Crystal Structural Analysis of DL-Mandelate Salt of Carvedilol and Its Correlation with Physicochemical Properties

 , and
, and 
Abstract
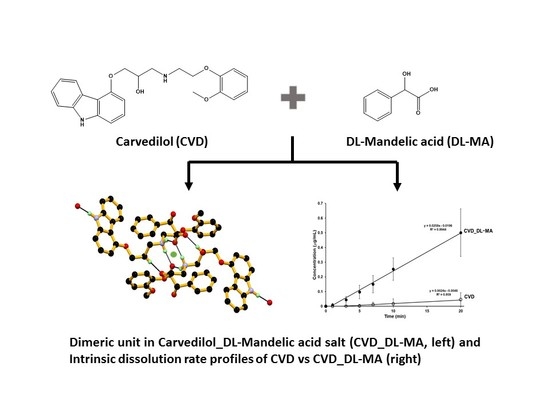
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of CVD Salts
2.3. Single-Crystal X-Ray Diffraction
2.4. Powder X-Ray Diffraction (PXRD)
2.5. Differential Scanning Calorimetry (DSC) and Thermogravimetric (TG) Measurements
2.6. Fourier-Transform Infrared Spectroscopy (FT-IR)
2.7. Solubility Tests
2.7.1. Equilibrium Solubility Experiments
2.7.2. Intrinsic Dissolution Experiment
2.8. High-Performance Liquid Chromatography (HPLC) Conditions
3. Results and Discussion
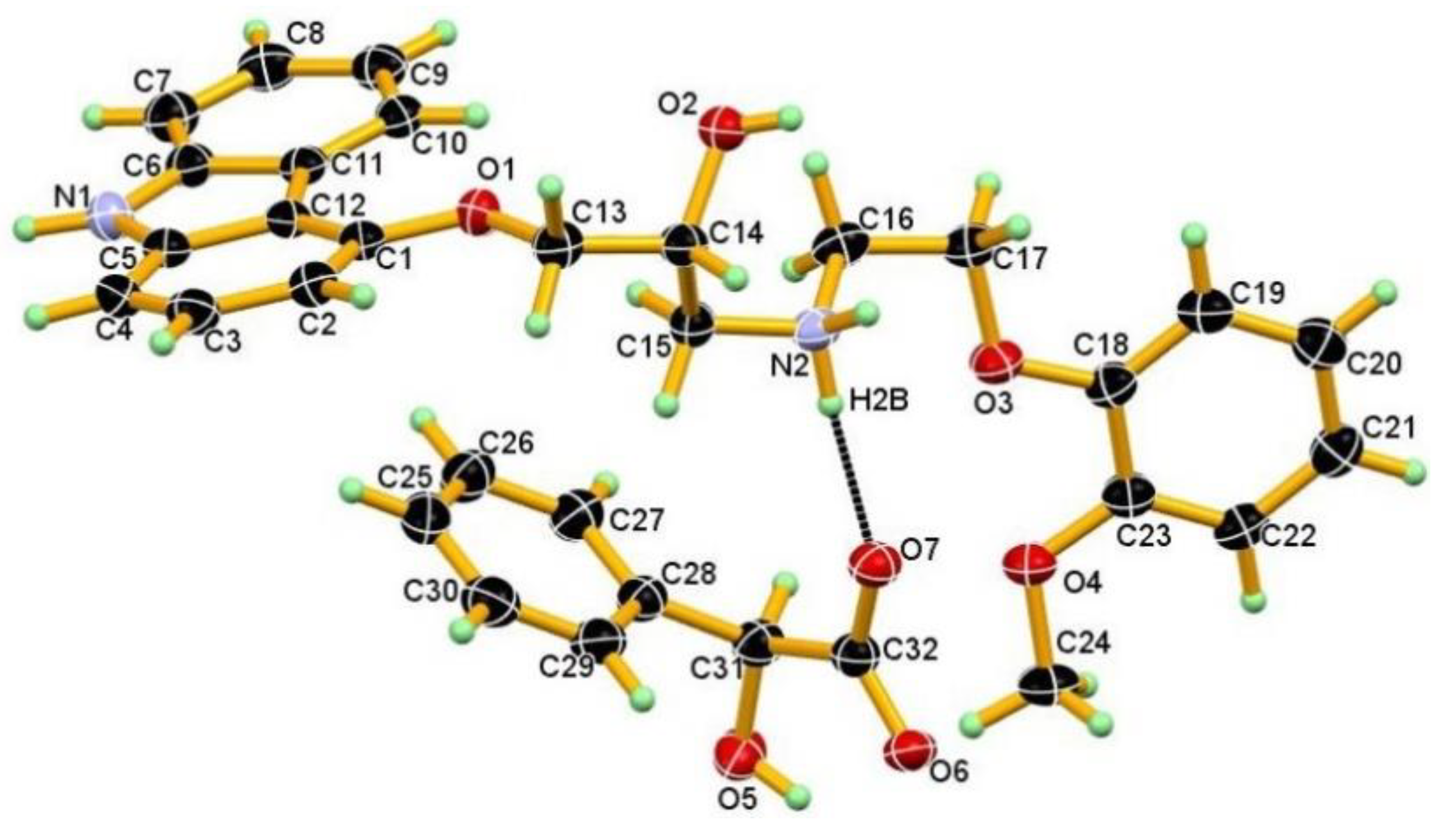
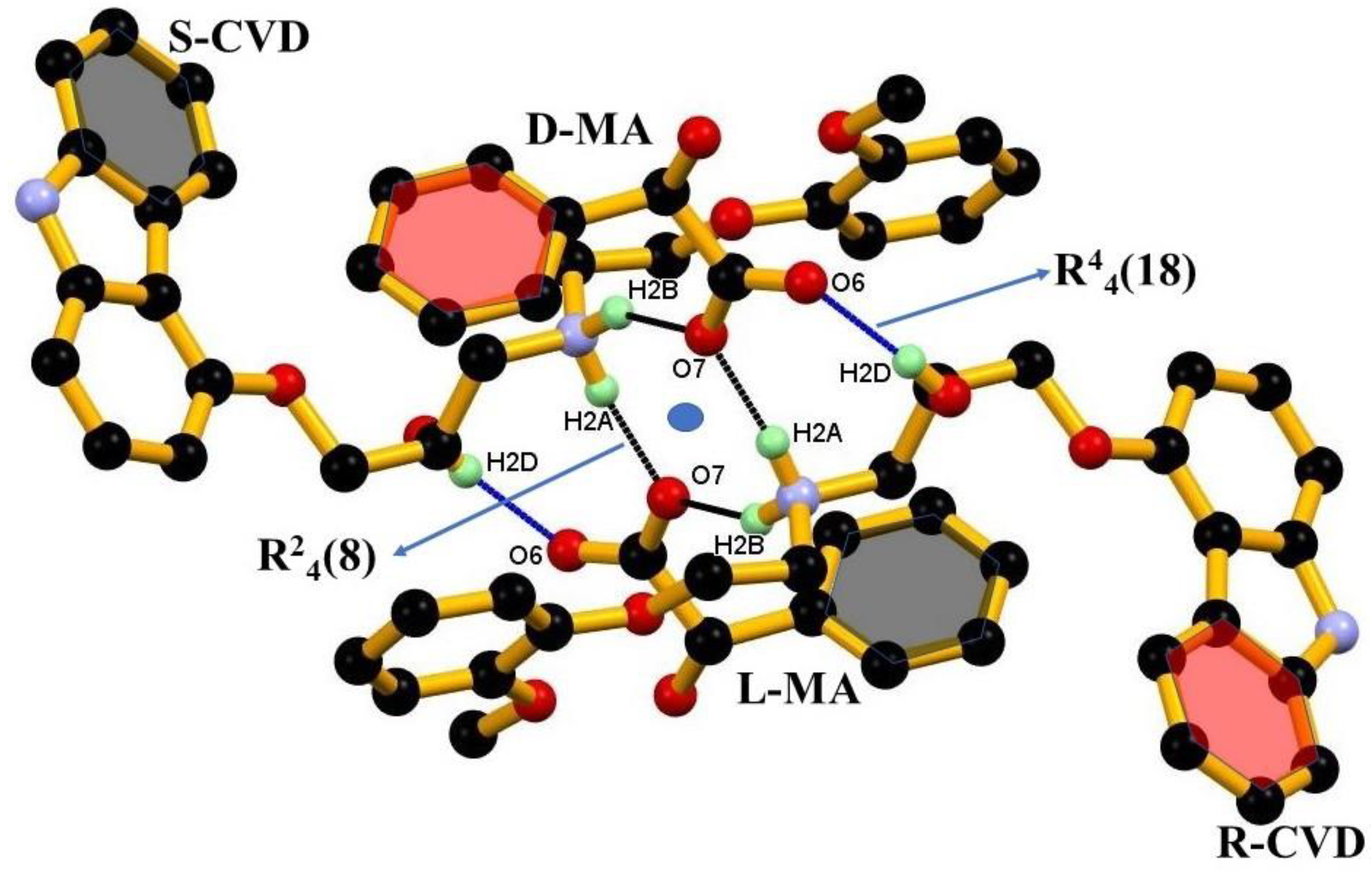
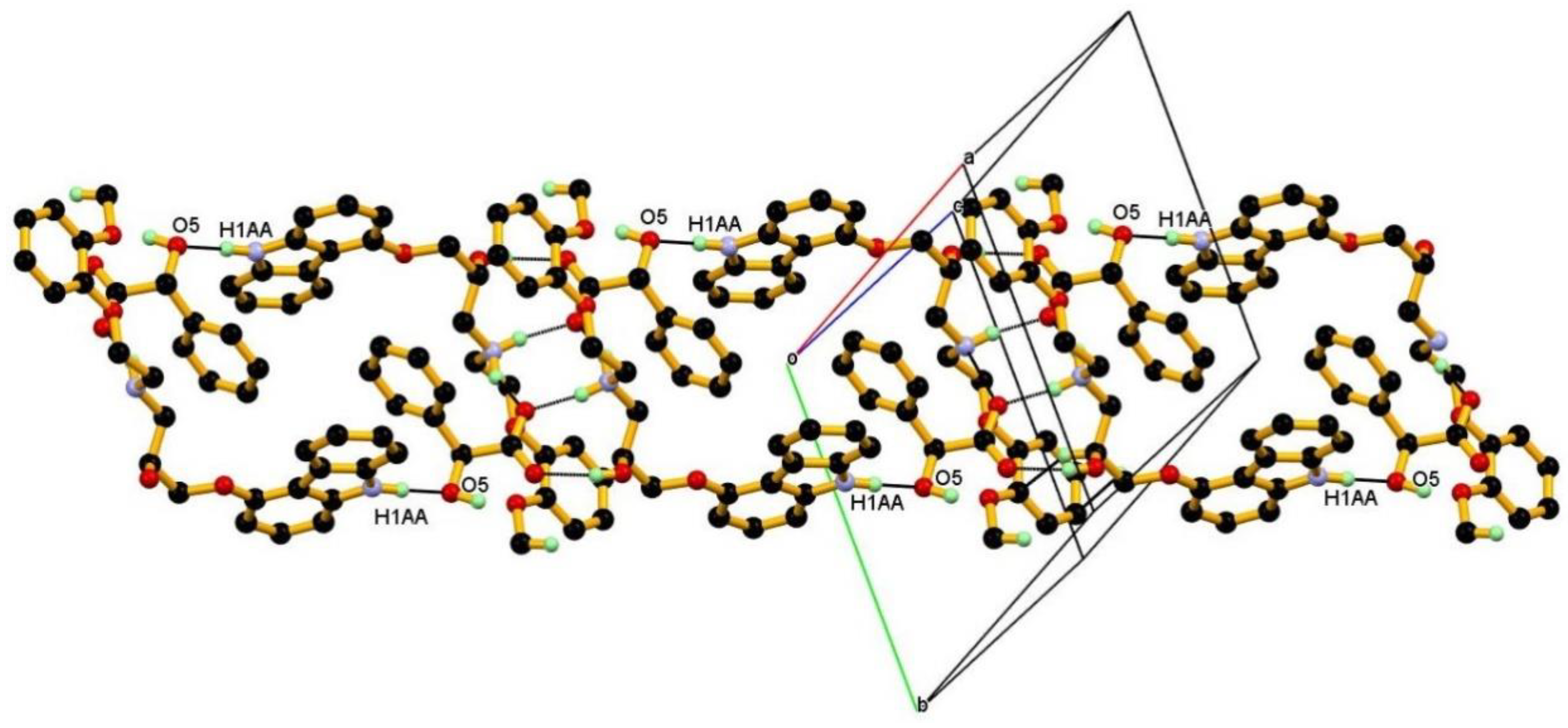
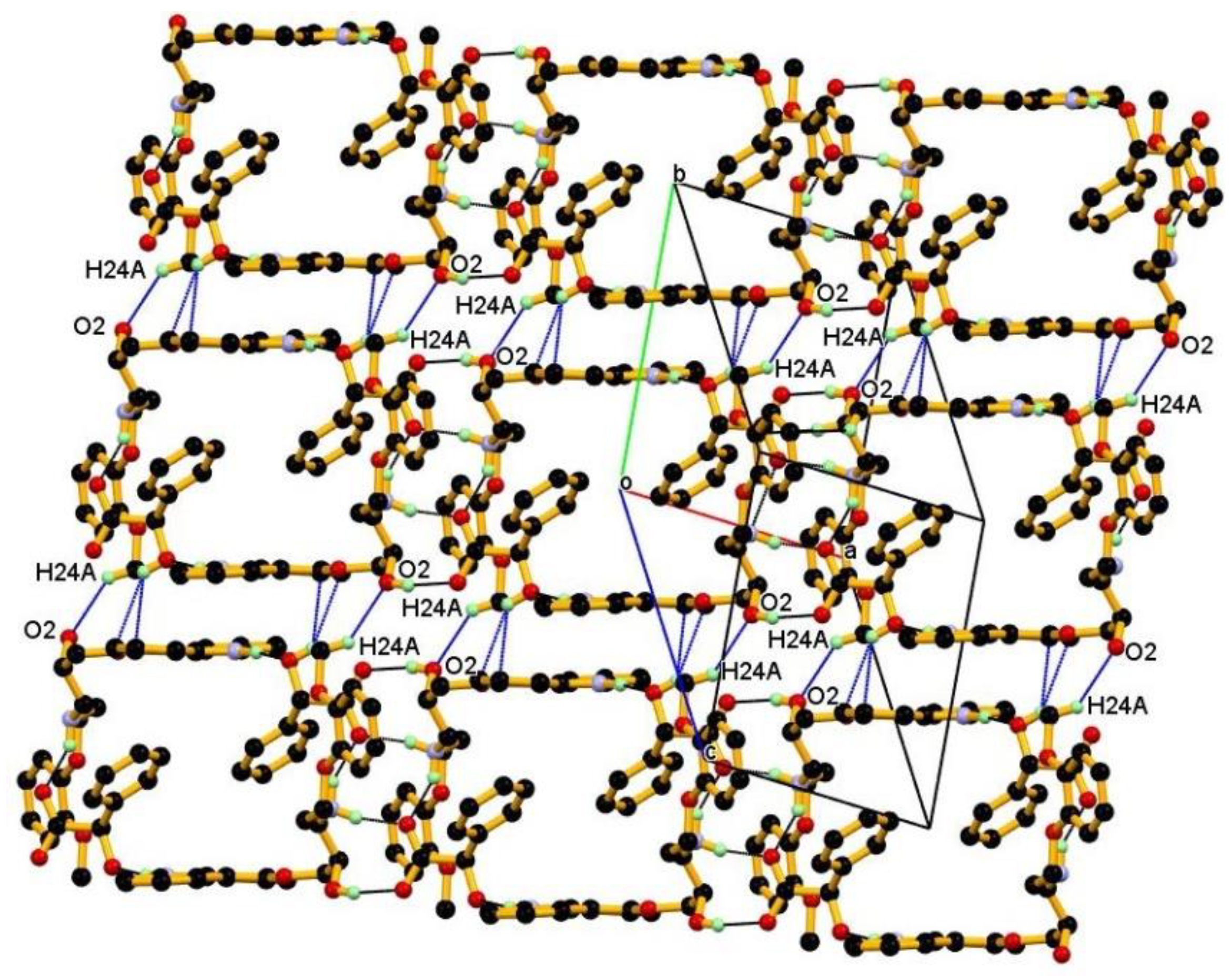
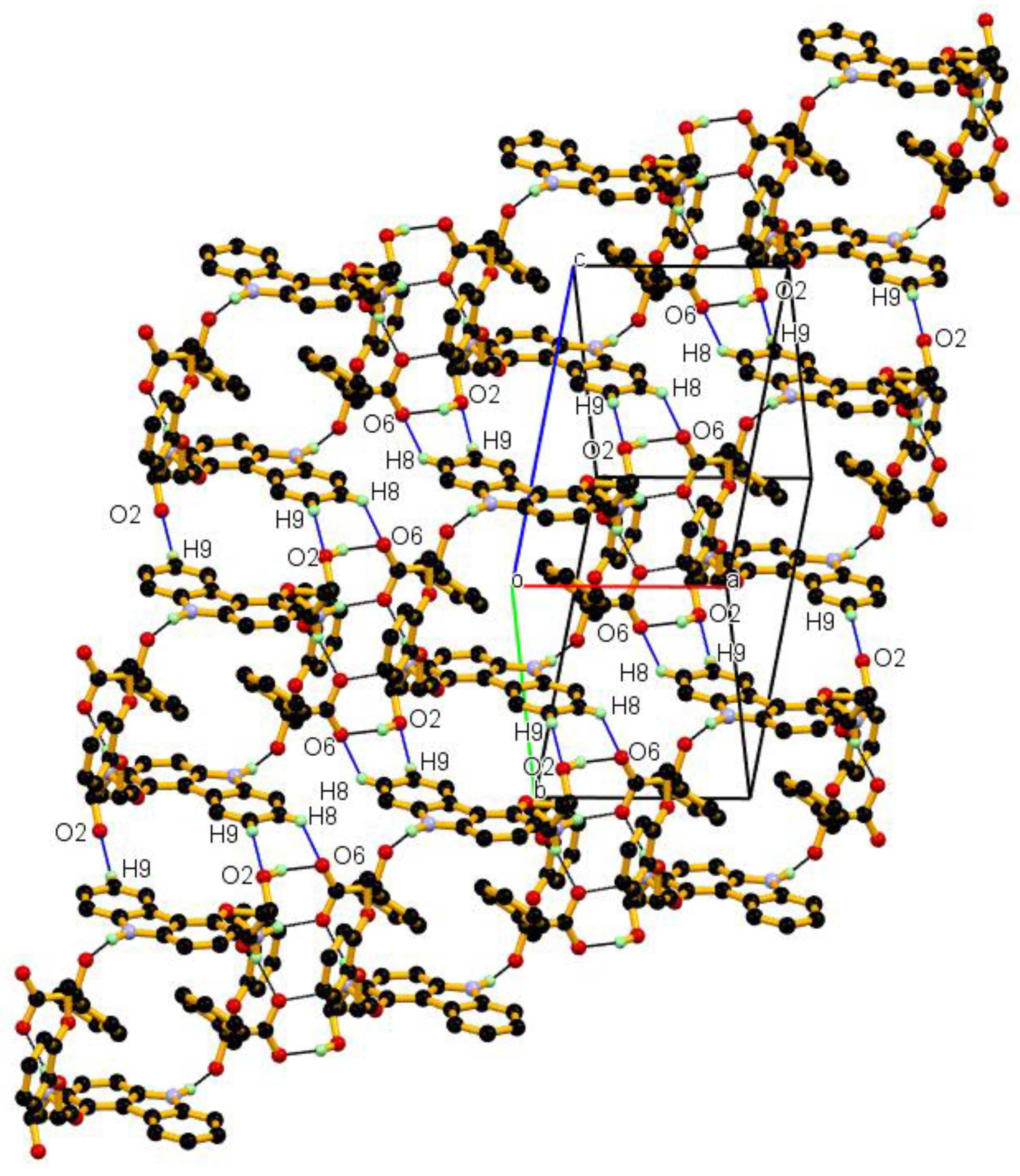
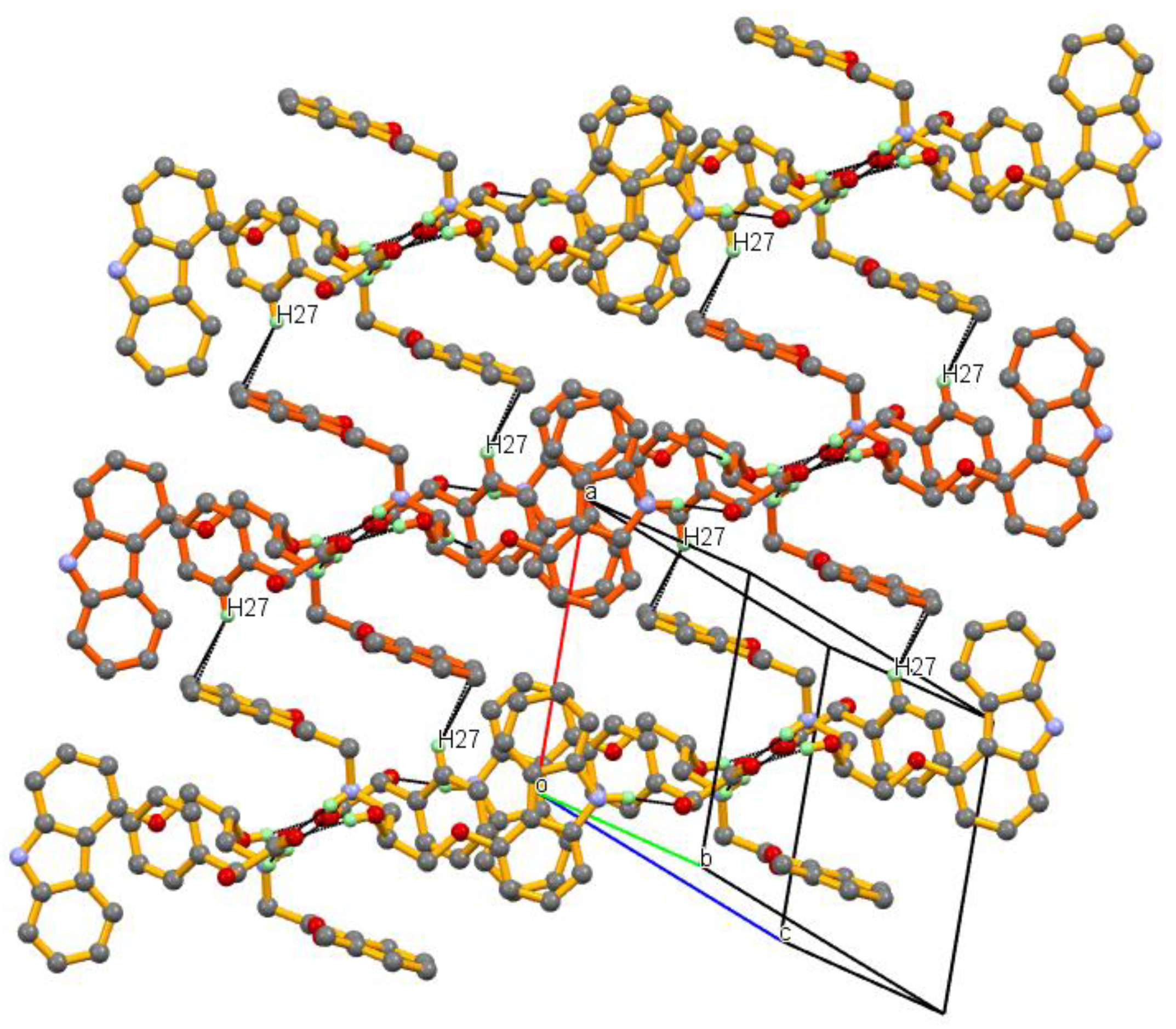
3.1. Crystal Structure
3.2. Crystal Structural Analysis and Its Correlation with Physicochemical Properties
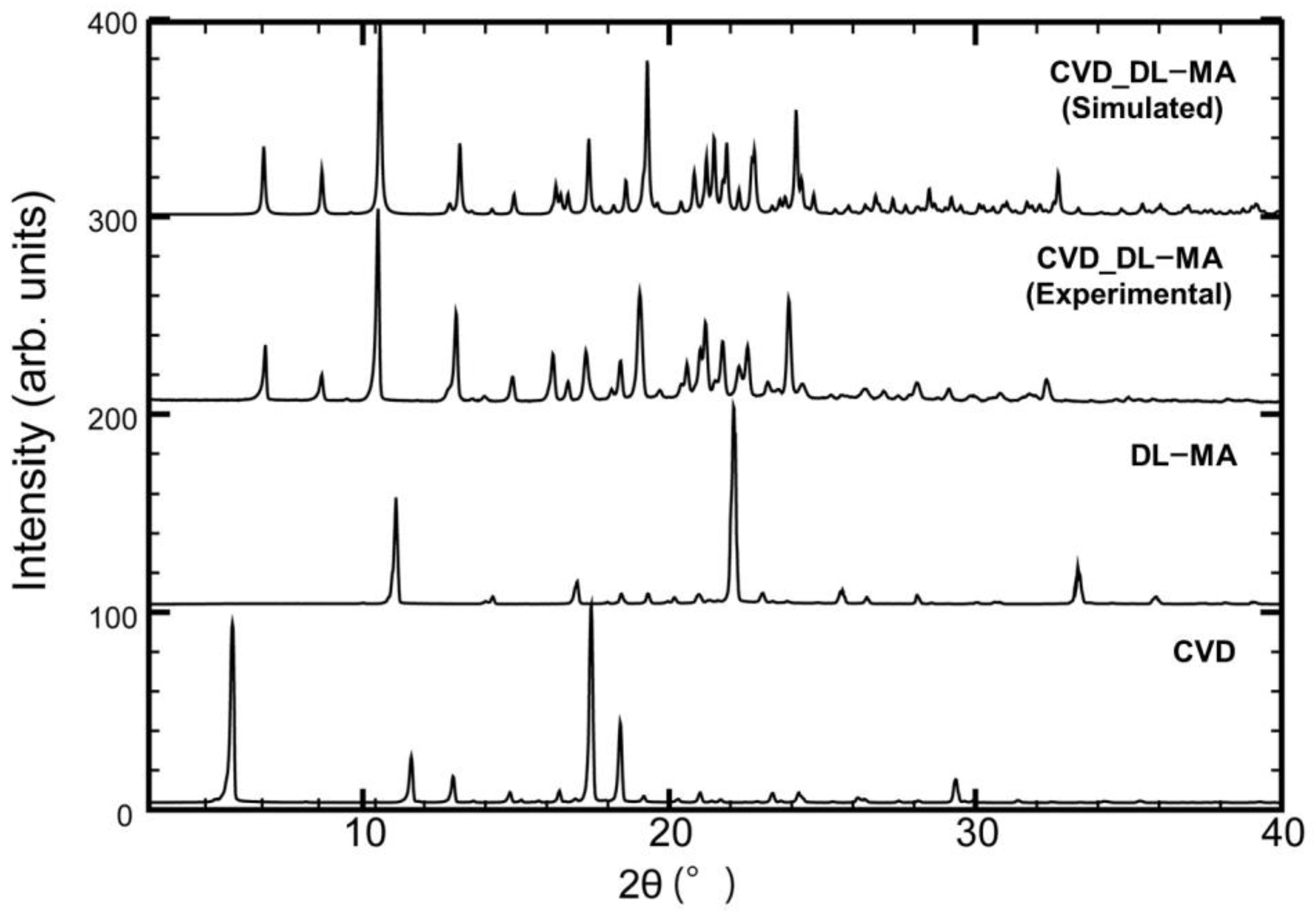
3.3. PXRD Measurements
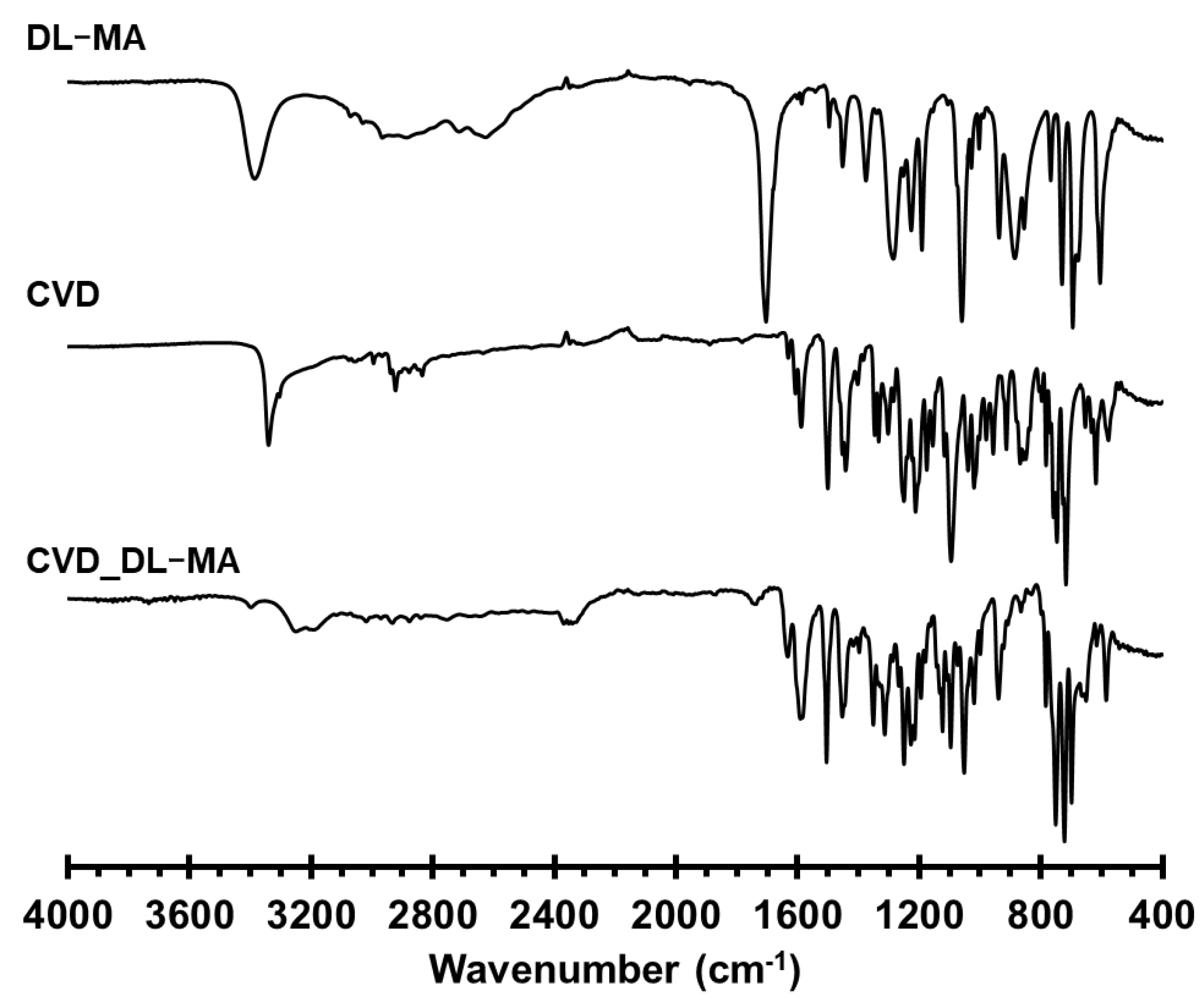
3.4. FT-IR Spectrum
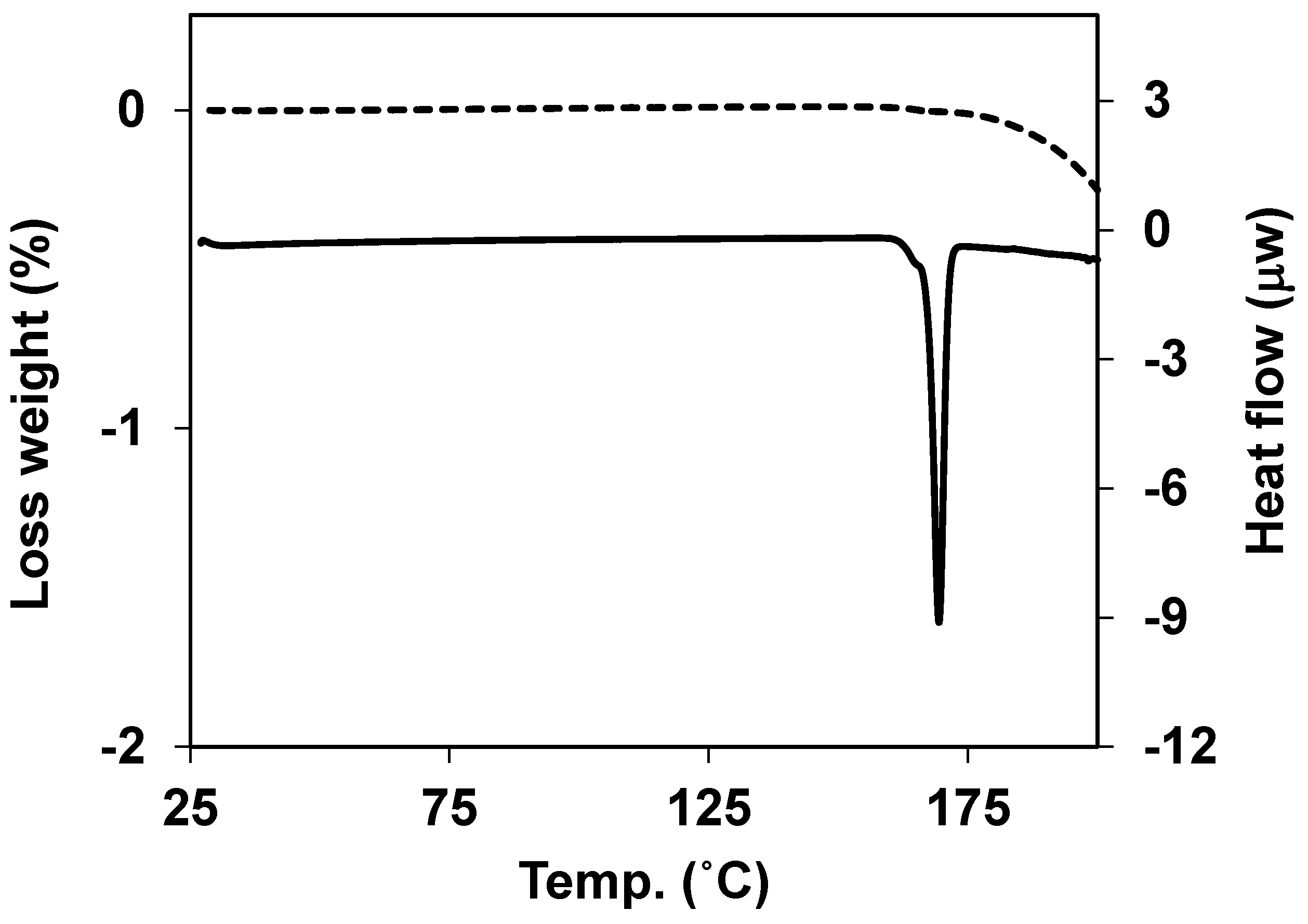
3.5. Thermal Properties
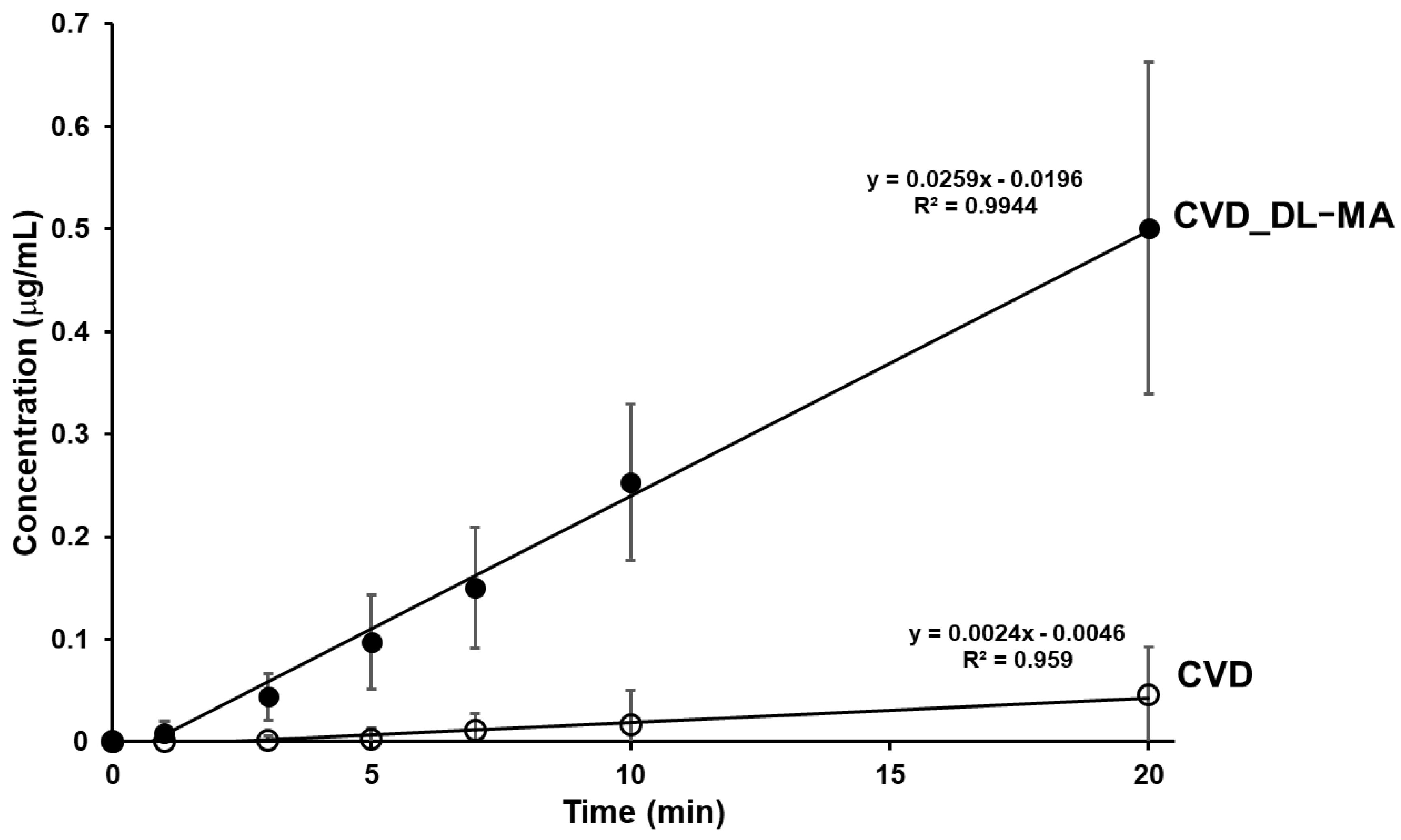
3.6. Dissolution Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aakeröy, C.B.; Salmon, D.J. Building co-crystals with molecular sense and supramolecular sensibility. CrystEngComm 2005, 7, 439–448. [Google Scholar] [CrossRef]
- Aitipamula, S.; Wong, A.B.H.; Chow, P.S.; Tan, R.B.H. Pharmaceutical salts of haloperidol with some carboxylic acids and artificial sweeteners: Hydrate formation, polymorphism, and physicochemical properties. Cryst. Growth Des. 2014, 14, 2542–2556. [Google Scholar] [CrossRef]
- Morissette, S.L.; Almarsson, O.; Peterson, M.L.; Remenar, J.F.; Read, M.J.; Lemmo, A.V.; Ellis, S.; Cima, M.J.; Gardner, C.R. High-throughput crystallization: Polymorphs, salts, co-crystals and solvates of pharmaceutical solids. Adv. Drug Deliv. Rev. 2004, 56, 275–300. [Google Scholar] [CrossRef] [PubMed]
