Applications of X-ray Powder Diffraction in Protein Crystallography and Drug Screening

 , , , ,
, , , ,
Abstract
:1. Introduction
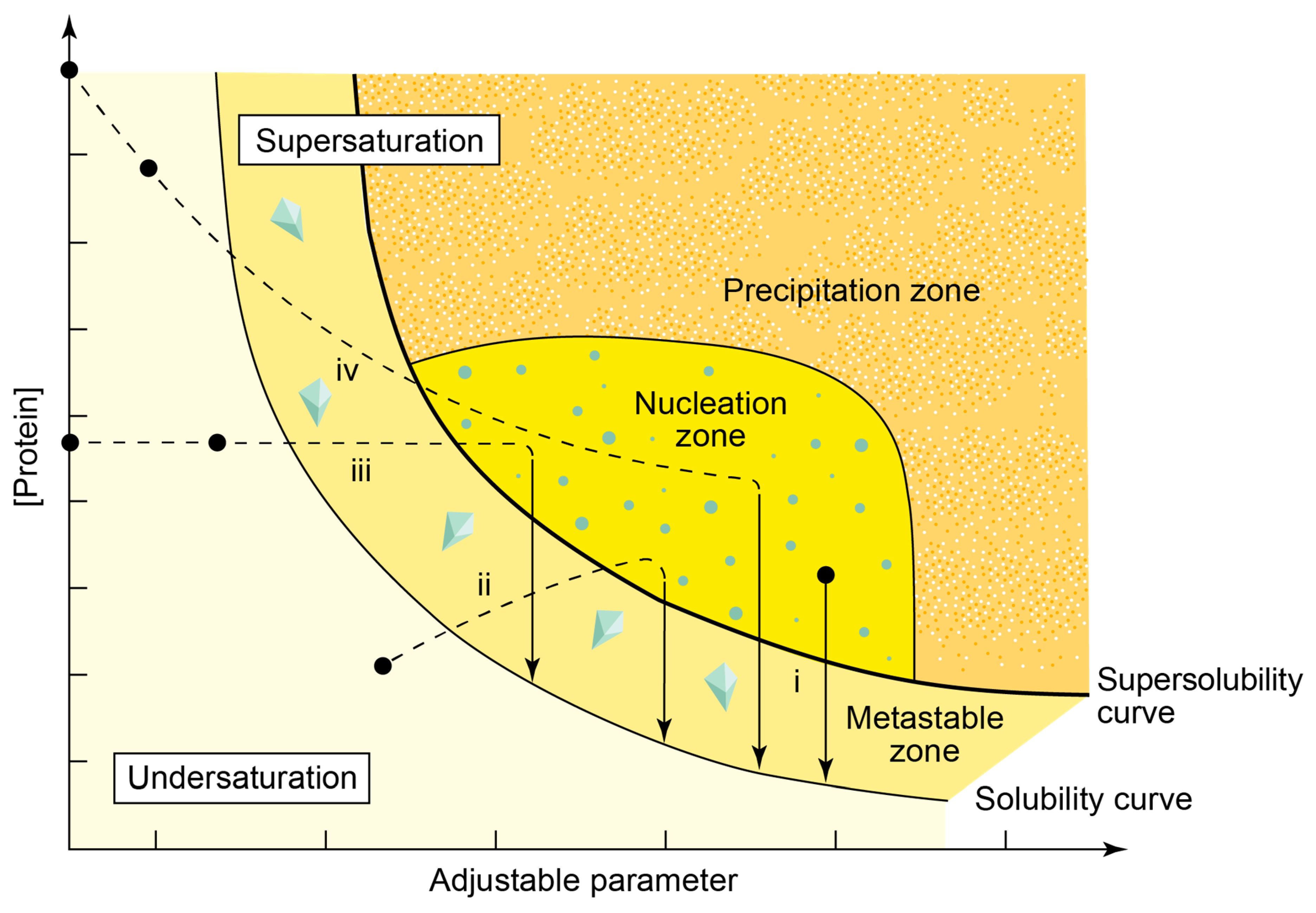
2. Challenging Samples: Macromolecular Assemblies & Subunits
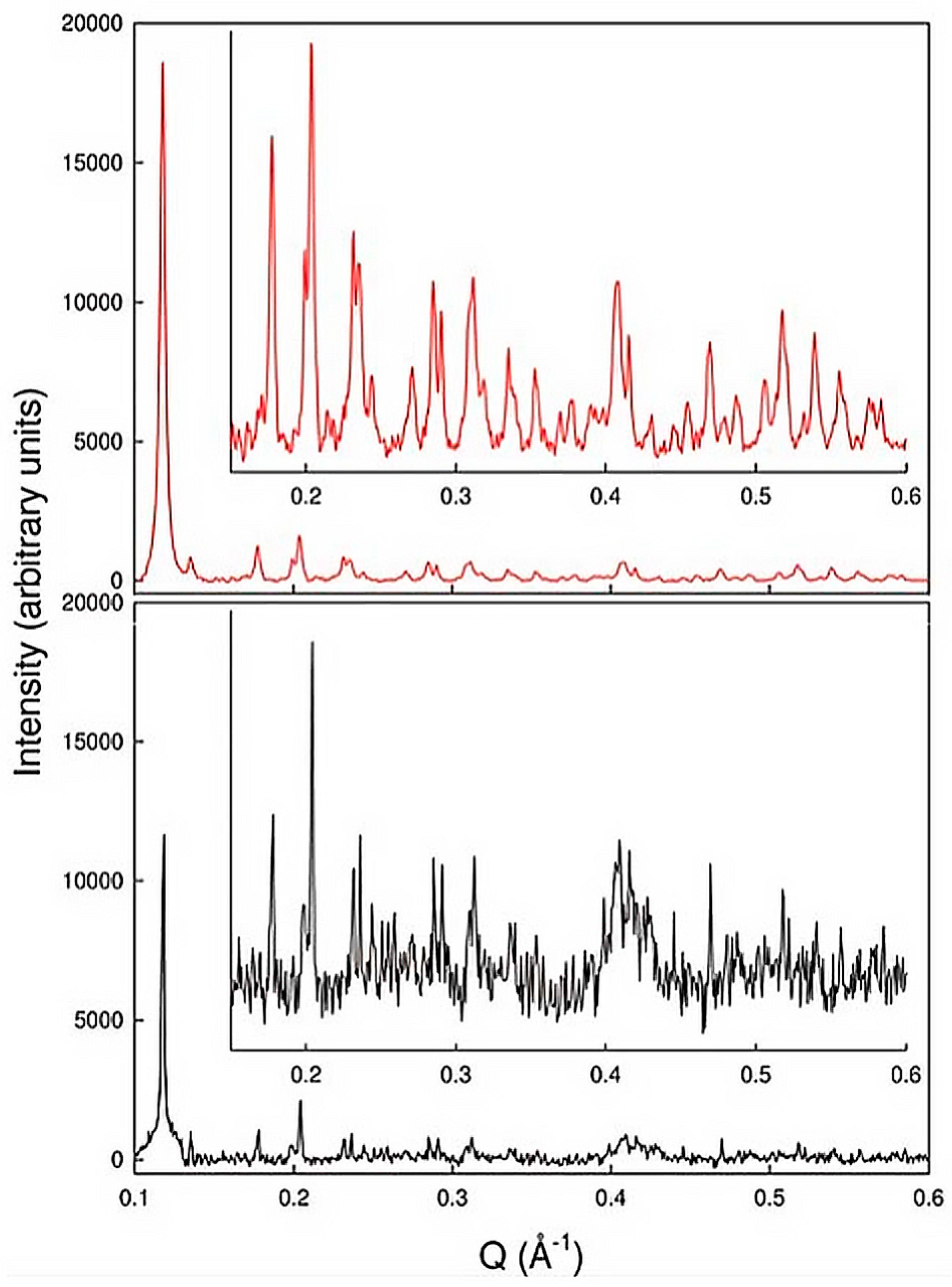
2.1. Polycrystalline Samples and First Virus Protein XRPD Studies
2.2. Preliminary Structural Data of Virus Proteins via XRPD
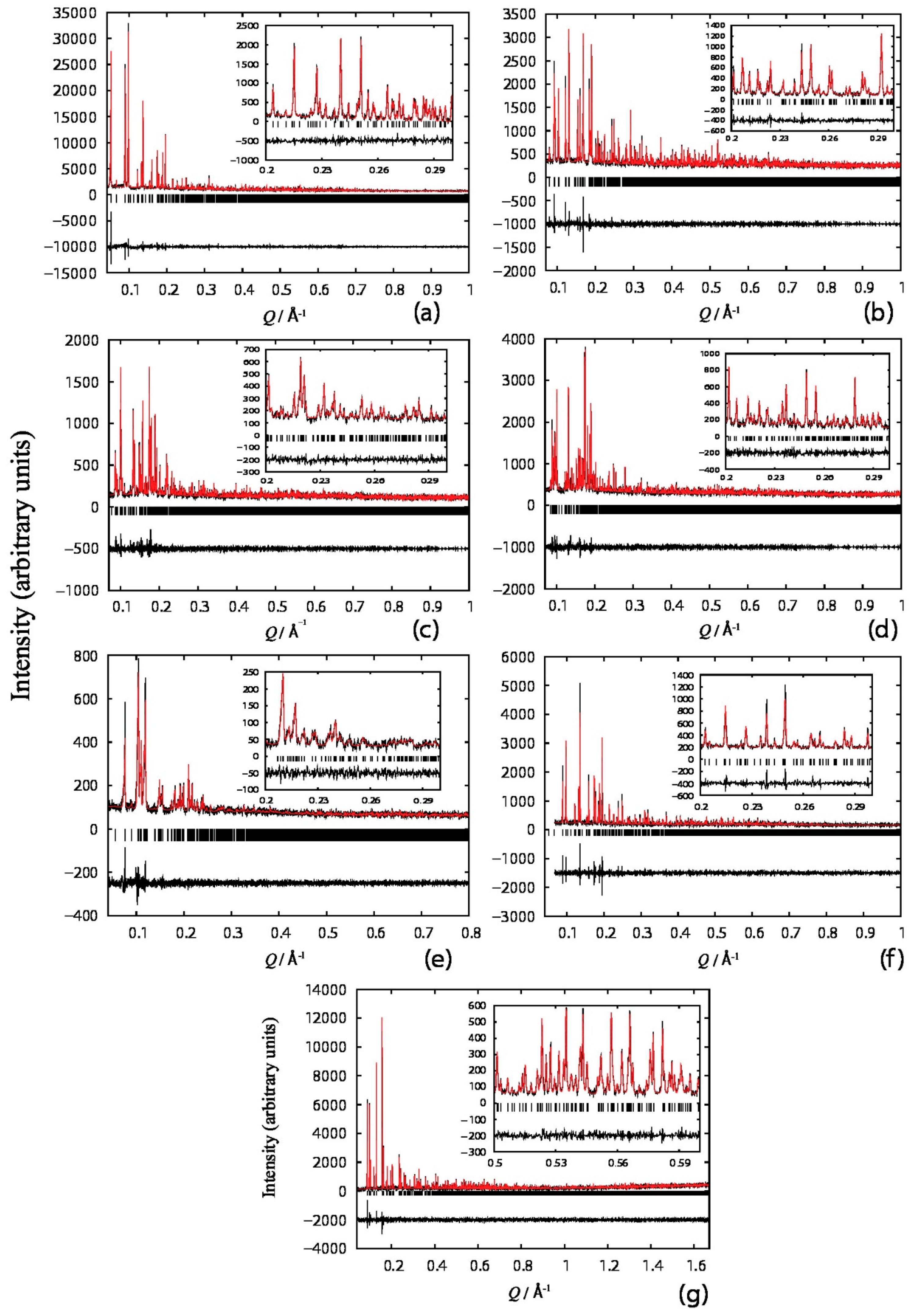
2.3. Protein Structure Solution via XRPD
3. Polymorph Identification
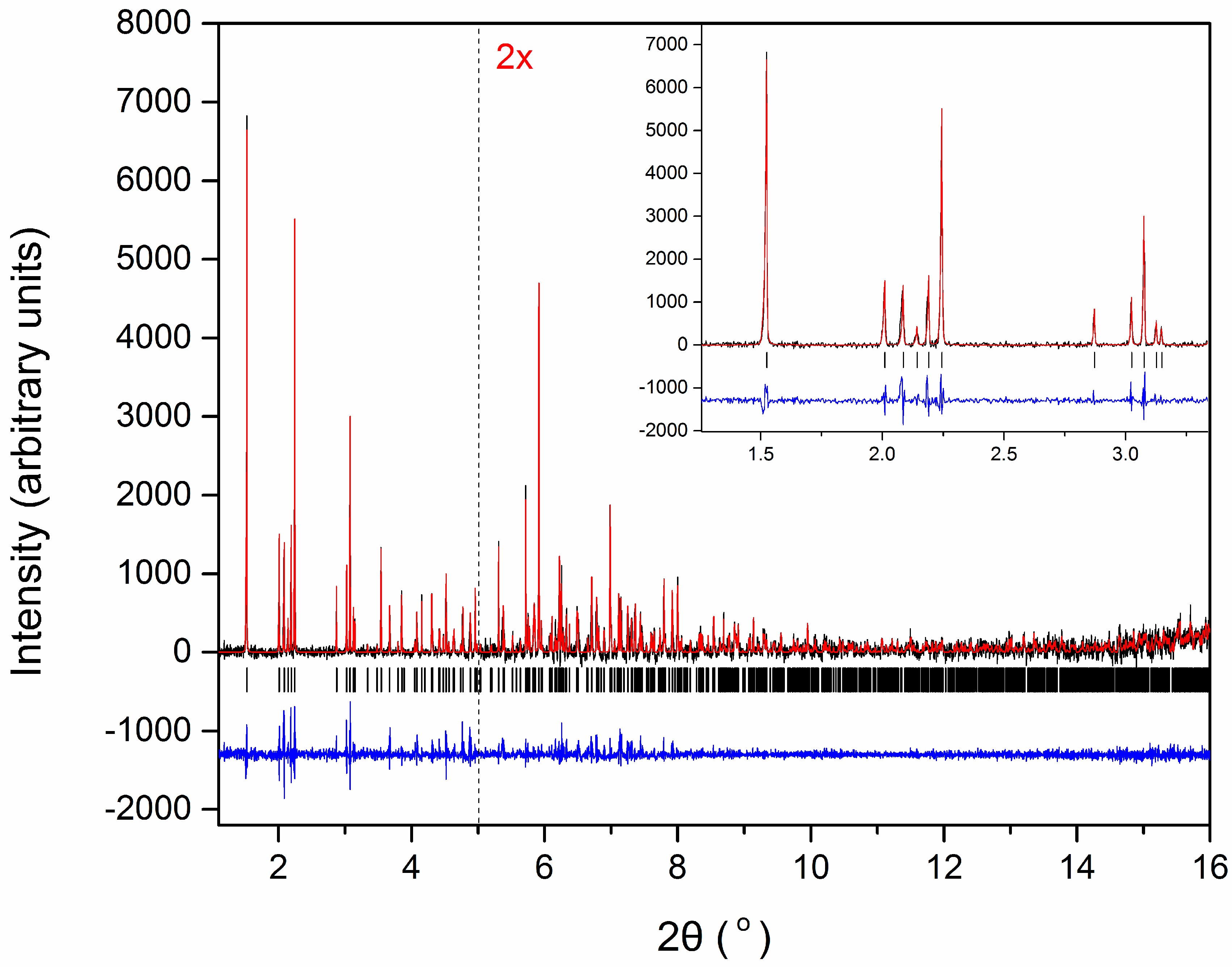
3.1. Macromolecular Polymorph Screening: The Case of Human Insulin
3.2. Distinct and Novel HI Polymorphs Identified via XRPD
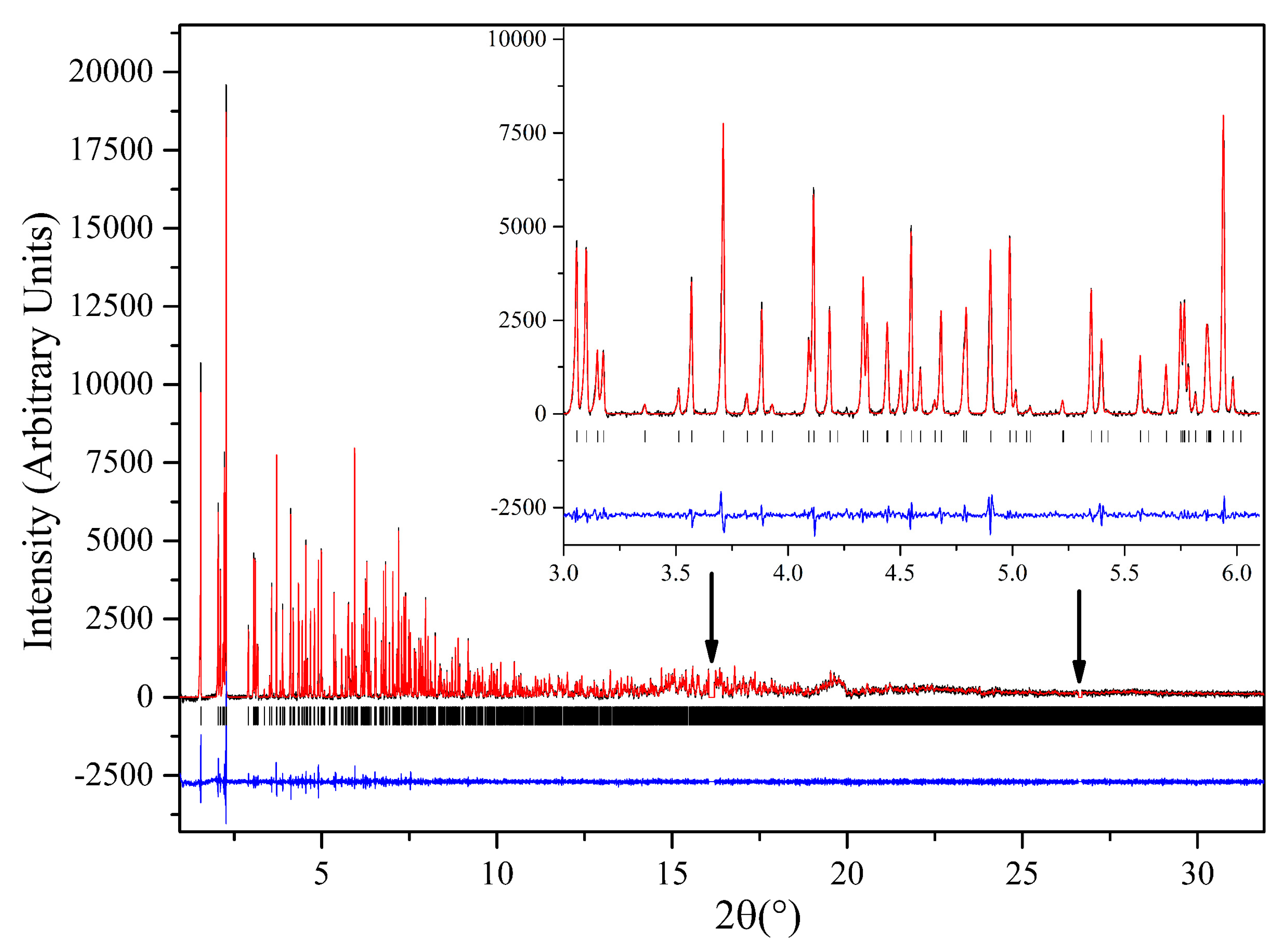
3.3. Macromolecular Polymorph Screening: The Case of Urate Oxidase
4. Drug Screening
4.1. Structure Refinement of a Pharmaceutical Peptide via XRPD
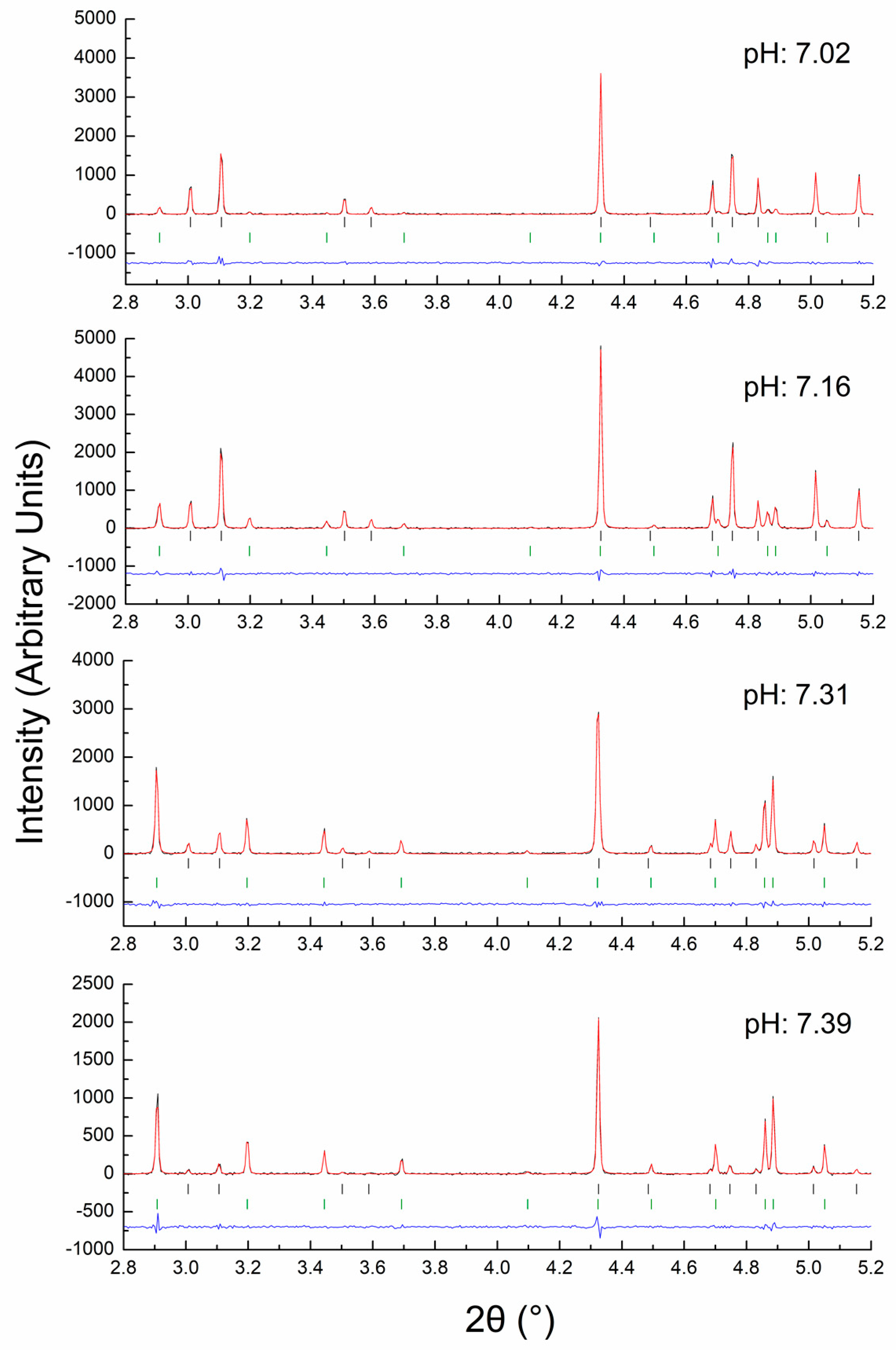
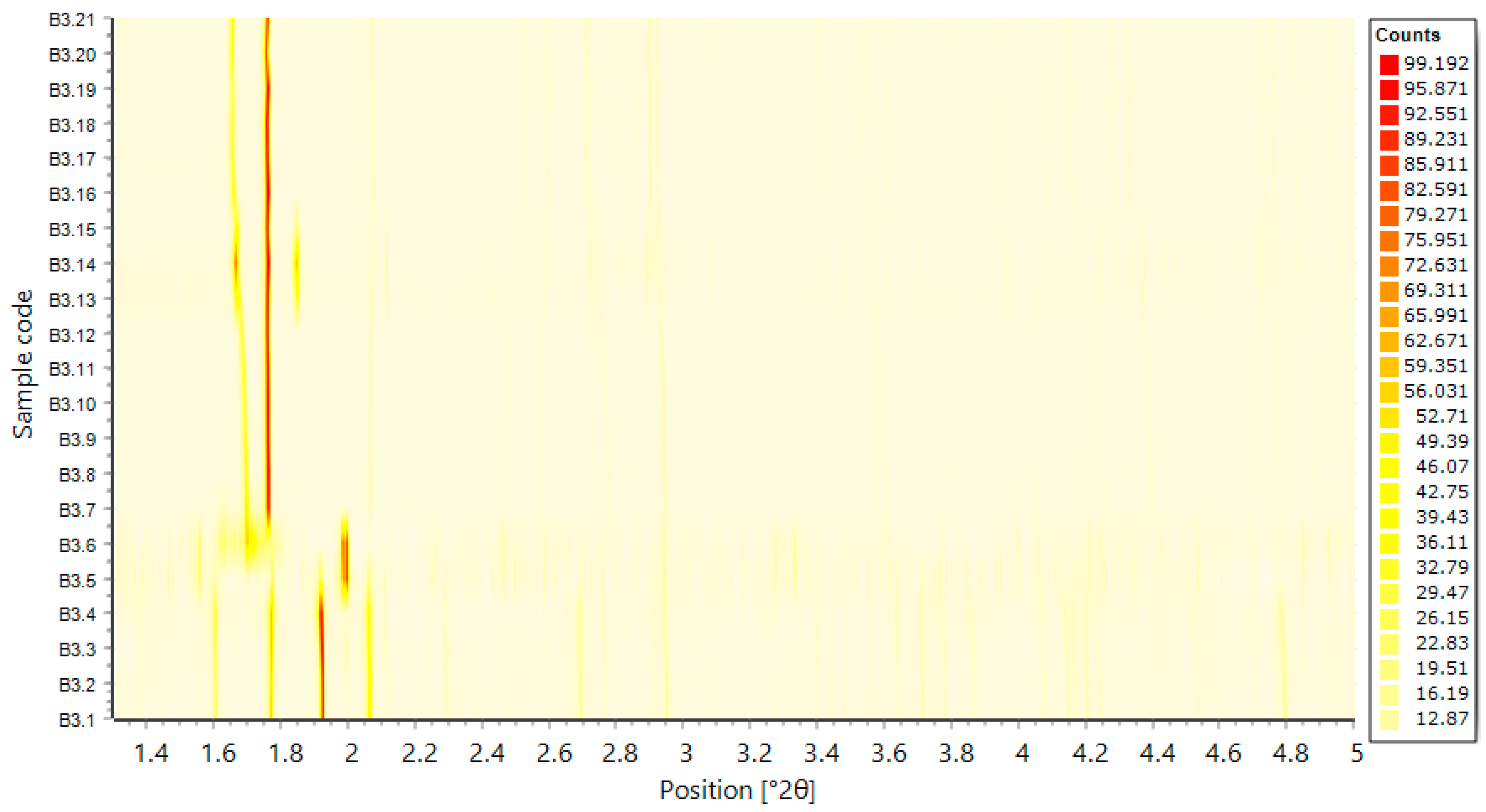
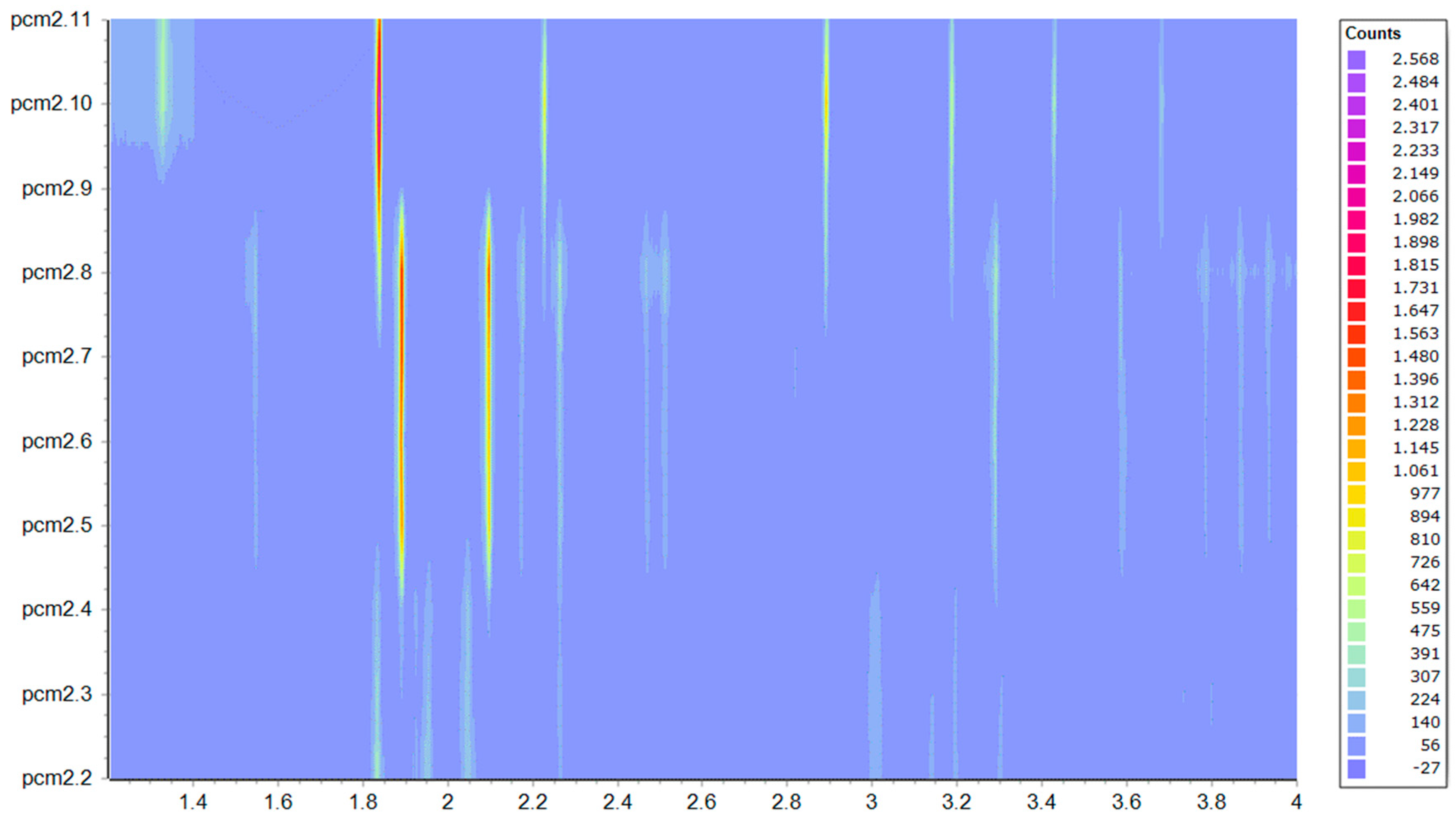
4.2. In Situ XRPD Measurements upon Variation of the Physicochemical Environment
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Russo Krauss, I.; Merlino, A.; Vergara, A.; Sica, F. An overview of biological macromolecule crystallization. Int. J. Mol. Sci. 2013, 14, 11643–11691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPherson, A. Introduction to protein crystallization. Methods 2004, 34, 254–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, G.; Axford, D.; Waterman, D.; Owen, R.L. Macromolecular microcrystallography. Crystallogr. Rev. 2011, 17, 105–142. [Google Scholar] [CrossRef]
- Debye, P.; Scherrer, P. Interferenzen an regellos orientierten Teilchen im Röntgenlicht. Phys. Z. 1916, 17, 277–291. [Google Scholar]
- Gingrich, N.S. The diffraction of x-rays by liquid elements. Rev. Mod. Phys. 1943, 15, 90–110. [Google Scholar] [CrossRef]
- Authier, A. Early Days of X-ray Crystallography; OUP: Oxford, UK, 2013. [Google Scholar]
- Dooryhee, E.; Greaves, G.N.; Steel, A.T.; Townsend, R.P.; Carr, S.W.; Thomas, J.M.; Catlow, C.R.A. Structural studies of high-area zeolitic adsorbents and catalysts by a combination of high-resolution X-ray powder diffraction and X-ray absorption spectroscopy. Faraday Discuss. Chem. Soc. 1990, 89, 119–136. [Google Scholar] [CrossRef]
- Cheetham, A.K.; Taylor, J.C. Profile analysis of neutron powder diffraction data: Its scope, limitations and applications in solid state chemistry. J. Solid State Chem. 1977, 21, 253–275. [Google Scholar] [CrossRef]
- Von Dreele, R.B. Protein Crystal Structure Analysis from High-Resolution X-ray Powder-Diffraction Data. Methods Enzymol. 2003, 368, 254–267. [Google Scholar]
- Margiolaki, I.; Wright, J.P.; Fitch, A.N.; Fox, G.C.; Von Dreele, R.B. Synchrotron X-ray Powder Diffraction Study of Hexagonal Turkey Egg-White Lysozyme. Acta Cryst. D 2005, 61, 423–432. [Google Scholar] [CrossRef]
- Margiolaki, I.; Wright, J.P.; Wilmanns, M.; Fitch, A.N.; Pinotsis, N. Second SH3 Domain of Ponsin Solved from Powder Diffraction. J. Am. Chem. Soc. 2007, 129, 11865–11871. [Google Scholar] [CrossRef]
- Margiolaki, I.; Giannopoulou, A.E.; Wright, J.P.; Knight, L.; Norrman, M.; Schluckebier, G.; Fitch, A.N.; Von Dreele, R.B. High-resolution powder X-ray data reveal the T6 hexameric form of bovine insulin. Acta Cryst. D 2013, 69, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Rietveld, H.M. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Young, R.A.; Mackie, P.E.; Von Dreele, R.B. Application of the pattern-fitting structure-refinement method of X-ray powder diffractometer patterns. J. Appl. Crystallogr. 1977, 10, 262–269. [Google Scholar] [CrossRef]
