest diff. peak and hole | 0.41 and −0.36 e.A−3 |
| D-H…A | d(D-H) | d(H…A) | d(D…A) | <(DHA) |
|---|---|---|---|---|
| N2-H2…O3 | 0.88 | 1.96 | 2.830(3) | 172 |
| O3-H3O…O1 | 0.84 | 1.97 | 2.775(3) | 160 |
| C7A-H7AA…S2A | 0.95 | 2.84 | 3.604(16) | 139 |
| C7B-H7BA…F1 | 0.95 | 2.35 | 3.080(9) | 133 |
| C4B-H4B…S1B | 0.95 | 2.84 | 3.738(8) | 158 |
| C10A-H10A…S2A | 0.99 | 2.50 | 3.378(7) | 147 |
| C17-H17…O1 | 0.95 | 2.56 | 3.368(4) | 143 |
| C26-H26…O1 | 0.95 | 2.55 | 3.435(4) | 154 |
| Energy | Aa | A | Ba | B |
|---|---|---|---|---|
| Etot. | −2235.837 | −2351.580 | −2235.838 | −2351.581 |
| ZPVE | 0.430 | 0.483 | 0.430 | 0.483 |
| Ecorr b | −2235.407 | −2351.097 | −2235.408 | −2351.097 |
| HOMO | −5.852 | −5.765 | −5.640 | −5.599 |
| LUMO | −1.782 | −1.711 | −1.696 | −1.710 |
| I | 5.852 | 5.765 | 5.640 | 5.599 |
| A | 1.782 | 1.711 | 1.696 | 1.710 |
| η | 4.071 | 4.054 | 3.944 | 3.889 |
| μ | −3.817 | −3.738 | −3.668 | −3.654 |
| S | 0.246 | 0.247 | 0.254 | 0.257 |
| ω | 1.790 | 1.723 | 1.705 | 1.717 |
| Compound | AChE Inhibition IC50 µg/mL | AChE Inhibition IC50 µM/mL |
|---|---|---|
| 5 | 62.25 | 124.34 |
| Galantamine | 0.98 | 3.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barakat, A.; Soliman, S.M.; Alshahrani, S.; Islam, M.S.; Ali, M.; Al-Majid, A.M.; Yousuf, S. Synthesis, X-ray Single Crystal, Conformational Analysis and Cholinesterase Inhibitory Activity of a New Spiropyrrolidine Scaffold Tethered Benzo[b]Thiophene Analogue. Crystals 2020, 10, 120. https://doi.org/10.3390/cryst10020120
Barakat A, Soliman SM, Alshahrani S, Islam MS, Ali M, Al-Majid AM, Yousuf S. Synthesis, X-ray Single Crystal, Conformational Analysis and Cholinesterase Inhibitory Activity of a New Spiropyrrolidine Scaffold Tethered Benzo[b]Thiophene Analogue. Crystals. 2020; 10(2):120. https://doi.org/10.3390/cryst10020120
Chicago/Turabian StyleBarakat, Assem, Saied M. Soliman, Saeed Alshahrani, Mohammad Shahidul Islam, M. Ali, Abdullah Mohammed Al-Majid, and Sammer Yousuf. 2020. "Synthesis, X-ray Single Crystal, Conformational Analysis and Cholinesterase Inhibitory Activity of a New Spiropyrrolidine Scaffold Tethered Benzo[b]Thiophene Analogue" Crystals 10, no. 2: 120. https://doi.org/10.3390/cryst10020120
APA StyleBarakat, A., Soliman, S. M., Alshahrani, S., Islam, M. S., Ali, M., Al-Majid, A. M., & Yousuf, S. (2020). Synthesis, X-ray Single Crystal, Conformational Analysis and Cholinesterase Inhibitory Activity of a New Spiropyrrolidine Scaffold Tethered Benzo[b]Thiophene Analogue. Crystals, 10(2), 120. https://doi.org/10.3390/cryst10020120