- Sathisaran, I.; Dalvi, S.V. Engineering cocrystals of poorly water-soluble drugs to enhance dissolution in aqueous medium. Pharmaceutics 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Schultheiss, N.; Newman, A. Pharmaceutical cocrystals and their physicochemical properties. Cryst. Growth Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.C. Cocrystallization for successful drug delivery. Expert Opin. Drug Deliv. 2013, 10, 201–213. [Google Scholar] [CrossRef]
- Vishweshwar, P.; McMahon, J.A.; Bis, J.A.; Zaworotko, M.J. Pharmaceutical co-crystals. J. Pharm. Sci. 2006, 95, 499–516. [Google Scholar] [CrossRef]
- Yousef, M.A.E.; Vangala, V.R. Pharmaceutical cocrystals: Molecules, crystals, formulations, medicines. Cryst. Growth Des. 2019, 19, 7420–7438. [Google Scholar] [CrossRef]
- Nauha, E.; Nissinen, M. Co-crystals of an agrochemical active—A pyridine-amine synthon for a thioamide group. J. Mol. Struct. 2011, 1006, 566–569. [Google Scholar] [CrossRef] [Green Version]
- Bučar, D.-K.; Filip, S.; Arhangelskis, M.; Lloyd, G.O.; Jones, W. Advantages of mechanochemical cocrystallisation in the solid-state chemistry of pigments: Colour-tuned fluorescein cocrystals. CrystEngComm 2013, 15, 6289–6291. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Li, Z.; Zhang, Q.; Peng, B.; Zhu, B.; Wang, J.-R.; Liu, L.; Mei, X. Fine-tuning the colors of natural pigment emodin with superior stability through cocrystal engineering. Cryst. Growth Des. 2018, 18, 6123–6132. [Google Scholar] [CrossRef]
- Bolton, O.; Simke, L.R.; Pagoria, P.F.; Matzger, A.J. High power explosive with good sensitivity: A 2:1 Cocrystal of CL-20:HMX. Cryst. Growth Des. 2012, 12, 4311–4314. [Google Scholar] [CrossRef]
- Millar, D.I.A.; Maynard-Casely, H.E.; Allan, D.R.; Cumming, A.S.; Lennie, A.R.; Mackay, A.J.; Oswald, I.D.H.; Tang, C.C.; Pulham, C.R. Crystal engineering of energetic materials: Co-crystals of CL-20. CrystEngComm 2012, 14, 3742–3749. [Google Scholar] [CrossRef] [Green Version]