- Alexander, L.E. Forty years of quantitative diffraction analysis. In Advances in X-ray Analysis, 2nd ed.; McMurdie, H.F., Barrett, C.S., Newkirk, J.B., Ruud, C.O., Eds.; Plenum Press: New York, NY, USA, 1976; Volume 20, pp. 1–13. [Google Scholar]
- Klug, H.P.; Alexander, L.E. X-ray Diffraction Procedures for Polycrystalline and Amorphous Materials, 2nd ed.; John Wiley: New York, NY, USA, 1974; pp. 271–418. [Google Scholar]
- Harris, K.D.M.; Tremayne, M. Crystal Structure Determination from Powder Diffraction Data. Chem. Mater. 1996, 8, 2554–2570. [Google Scholar] [CrossRef]
- Das, U.; Naskar, J.; Mukherjee, A.K. Conformational analysis of an acyclic tetrapeptide: Ab-initio structure determination from X-ray powder diffraction, Hirshfeld surface analysis and electronic structure. J. Pept. Sci. 2015, 21, 845–852. [Google Scholar] [CrossRef]
- Gu, L.; Liu, C.; Guo, Z. Structural Insights into Aβ42 Oligomers Using Site-directed Spin Labeling. J. Biol. Chem. 2013, 288, 18673–18683. [Google Scholar] [CrossRef] [Green Version]
- Von Dreele, R.B. Combined Rietveld and stereochemical restraint refinement of a protein crystal structure. J. Appl. Crystallogr. 1999, 32, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Von Dreele, R.B.; Stephens, P.W.; Smith, G.D.; Blessing, R.H. The first protein crystal structure determined from high-resolution X-ray powder diffraction data: A variant of T3R3 human insulin–zinc complex produced by grinding. Acta Cryst. D 2000, 56, 1549–1553. [Google Scholar] [CrossRef]
- Von Dreele, R.B. Multipattern Rietveld refinement of protein powder data: An approach to higher resolution. J. Appl. Cryst. 2007, 40, 133–143. [Google Scholar] [CrossRef]
- Margiolaki, I.; Wright, J.P. Powder crystallography on macromolecules. Acta Cryst. A 2008, 64, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Collings, I.; Watier, Y.; Giffard, M.; Dagogo, S.; Kahn, R.; Bonneté, F.; Wright, J.P.; Fitch, A.N.; Margiolaki, I. Polymorphism of microcrystalline urate oxidase from Aspergillus flavus. Acta Cryst. D 2010, 66, 539–548. [Google Scholar] [CrossRef]
- Karavassili, F.; Margiolaki, I. Macromolecular Powder Diffraction: Ready for genuine biological problems. Protein Pept. Lett. 2016, 23, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Karavassili, F.; Valmas, A.; Fili, S.; Georgiou, C.; Margiolaki, I. In quest for improved drugs against diabetes: The added value of X-ray powder diffraction methods. Biomolecules 2017, 7, 63. [Google Scholar] [CrossRef]
- Margiolaki, I. Macromolecular powder diffraction. In International Tables for Crystallography, 1st ed; Gilmore, C.J., Kaduk, J.A., Schenk, H., Eds.; John Wiley & Sons. Inc.: New York, NY, USA, 2019; Volume H, pp. 718–736. [Google Scholar]
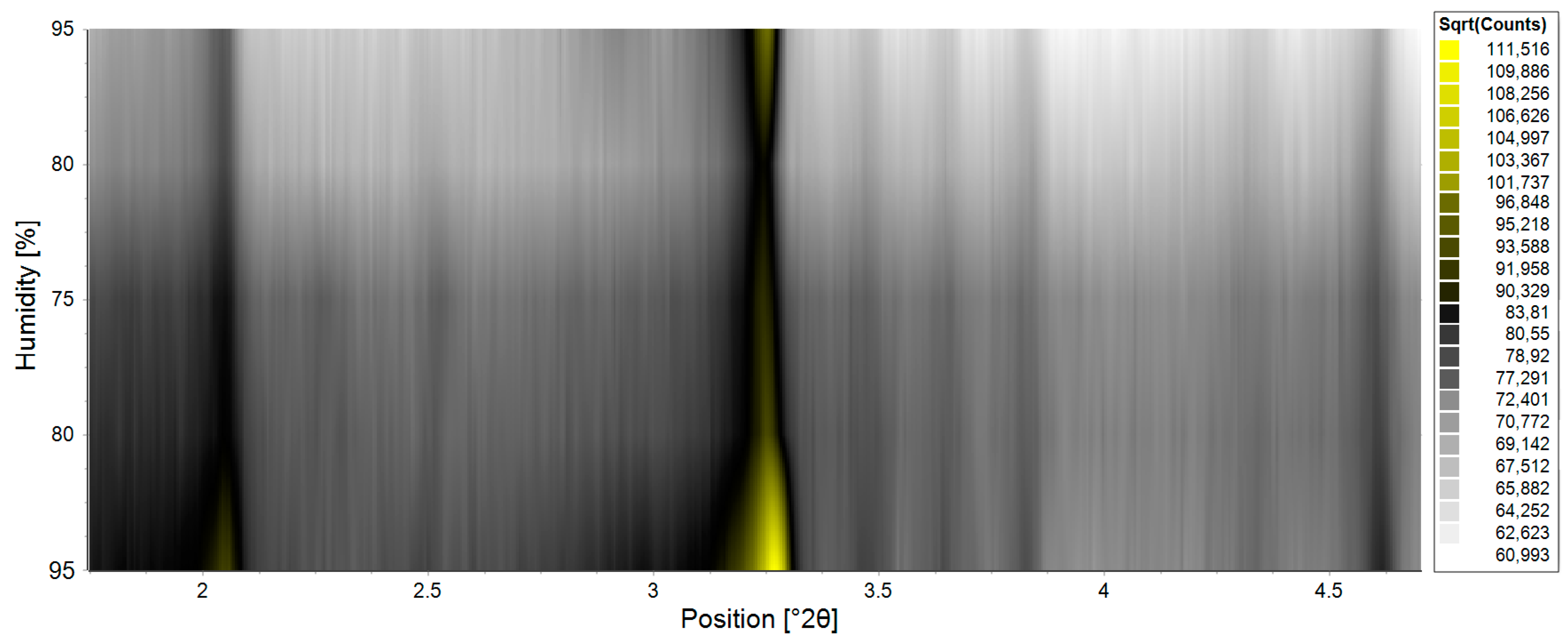
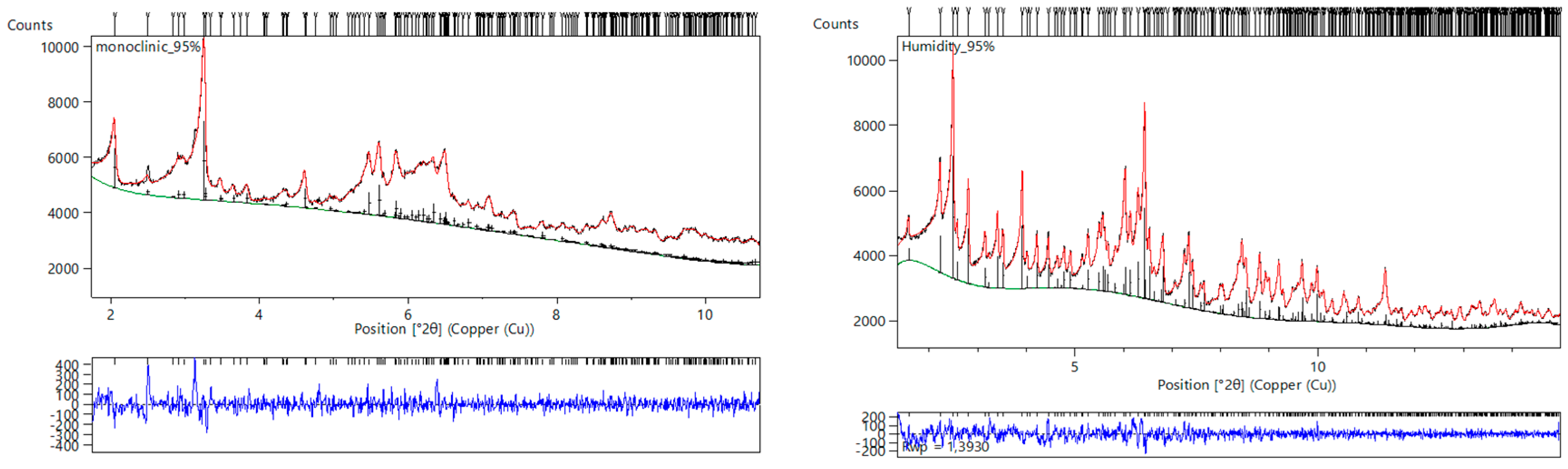
- Trampari, S.; Valmas, A.; Logotheti, S.; Saslis, S.; Fili, S.; Spiliopoulou, M.; Beckers, D.; Degen, T.; Nénert, G.; Fitch, A.N.; et al. In situ detection of a novel lysozyme monoclinic crystal form upon controlled relative humidity variation. J. Appl. Cryst. 2018, 51, 1671–1683. [Google Scholar] [CrossRef]
- Logotheti, S.; Valmas, A.; Trampari, S.; Fili, S.; Saslis, S.; Spiliopoulou, M.; Beckers, D.; Degen, T.; Nénert, G.; Fitch, A.N.; et al. Unit-cell response of tetragonal hen egg white lysozyme upon controlled relative humidity variation. J. Appl. Cryst. 2019, 52, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Karavassili, F.; Giannopoulou, A.E.; Kotsiliti, E.; Knight, L.; Norrman, M.; Schluckebier, G.; Drube, L.; Fitch, A.N.; Wright, J.P.; Margiolaki, I. Structural studies of human insulin cocrystallized with phenol or resorcinol via powder diffraction. Acta Cryst. D 2012, 68, 1632–1641. [Google Scholar] [CrossRef]