- Bolton, O.; Matzger, A.J. Improved stability and smart-material functionality realized in an energetic cocrystal. Angew. Chem. Int. Ed. 2011, 50, 8960–8963. [Google Scholar] [CrossRef]
- Thayer, A.M. Form and function. Chem. Eng. News 2007, 85, 17–30. [Google Scholar] [CrossRef]
- Prohotsky, D.L.; Zhao, F. A survey of top 200 drugs-inconsistent practice of drug strength expression for drugs containing salt forms. J. Pharm. Sci. 2012, 101, 1–6. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Liversidge, G.G.; Cundy, K.C. Particle size reduction for improvement of oral bioavailability of hydrophobic drugs: I. Absolute oral bioavailability of nanocrystalline danazol in beagle dogs. Int. J. Pharm. 1995, 125, 91–97. [Google Scholar] [CrossRef]
- Müller, R.H.; Peters, K. Nanosuspensions for the formulation of poorly soluble drugs: I. Preparation by a size-reduction technique. Int. J. Pharm. 1998, 160, 229–237. [Google Scholar] [CrossRef]
- Shaikh, J.; Ankola, D.D.; Beniwal, V.; Singh, D.; Kumar, M.N. Nanoparticle encapsulation improves oral bioavailability of curcumin by at least 9-fold when compared to curcumin administered with piperine as absorption enhancer. Eur. J. Pharm. Sci. 2009, 37, 223–230. [Google Scholar] [CrossRef]
- Ohara, T.; Kitamura, S.; Kitagawa, T.; Terada, K. Dissolution mechanism of poorly water-soluble drug from extended release solid dispersion system with ethylcellulose and hydroxypropylmethylcellulose. Int. J. Pharm. 2005, 302, 95–102. [Google Scholar] [CrossRef]
- Hasegawa, S.; Hamaura, T.; Furuyama, N.; Kusai, A.; Yonemochi, E.; Terada, K. Effects of water content in physical mixture and heating temperature on crystallinity of troglitazone-PVP K30 solid dispersions prepared by closed melting method. Int. J. Pharm. 2005, 302, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Jambhekar, S.S.; Breen, P. Cyclodextrins in pharmaceutical formulations II: Solubilization, binding constant, and complexation efficiency. Drug Discov. Today 2016, 21, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Rajput, L.; Sanphui, P.; Desiraju, G.R. New Solid Forms of the Anti-HIV Drug Etravirine: Salts, Cocrystals, and Solubility. Cryst. Growth Des. 2013, 13, 3681–3690. [Google Scholar] [CrossRef]
- Banerjee, R.; Bhatt, P.M.; Ravindra, N.V.; Desiraju, G.R. Saccharin salts of active pharmaceutical ingredients, their crystal structures, and increased water solubilities. Cryst. Growth Des. 2005, 5, 2299–2309. [Google Scholar] [CrossRef]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating cocrystals: A review of pharmaceutical cocrystal preparation routes and applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Cao, H.-L.; Zhou, J.-R.; Cai, F.-Y.; Lü, J.; Cao, R. Two-component pharmaceutical cocrystals regulated by supramolecular synthons comprising primary N···H···O interactions. Cryst. Growth Des. 2019, 19, 3–16. [Google Scholar] [CrossRef]
- Thorat, S.H.; Patwadkar, M.V.; Gonnade, R.G.; Vaidhyanathan, R. Capturing a novel metastable polymorph of the anticancer drug gefitinib. CrystEngComm 2014, 16, 8638–8641. [Google Scholar] [CrossRef]
- Zhu, B.; Fang, X.; Zhang, Q.; Mei, X.; Ren, G. Study of crystal structures, properties, and form transformations among a polymorph, hydrates, and solvates of apatinib. Cryst. Growth Des. 2019, 19, 3060–3069. [Google Scholar] [CrossRef]
- Colucci, W.S.; Packer, M.; Bristow, M.R.; Gilbert, E.M.; Cohn, J.N.; Fowler, M.B.; Krueger, S.K.; Hershberger, R.; Uretsky, B.F.; Bowers, J.A.; et al. Carvedilol inhibits clinical progression in patients with mild symptoms of heart failure. Circulation 1996, 94, 2800–2806. [Google Scholar] [CrossRef]
- Packer, M.; Bristow, M.R.; Cohn, J.N.; Colucci, W.S.; Fowler, M.B.; Gilbert, E.M.; Shusterman, N.H. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. N. Engl. J. Med. 1996, 334, 1349–1355. [Google Scholar] [CrossRef]
- Dargie, H.J. Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: The CAPRICORN randomised trial. Lancet 2001, 357, 1385–1390. [Google Scholar] [CrossRef]