- Valmas, A.; Magiouf, K.; Fili, S.; Norrman, M.; Schluckebier, G.; Beckers, D.; Degen, T.; Wright, J.; Fitch, A.; Gozzo, F.; et al. Novel crystalline phase and first-order phase transitions of human insulin complexed with two distinct phenol derivatives. Acta Cryst. D 2015, 71, 819–828. [Google Scholar] [CrossRef]
- Fili, S.; Valmas, A.; Norrman, M.; Schluckebier, G.; Beckers, D.; Degen, T.; Wright, J.; Fitch, A.; Gozzo, F.; Giannopoulou, A.E.; et al. Human insulin polymorphism upon ligand binding and pH variation: The case of 4-ethylresorcinol. IUCrJ 2015, 2, 534–544. [Google Scholar] [CrossRef]
- Caira, M.R. Currents Applications of Powder X-ray Diffraction in Drug Discovery and Development. Am. Pharm. Rev. 2014, 17, 54. [Google Scholar]
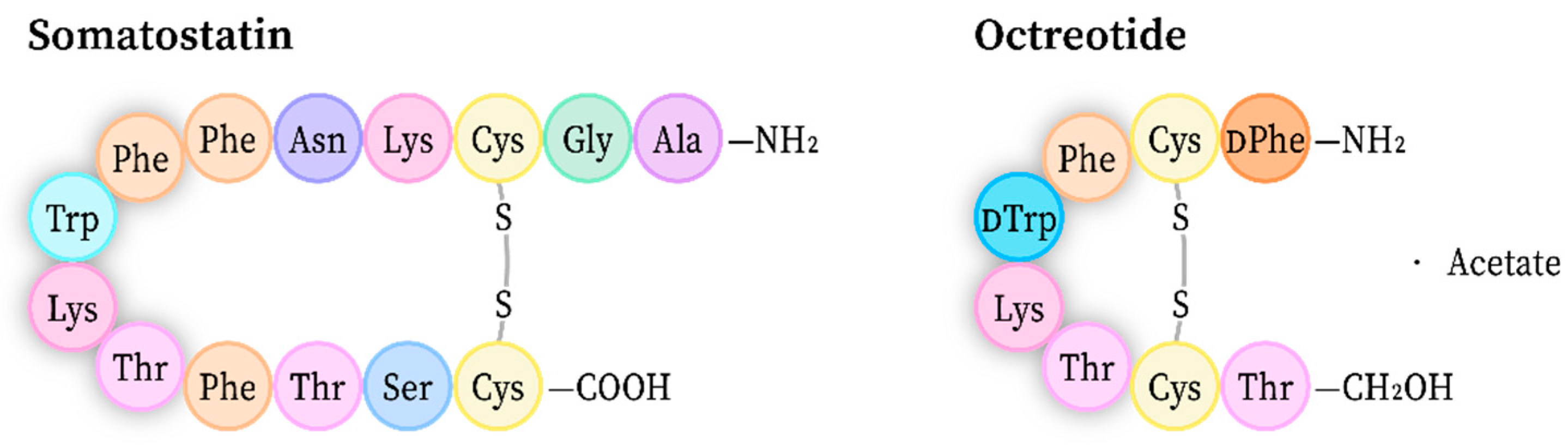
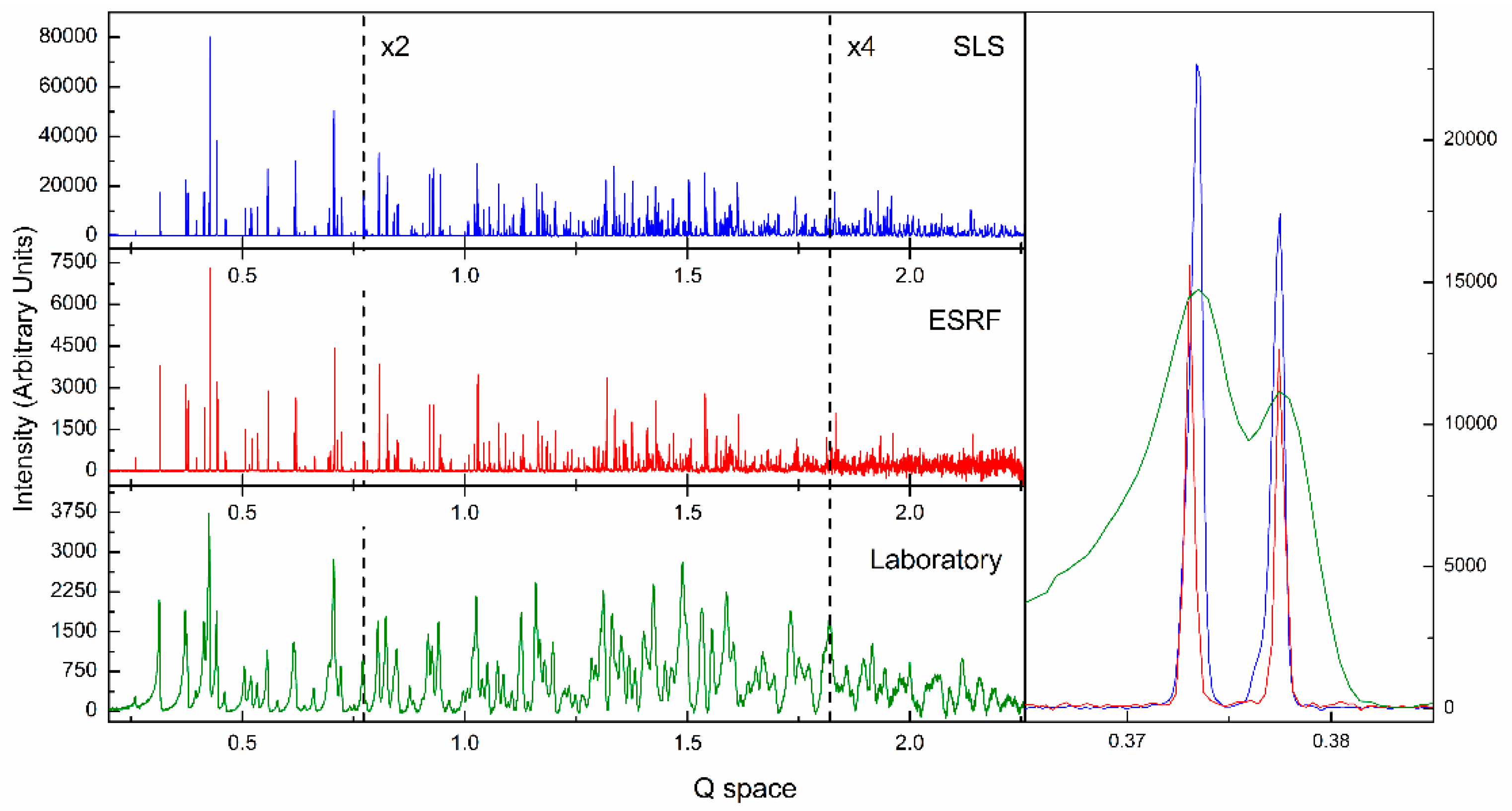
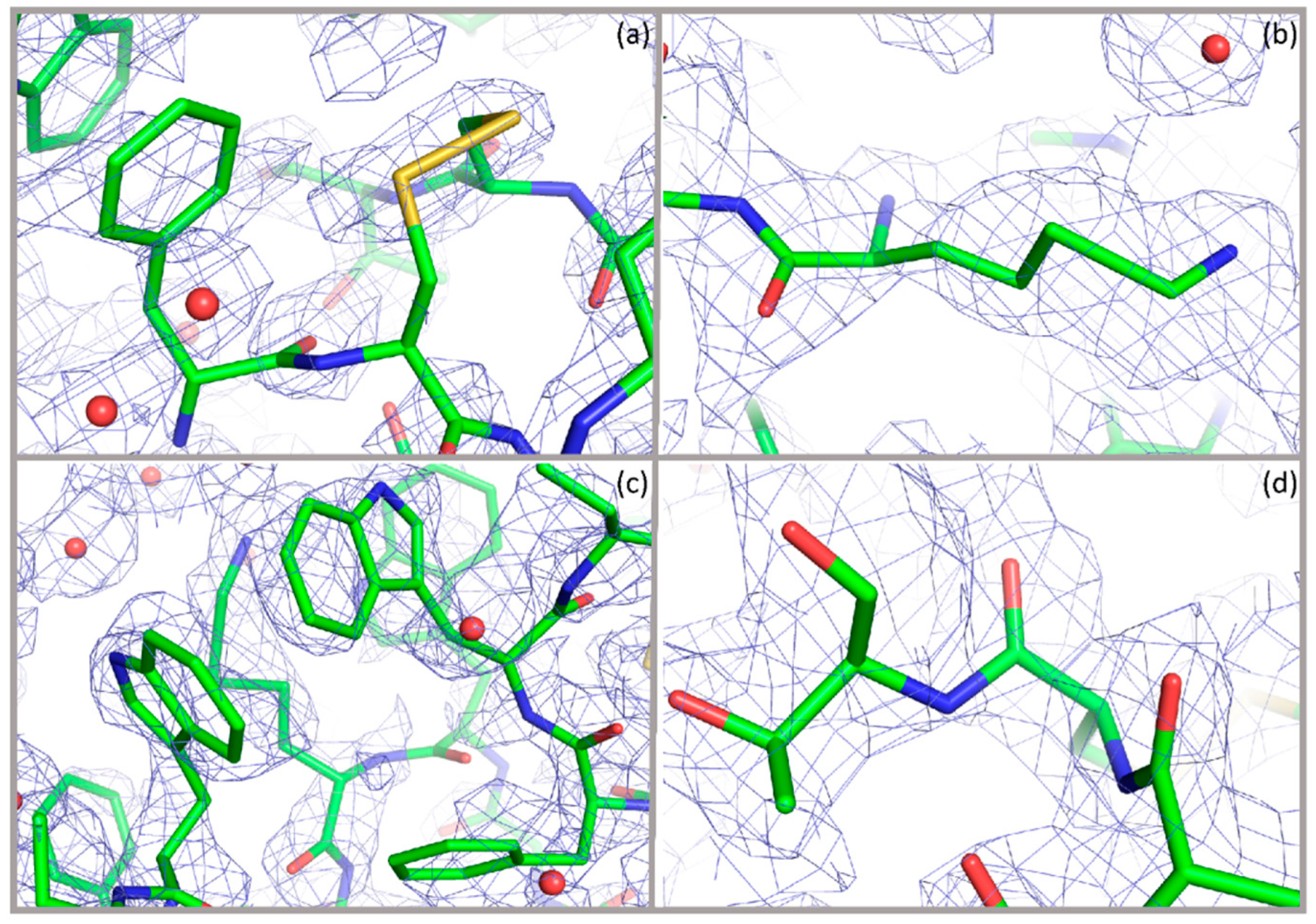
- Fili, S.; Valmas, A.; Spiliopoulou, M.; Kontou, P.; Fitch, A.N.; Beckers, D.; Degen, T.; Barlos, K.; Barlos, K.K.; Karavassili, F.; et al. Revisiting the structure of a synthetic Somatostatin analogue for peptide drug design. Acta Cryst. B 2019, 75, 611–620. [Google Scholar] [CrossRef]
- Basu, S.K.; Govardhan, C.P.; Jung, C.W.; Margolin, A.L. Protein crystals for the delivery of biopharmaceuticals. Expert Opin. Biol. Ther. 2004, 4, 301–317. [Google Scholar] [CrossRef]
- Chyall, L.J. Current Applications of X-ray Powder Diffraction in the Pharmaceutical Industry. Am. Pharm. Rev. 2012, 15, 70. [Google Scholar]
- Das, R.; Ali, E.; Abd Hamid, S.B. Current applications of X-ray powder diffraction—A review. Rev. Adv. Mater. Sci. 2014, 38, 95–109. [Google Scholar]
- Yin, S.X.; Scaringe, R.P.; Malley, M.F.; Gougoutas, J.Z. In-situ variable temperature powder X-ray diffraction and thermal analysis-applications in the pharmaceutical industry. Am. Pharm. Rev. 2005, 8, 56–58, 60, 62, 67. [Google Scholar]
- Ungár, T. The Meaning of Size Obtained from Broadened X-ray Diffraction Peaks. Adv. Eng. Mater. 2003, 5, 323–329. [Google Scholar] [CrossRef]
- Dale, G.E.; Oefner, C.; D’Arcy, A. The protein as a variable in protein crystallization. J. Struct. Biol. 2003, 142, 88–97. [Google Scholar] [CrossRef]
- Verdaguer, N.; Garriga, D.; Fita, I. X-Ray crystallography of viruses. In Structure and Physics of Viruses; Springer: Dordrecht, The Netherlands, 2013; pp. 117–144. [Google Scholar]
- Carrillo-Tripp, M.; Shepherd, C.M.; Borelli, I.A.; Venkataraman, S.; Lander, G.; Natarajan, P.; Johnson, J.E.; Brooks, C.L., III; Reddy, V.S. VIPERdb2: An enhanced and web API enabled relational database for structural virology. Nucleic Acids Res. 2009, 37, D436–D442. [Google Scholar] [CrossRef]
- Verdaguer, N.; Ferrero, D.; Murthy, M.R. Viruses and viral proteins. IUCrJ 2014, 1, 492–504. [Google Scholar] [CrossRef]
- Berman, J.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Bassetto, M.; Massarotti, A.; Coluccia, A.; Brancale, A. Structural biology in antiviral drug discovery. Curr. Opin. Pharmacol. 2016, 30, 116–130. [Google Scholar] [CrossRef]
- D’Arcy, A.L.L.A.N. Crystallizing proteins—A rational approach? Acta Cryst. D 1994, 50, 469–471. [Google Scholar] [CrossRef]
- Abola, E.; Kuhn, P.; Earnest, T.; Stevens, R.C. Automation of X-ray crystallography. Nat. Struct. Biol. 2000, 7, 973–977. [Google Scholar] [CrossRef]
- Blundell, T.L.; Jhoti, H.; Abell, C. High-throughput crystallography for lead discovery in drug design. Nat. Rev. Drug Discov. 2002, 1, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Boettner, M.; Prinz, B.; Holz, C.; Stahl, U.; Lang, C. Highthroughput screening for expression of heterologous proteins in the yeast Pichia pastoris. J. Biotechnol. 2002, 99, 51–62. [Google Scholar] [CrossRef]