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Sun, P.; Nie, S.; Pan, W. Preparation and evaluation of SEDDS and SMEDDS containing carvedilol. Drug Dev. Ind. Pharm. 2005, 31, 785–794. [Google Scholar] [CrossRef]
- Mahmoud, E.A.; Bendas, E.R.; Mohamed, M.I. Preparation and evaluation of self-nanoemulsifying tablets of carvedilol. AAPS PharmSciTech 2009, 10, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Xu, H.; Tian, B.; Yuan, K.; Pan, H.; Ma, S.; Yang, X.; Pan, W. Fabrication of carvedilol nanosuspensions through the anti-solvent precipitation-ultrasonication method for the improvement of dissolution rate and oral bioavailability. AAPS PharmSciTech 2012, 13, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhi, Z.; Li, X.; Gao, J.; Song, Y. Carboxylated mesoporous carbon microparticles as new approach to improve the oral bioavailability of poorly water-soluble carvedilol. Int. J. Pharm. 2013, 454, 403–411. [Google Scholar] [CrossRef]
- Hirlekar, R.; Kadam, V. Preparation and characterization of inclusion complexes of carvedilol with methyl-β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2009, 63, 219–224. [Google Scholar] [CrossRef]
- Diniz, L.F.; Carvalho, P.S.; da Nova Mussel, W.; Yoshida, M.I.; Diniz, R.; Fernandes, C. Racemic salts and solid solutions of enantiomers of the antihypertensive drug carvedilol. Cryst. Growth Des. 2019, 19, 4498–4509. [Google Scholar] [CrossRef]
- Hiendrawan, S.; Widjojokusumo, E.; Veriansyah, B.; Tjandrawinata, R.R. Pharmaceutical salts of carvedilol: Polymorphism and physicochemical properties. AAPS PharmSciTech 2017, 18, 1417–1425. [Google Scholar] [CrossRef]
- Prado, L.D.; Rocha, H.V.A.; Resende, J.A.L.C.; Ferreira, G.B.; de Figuereido Teixeira, A.M.R. An insight into carvedilol solid forms: Effect of supramolecular interactions on the dissolution profiles. CrystEngComm 2014, 16, 3168–3179. [Google Scholar] [CrossRef]
- Bolla, G.; Nangia, A. Clofazimine Mesylate: A High Solubility Stable Salt. Cryst. Growth Des. 2012, 12, 6250–6259. [Google Scholar] [CrossRef]