- Chayen, N.E.; Saridakis, E. Protein crystallization for genomics: Towards high-throughput optimization techniques. Acta Cryst. D 2002, 58, 921–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieckman, L.; Gu, M.; Stols, L.; Donnelly, M.I.; Collart, F.R. High throughput methods for gene cloning and expression. Protein Expr. Purif. 2002, 25, 1–7. [Google Scholar] [CrossRef]
- Karain, W.I.; Bourenkov, G.P.; Blume, H.; Bartunik, H.D. Automated mounting, centering and screening of crystals for highthroughput protein crystallography. Acta Cryst. D 2002, 58, 1519–1522. [Google Scholar] [CrossRef]
- Rupp, B. Origin and use of crystallization phase diagrams. Acta Cryst. F 2015, 71, 247–260. [Google Scholar] [CrossRef] [Green Version]
- Gavira, J.A.; Garcia-Ruiz, J.M. Agarose as crystallisation media for proteins II: Trapping of gel fibres into the crystals. Acta Cryst. D 2002, 58, 1653–1656. [Google Scholar] [CrossRef] [Green Version]
- Moreno, A.; Quiroz-García, B.; Yokaichiya, F.; Stojanoff, V.; Rudolph, P. Protein crystal growth in gels and stationary magnetic fields. Cryst. Res. Technol. 2007, 42, 231–236. [Google Scholar] [CrossRef]
- Chayen, N.E.; Saridakis, E. Protein crystallization: From purified protein to diffraction-quality crystal. Nat. Methods 2008, 5, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Chayen, N.E.; Boggon, T.J.; Cassetta, A.; Deacon, A.; Gleichmann, T.; Habash, J.; Harrop, S.J.; Helliwell, J.R.; Nieh, Y.P.; Peterson, M.R.; et al. Trends and challenges in experimental macromolecular crystallography. Q. Rev. Biophys. 1996, 29, 227–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ataka, M. Protein crystal growth: An approach based on phase diagram determination. Phase Transit. 1993, 45, 205–219. [Google Scholar] [CrossRef]
- Stura, E.A.; Wilson, I.A. Application of the streak seeding technique in protein crystallization. J. Cryst. Growth 1991, 110, 270–282. [Google Scholar] [CrossRef]
- Chayen, N.E. Automation of non-conventional crystallization techniques for screening and optimization. In Macromolecular Crystallography: Conventional and High-Throughput Methods, 2nd ed.; Sanderson, M.R., Skelly, J.V., Eds.; Oxford University Press: Oxford, UK, 2007; pp. 45–58. [Google Scholar]
- Govada, L.; Chayen, N.E. Choosing the method of crystallization to obtain optimal results. Crystals 2019, 9, 106. [Google Scholar] [CrossRef] [Green Version]
- Chayen, N.E. Turning protein crystallisation from an art into a science. Curr. Opin. Struct. Biol. 2004, 14, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Corey, R.B.; Wyckoff, R.W.G. Long spacings in macromolecular solids. J. Biol. Chem. 1936, 114, 407–414. [Google Scholar]
- Wyckoff, R.W.G.; Corey, R.B. X-ray diffraction patterns of crystalline tobacco mosaic proteins. J. Biol. Chem. 1936, 116, 51–55. [Google Scholar]
- Bernal, J.D.; Fankuchen, I. X-ray and crystallographic studies of plant virus preparations: I. Introduction and preparation of specimens II. Modes of aggregation of the virus particles. J. Gen. Physiol. 1941, 25, 111–165. [Google Scholar]
- Xiaa, D.; Yu-Kunb, S.; McCraec, M.A.; Rossmann, M.G. X-ray powder pattern analysis of cytoplasmic polyhedrosis virus inclusion bodies. Virology 1991, 180, 153–158. [Google Scholar] [CrossRef]
- Watier, Y. Powder Diffraction Studies of Proteins. Ph.D. Thesis, University Joseph Fourier, Grenoble, France, 2011. [Google Scholar]
- Sliz, P.; Harrison, S.C.; Rosenbaum, G. How does radiation damage in protein crystals depend on X-ray dose? Structure 2003, 11, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Papageorgiou, N.; Watier, Y.; Saunders, L.; Coutard, B.; Lantez, V.; Gould, E.A.; Fitch, A.N.; Wright, J.P.; Canard, B.; Margiolaki, I. Preliminary insights into the non-structural protein 3 macro domain of the Mayaro virus by powder diffraction. Z. Kristallogr. Cryst. Mater. 2010, 225, 576–580. [Google Scholar] [CrossRef]
- Margiolaki, I. Proteins and Powders: An Overview. In Uniting Electron Crystallography and Powder Diffraction; Kolb, U., Shankland, K., Meshi, L., Avilov, A., David, W.I.F., Eds.; Springer: Dordrecht, The Netherlands, 2012; Volume 13, pp. 137–147. [Google Scholar]
- Fili, S.; Valmas, A.; Christopoulou, M.; Spiliopoulou, M.; Nikolopoulos, N.; Lichière, J.; Logotheti, S.; Karavassili, F.; Rosmaraki, E.; Fitch, A.; et al. Coxsackievirus B3 protease 3C: Expression, purification, crystallization and preliminary structural insights. Acta Cryst. F 2016, 72, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Valmas, A.; Fili, S.; Nikolopoulos, N.; Spiliopoulou, M.; Christopoulou, M.; Karavassili, F.; Kosinas, C.; Bastalias, K.; Rosmaraki, E.; Lichiére, J.; et al. Dengue virus 3 NS5 methyltransferase domain: Expression, purification, crystallization and first structural data from microcrystalline specimens. Z. Kristallogr. Cryst. Mater. 2017, 233, 309–316. [Google Scholar] [CrossRef]
- Lee, C.C.; Kuo, C.J.; Ko, T.P.; Hsu, M.F.; Tsui, Y.C.; Chang, S.C.; Yang, S.; Chen, S.J.; Chen, H.C.; Hsu, M.C.; et al. Structural basis of inhibition specificities of 3C and 3C-like proteases by zinc-coordinating and peptidomimetic compounds. J. Biol. Chem. 2009, 284, 7646–7655. [Google Scholar] [CrossRef] [Green Version]
- Marcotte, L.L.; Wass, A.B.; Gohara, D.W.; Pathak, H.B.; Arnold, J.J.; Filman, D.J.; Cameron, C.E.; Hogle, J.M. Crystal structure of poliovirus 3CD protein: Virally encoded protease and precursor to the RNA-dependent RNA polymerase. J. Virol. 2007, 81, 3583–3596. [Google Scholar] [CrossRef] [Green Version]
- Pawley, G.S. Unit-cell refinement from powder diffraction scans. J. Appl. Cryst. 1981, 14, 357–361. [Google Scholar] [CrossRef]
- Malavige, G.N.; Fernando, S.; Fernando, D.J.; Seneviratne, S.L. Dengue viral infections. Postgrad. Med. J. 2004, 80, 588–601. [Google Scholar] [CrossRef]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef]
- Egloff, M.P.; Benarroch, D.; Selisko, B.; Romette, J.L.; Canard, B. An RNA cap (nucleoside-2′-O-) methyltransferase in the flavivirus RNA polymerase NS5: Crystal structure and functional characterization. EMBO J. 2002, 21, 2757–2768. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.L.; Xu, T.; Chen, Y.L.; Malet, H.; Egloff, M.P.; Canard, B.; Vasudevan, S.G.; Lescar, J. Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J. Virol. 2007, 81, 4753–4765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutard, B.; Decroly, E.; Li, C.; Sharff, A.; Lescar, J.; Bricogne, G.; Barral, K. Assessment of Dengue virus helicase and methyltransferase as targets for fragment-based drug discovery. Antivir. Res. 2014, 106, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Soh, T.S.; Zheng, J.; Chan, K.W.K.; Phoo, W.W.; Lee, C.C.; Tay, M.Y.; Swaminathan, K.; Cornvik, T.C.; Lim, S.P.; et al. A crystal structure of the dengue virus NS5 protein reveals a novel inter-domain interface essential for protein flexibility and virus replication. PLoS Pathog. 2015, 11, e1004682. [Google Scholar] [CrossRef] [PubMed]
- Benmansour, F.; Trist, I.; Coutard, B.; Decroly, E.; Querat, G.; Brancale, A.; Barral, K. Discovery of novel dengue virus NS5 methyltransferase non-nucleoside inhibitors by fragment-based drug design. Eur. J. Med. Chem. 2017, 125, 865–880. [Google Scholar] [CrossRef]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Burla, M.C.; Polidori, G. On the number of statistically independent observations in a powder diffraction pattern. J. Appl. Crystallogr. 1995, 28, 738–744. [Google Scholar] [CrossRef]