- Gunnam, A.; Nangia, A.K. High-Solubility Salts of the Multiple Sclerosis Drug Teriflunomide. Cryst. Growth Des. 2019, 19, 5407–5417. [Google Scholar] [CrossRef]
- Putra, O.D.; Umeda, D.; Nugraha, Y.P.; Furuishi, T.; Nagase, H.; Fukuzawa, K.; Uekusa, H.; Yonemochi, E. Solubility improvement of epalrestat by layered structure formation via cocrystallization. CrystEngComm 2017, 19, 2614–2622. [Google Scholar] [CrossRef]
- Putra, O.D.; Umeda, D.; Nugraha, Y.P.; Nango, K.; Yonemochi, E.; Uekusa, H. Simultaneous improvement of epalrestat photostability and solubility via cocrystallization: A case study. Cryst. Growth Des. 2018, 18, 373–379. [Google Scholar] [CrossRef]
- Dwichandra Putra, O.; Umeda, D.; Fujita, E.; Haraguchi, T.; Uchida, T.; Yonemochi, E.; Uekusa, H. solubility improvement of benexate through salt formation using artificial sweetener. Pharmaceutics 2018, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Ainurofiq, A.; Mauludin, R.; Mudhakir, D.; Umeda, D.; Soewandhi, S.N.; Putra, O.D.; Yonemochi, E. Improving mechanical properties of desloratadine via multicomponent crystal formation. Eur. J. Pharm. Sci. 2018, 111, 65–72. [Google Scholar] [CrossRef]
- Teraoka, R.; Fukami, T.; Furuishi, T.; Nagase, H.; Ueda, H.; Tode, C.; Yutani, R.; Kitagawa, S.; Sakane, T. Improving the solid-state photostability of furosemide by its cocrystal formation. Chem. Pharm. Bull. 2019, 67, 940–944. [Google Scholar] [CrossRef] [Green Version]
- Higashi, T. Calculated Using ABSCOR: Empirical Absorption Correction Based on Fourier Series Approximation; Rigaku: The Woodland, TX, USA, 1994. [Google Scholar]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Crystallogr. 2005, 38, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Parmar, V.K.; Shah, S.A. Hydrochloride salt co-crystals: Preparation, characterization and physicochemical studies. Pharm. Dev. Technol. 2013, 18, 443–453. [Google Scholar] [CrossRef]