- David, W.I.F. On the equivalence of the Rietveld method and the correlated integrated intensities method in powder diffraction. J. Appl. Crystallogr. 2004, 37, 621–628. [Google Scholar] [CrossRef]
- Wright, J.P.; Forsyth, J.B. PRODD, Profile Refinement of Diffraction Data using the Cambridge Crystallographic Subroutine Library. In Rutherford Appleton Laboratory Report RAL-TR-2000-012; CCLRC: Didcot, Oxon, UK, 2000. [Google Scholar]
- Wright, J.P. Extraction and use of correlated integrated intensities with powder diffraction data. Z. Kristallogr. Cryst. Mater. 2004, 219, 791–802. [Google Scholar] [CrossRef]
- Le Bail, A.; Duroy, H.; Fourquet, J.L. Ab-initio structure determination of LiSbWO6 by X-ray powder diffraction. Mater. Res. Bull. 1988, 23, 447–452. [Google Scholar] [CrossRef]
- Rossmann, M.G. The molecular replacement method. Acta Cryst. A 1990, 46, 73–82. [Google Scholar] [CrossRef]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Larson, A.C.; Von Dreele, R.B. General Structure Analysis System (GSAS). In Los Alamos National Laboratory Report LAUR; UC Regents: Los Alamos, CA, USA, 2004; pp. 86–748. [Google Scholar]
- Von Dreele, R.B. Binding of N-acetylglucosamine to chicken egg lysozyme: A powder diffraction study. Acta Cryst. D 2001, 57, 1836–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Dreele, R.B. Binding of N-acetylglucosamine oligosaccharides to hen egg-white lysozyme: A powder diffraction study. Acta Cryst. D 2005, 61, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Doebbler, J.A.; Von Dreele, R.B. Application of molecular replacement to protein powder data from image plates. Acta Cryst. D 2009, 65, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, J.; Cheng, A.; DeYoung, S.M.; Chen, X.; Dold, L.H.; Saltiel, A.R. CAP interacts with cytoskeletal proteins and regulates adhesion-mediated ERK activation and motility. EMBO J. 2006, 25, 5284–5293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehmlich, K.; Pinotsis, N.; Hayeß, K.; van der Ven, P.F.; Milting, H.; El Banayosy, A.; Körfer, R.; Wilmanns, M.; Ehler, E.; Fürst, D.O. Paxillin and ponsin interact in nascent costameres of muscle cells. J. Mol. Biol. 2007, 369, 665–682. [Google Scholar] [CrossRef]
- Larson, A.C.; Von Dreele, R.B. General structure analysis system (GSAS). In Los Alamos National Laboratory Report LAUR; UC Regents: Los Alamos, CA, USA, 1994; pp. 86–748. [Google Scholar]
- Basso, S.; Fitch, A.N.; Fox, G.C.; Margiolaki, I.; Wright, J.P. High-throughput phase-diagram mapping via powder diffraction: A case study of HEWL versus pH. Acta Cryst. D 2005, 61, 1612–1625. [Google Scholar] [CrossRef] [Green Version]
- Haleblian, J.; McCrone, W. Pharmaceutical applications of polymorphism. J. Pharm. Sci. 1969, 58, 911–929. [Google Scholar] [CrossRef]
- Triandafillidis, D.T.; Parthenios, N.; Spiliopoulou, M.; Valmas, A.; Gozzo, F.; Reinle-Schmitt, M.; Beckers, D.; Degen, T.; Pop, M.; Fitch, A.; et al. Insulin polymorphism induced by two polyphenols: New crystal forms and advances in macromolecular powder diffraction. J. Pharm. Sci. 2020. submitted. [Google Scholar]
- Katrincic, L.M.; Sun, Y.T.; Carlton, R.A.; Diederich, A.M.; Mueller, R.L.; Vogt, F.G. Characterization, selection, and development of an orally dosed drug polymorph from an enantiotropically related system. Int. J. Pharm. 2009, 366, 1–13. [Google Scholar] [CrossRef]
- Newman, A. XRD-X-ray Powder Diffraction in Solid Form Screening and Selection. Am. Pharm. Rev. 2011, 14, 44. [Google Scholar]