- Arenas-García, J.I.; Herrera-Ruiz, D.; Mondragón-Vásquez, K.; Morales-Rojas, H.; Höpfl, H. Co-crystals of active pharmaceutical ingredients—Acetazolamide. Cryst. Growth Des. 2010, 10, 3732–3742. [Google Scholar] [CrossRef]
- Stevanus, H.; Bambang, V.; Edward, W.; Sundani Nurono, S.; Saleh, W.; Raymond, R.T. Simultaneous cocrystallization and micronization of paracetamol-dipicolinic acid cocrystal by supercritical antisolvent (SAS). Int. J. Pharm. Pharm. Sci. 2016, 8, 89–98. [Google Scholar]
- Mishra, J.; Lobmann, K.; Grohganz, H.; Rades, T. Influence of preparation technique on co-amorphization of carvedilol with acidic amino acids. Int. J. Pharm. 2018, 552, 407–413. [Google Scholar] [CrossRef]
- Swapna, B.; Maddileti, D.; Nangia, A. Cocrystals of the tuberculosis drug isoniazid: Polymorphism, isostructurality, and stability. Cryst. Growth Des. 2014, 14, 5991–6005. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | CVD_DL-MA |
|---|---|
| Empirical formula | C32 H34 N2 O7 |
| Formula weight | 558.61 |
| Temperature | 123(2) K |
| Wavelength | 1.54187 Å |
| Crystal system | Triclinic |
| Space group | P −1 |
| Unit cell dimensions | a = 9.8416(5) Å, α = 108.595(8)° |
| b = 11.4689(5) Å, β = 95.182(7)° | |
| c = 14.0746(7) Å, γ = 107.323(8)° | |
| Volume | 1406.95(15) Å3 |
| Z | 2 |
| Density (calculated) | 1.319 Mg/m3 |
| Absorption coefficient | 0.764 mm−1 |
| F(000) | 592 |
| Crystal size | 0.210 × 0.150 × 0.100 mm3 |
| Theta range for data collection | 3.385° to 68.184° |
| Index ranges | −11<=h<=11, −13<=k<=13, −16<=l<=16 |
| Reflections collected | 16381 |
| Independent reflections | 5033 [R(int) = 0.0359] |
| Completeness to theta = 67.687° | 97.9% |
| Absorption correction | Semi-empirical from equivalents |
| Max. and min. transmission | 0.926 and 0.605 |
| Refinement method | Full-matrix least-squares on F2 |
| Data / restraints / parameters | 5033/0/380 |
| Goodness-of-fit on F2 | 1.056 |
| Final R indices [I>2sigma(I)] | R1 = 0.0468, wR2 = 0.1252 |
| R indices (all data) | R1 = 0.0626, wR2 = 0.1352 |
| Extinction coefficient | n/a |
| Largest diff. peak and hole | 0.356 and −0.227 e.Å−3 |