- Láng, P.; Kiss, V.; Ambrus, R.; Farkas, G.; Szabó-Révész, P.; Aigner, Z.; Várkonyi, E. Polymorph screening of an active material. J. Pharm. Biomed. Anal. 2013, 84, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Van Eupen, J.T.H.; Westheim, R.; Deij, M.A.; Meekes, H.; Bennema, P.; Vlieg, E. The solubility behaviour and thermodynamic relations of the three forms of Venlafaxine free base. Int. J. Pharm. 2009, 368, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Stahly, G.P. Diversity in single-and multiple-component crystals. The search for and prevalence of polymorphs and cocrystals. Cryst. Growth Des. 2007, 7, 1007–1026. [Google Scholar] [CrossRef] [Green Version]
- Basavoju, S.; Boström, D.; Velaga, S.P. Pharmaceutical cocrystal and salts of norfloxacin. Cryst. Growth Des. 2006, 6, 2699–2708. [Google Scholar] [CrossRef]
- Engers, D.; Teng, J.; Jimenez-Novoa, J.; Gent, P.; Hossack, S.; Campbell, C.; Thomson, J.; Ivanisevic, I.; Templeton, A.; Byrn, S.; et al. A Solid-State Approach to Enable Early Development Compounds: Selection and Animsal Bioavailability Studies of an Itraconazole Amorphous Solid Dispersion. J. Pharm. Sci. 2010, 99, 3901–3922. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, M.; Gougoutas, J.Z.; Scaringe, R.P.; Vitéz, I.; Yin, S. Detection of polymorphism by powder X-ray diffraction: Interference by preferred orientation. Am. Pharm. Rev. 2004, 7, 10–17. [Google Scholar]
- Banting, F.G.; Best, C.H. The internal secretion of the pancreas. J. Lab. Clin. Med. 1922, 7, 251–266. [Google Scholar]
- Adams, M.J.; Blundell, T.L.; Dodson, E.J.; Dodson, G.G.; Vijayan, M.; Baker, E.N.; Harding, M.M.; Hodgkin, D.C.; Rimmer, B.; Sheat, S. Structure of Rhombohedral 2 Zinc Insulin Crystals. Nature 1969, 224, 491–495. [Google Scholar] [CrossRef]
- Brange, J.; Langkjœr, L. Insulin structure and stability. In Stability and Characterization of Protein and Peptide Drugs; Springer: Boston, MA, USA, 2013; pp. 315–350. [Google Scholar]
- Chang, S.G.; Choi, K.D.; Jang, S.H.; Shin, H.C. Role of Disulfide Bonds in the Structure and Activity of Human Insulin. Mol. Cells 2003, 16, 323–330. [Google Scholar]
- Abel, J.J. Crystalline insulin. Proc. Natl. Acad. Sci. USA 1926, 12, 132–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, D.A. Crystalline insulin. Biochem. J. 1934, 28, 1592–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halban, P.A.; Mutkoski, R.; Dodson, G.; Orci, L. Resistance of the insulin crystal to lysosomal proteases: Implications for pancreatic B-cell crinophagy. Diabetologia 1987, 30, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Hagedorn, H.C.; Jensen, B.N.; Krarup, N.B.; Wodstrup, I. Protamine insulinate. J. Intern. Med. 1936, 90, 678–684. [Google Scholar]
- Hallas-Møller, K.; Jersild, M.; Petersen, K.; Schlichtkrull, J. Zinc insulin preparations for single daily injection: Clinical studies of new preparations with prolonged action. JAMA 1952, 150, 1667–1671. [Google Scholar] [CrossRef]
- Hartman, I. Insulin analogs: Impact on treatment success, satisfaction, quality of life, and adherence. Clin. Med. Res. 2008, 6, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Joshi, S.R.; Parikh, R.M.; Das, A.K. Insulin History, Biochemistry, Physiology and Pharmacology. Suppl. JAPI 2007, 55, 19–25. [Google Scholar]
- Van Lierop, B.; Ong, S.C.; Belgi, A.; Delaine, C.; Andrikopoulos, S.; Haworth, N.L.; Menting, J.G.; Lawrence, M.C.; Robinson, A.J.; Forbes, B.E. Insulin in motion: The A6-A11 disulfide bond allosterically modulates structural transitions required for insulin activity. Sci. Rep. 2017, 7, 17239–17254. [Google Scholar] [CrossRef]