| D-H···A | D-H (Å) | H···A (Å) | D···A (Å) | D-H···A (°) | Symmetry codes |
|---|---|---|---|---|---|
| N1-H1AA∙∙∙O5 | 0.99(3) | 1.87(3) | 2.848(2) | 169(2) | −x, −y, −z |
| N2-H2A∙∙∙O7 | 0.91 | 1.80 | 2.699(2) | 168 | 1 − x,1 − y,1 − z |
| N2-H2B∙∙∙O7 | 0.91 | 1. 91 | 2.741(2) | 151 | x, y, z |
| O2-H2B∙∙∙O6 | 0.84 | 1.89 | 2.7164(18) | 168 | 1 − x,1 − y,1 − z |
| C24-H24A∙∙∙O2 | 0.98 | 2.66 | 3.418(3) | 134 | x, 1 + y, z |
| O5-5HA∙∙∙Cg2 | 0.90(3) | 2.74(3) | 3.1361(19) | 108(2) | x, 1 + y, z |
| C8-H8∙∙∙O6 | 0.95 | 2.65 | 3.383(2) | 134 | −1 + x, −1 + y, z |
| C9-H9∙∙∙O2 | 0.95 | 2.68 | 3.597 | 163 | −x, −y, 1 − z |
| C27-H27∙∙∙Cg4 | 0.95 | 2.85 | 3.763(2) | 163 | −x, 1 − y, 1 − z |
| C13-H13A∙∙∙Cg4 | 0.99 | 2.86 | 3.603(2) | 133 | 1 − x, 1 − y, 1 − z |
| C15-H15A∙∙∙Cg8 | 0.99 | 2.69 | 3.403 | 129 | x, y, z |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hata, N.; Furuishi, T.; Tamboli, M.I.; Ishizaki, M.; Umeda, D.; Fukuzawa, K.; Yonemochi, E. Crystal Structural Analysis of DL-Mandelate Salt of Carvedilol and Its Correlation with Physicochemical Properties. Crystals 2020, 10, 53. https://doi.org/10.3390/cryst10010053
Hata N, Furuishi T, Tamboli MI, Ishizaki M, Umeda D, Fukuzawa K, Yonemochi E. Crystal Structural Analysis of DL-Mandelate Salt of Carvedilol and Its Correlation with Physicochemical Properties. Crystals. 2020; 10(1):53. https://doi.org/10.3390/cryst10010053
Chicago/Turabian StyleHata, Nanami, Takayuki Furuishi, Majid I. Tamboli, Momiji Ishizaki, Daiki Umeda, Kaori Fukuzawa, and Etsuo Yonemochi. 2020. "Crystal Structural Analysis of DL-Mandelate Salt of Carvedilol and Its Correlation with Physicochemical Properties" Crystals 10, no. 1: 53. https://doi.org/10.3390/cryst10010053
APA StyleHata, N., Furuishi, T., Tamboli, M. I., Ishizaki, M., Umeda, D., Fukuzawa, K., & Yonemochi, E. (2020). Crystal Structural Analysis of DL-Mandelate Salt of Carvedilol and Its Correlation with Physicochemical Properties. Crystals, 10(1), 53. https://doi.org/10.3390/cryst10010053