- Norrman, M.; Schluckebier, G. Crystallographic characterization of two novel crystal forms of human insulin induced by chaotropic agents and a shift in pH. BMC Struct. Biol. 2007, 7, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.D.; Ciszak, E.; Magrum, L.A.; Pangborn, W.A.; Blessing, R.H. R6 hexameric insulin complexed with m-cresol or resorcinol. Acta Cryst. D 2000, 56, 1541–1548. [Google Scholar] [CrossRef]
- Norrman, M.; Ståhl, K.; Schluckebier, G.; Al-Karadaghi, S. Characterization of insulin microcrystals using powder diffraction and multivariate data analysis. J. Appl. Cryst. 2006, 39, 391–400. [Google Scholar] [CrossRef] [Green Version]
- Bhatnagar, S.; Srivastava, D.; Jayadev, M.S.K.; Dubey, A.K. Molecular variants and derivatives of insulin for improved glycemic control in diabetes. Prog. Biophys. Mol. Biol. 2006, 91, 199–228. [Google Scholar] [CrossRef] [PubMed]
- Dunn, M.F. Zinc-Ligand Interactions Modulate Assembly and Stability of the Insulin Hexamer—A Review. BioMetals 2005, 18, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Kaarsholm, N.C.; Ko, H.C.; Dunn, M.F. Comparison of solution structural flexibility and zinc binding domains for insulin, proinsulin, and miniproinsulin. Biochemistry 1989, 28, 4427–4435. [Google Scholar] [CrossRef]
- Derewenda, U.; Derewenda, Z.; Dodson, E.J.; Dodson, G.G.; Reynolds, C.D.; Smith, G.D.; Sparks, C.; Swenson, D. Phenol stabilizes more helix in a new symmetrical zinc insulin hexamer. Nature 1989, 338, 594–596. [Google Scholar] [CrossRef]
- Smith, G.D.; Pangborn, W.A.; Blessing, R.H. The structure of T6 human insulin at 1.0 Å resolution. Acta Cryst. D 2003, 59, 474–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahuel-Clermont, S.; French, C.A.; Kaarsholm, N.C.; Dunn, M.F. Mechanisms of Stabilization of the Insulin Hexamer through Allosteric Ligand Interactions. Biochemistry 1997, 36, 5837–5845. [Google Scholar] [CrossRef] [PubMed]
- Wold, S.; Esbensen, K.; Geladi, P. Principal Component Analysis. Chemom. Intell. Lab. Syst. 1987, 2, 37–52. [Google Scholar] [CrossRef]
- Zijlstra, E.; Heinemann, L.; Plum-Mörschel, L. Oral insulin reloaded: A structured approach. J. Diabetes Sci. Technol. 2014, 8, 458–465. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, S.; Arabi, A.; El-Menyar, A.; Abdulkarim, S.; Aljundi, A.; Alqahtani, A.; Arafa, S.; Al Suwaidi, J. Impact of polypharmacy on adherence to evidence-based medication in patients who underwent percutaneous coronary intervention. Curr. Vasc. Pharmacol. 2016, 14, 388–393. [Google Scholar] [CrossRef]
- Banerjee, A.; Ibsen, K.; Brown, T.; Chen, R.; Agatemor, C.; Mitragotri, S. Ionic liquids for oral insulin delivery. Proc. Natl. Acad. Sci. USA 2018, 115, 7296–7301. [Google Scholar] [CrossRef] [Green Version]
- Grayson, B.E.; Woods, S.C. Insulin. In Handbook of Biologically Active Peptides; Academic Press, Elsevier: Oxford, UK, 2013; pp. 1123–1128. [Google Scholar]
- Gursky, O.; Badger, J.; Li, Y.; Caspar, D.L. Conformational changes in cubic insulin crystals in the pH range 7–11. Biophys. J. 1992, 63, 1210–1220. [Google Scholar] [CrossRef] [Green Version]
- Whittingham, J.L.; Chaudhurri, S.; Dodson, E.J.; Moody, P.C.E.; Dodson, G.G. X-ray Crystallographic Studies on Hexameric Insulins in the Presence of Helix-Stabilizing Agents, Thiocyanate, Methylparaben and Phenol. Biochemistry 1995, 34, 15553–15563. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A. Crystallization of Proteins by Variation of pH or Temperature. Methods Enzymol. 1985, 114, 125–127. [Google Scholar] [PubMed]
- Farr, R.; Perryman, A.; Samudzi, C. Re-clustering the database for crystallization of macromolecules. J. Cryst. Growth 1998, 183, 653–668. [Google Scholar] [CrossRef]
- Abdalla, M.; Eltayb, W.A.; Samad, A.; SHM, E.; Dafaalla, T. Important factors influencing protein crystallization. Glob. J. Biotechnol. Biomater. Sci. 2016, 2, 025–028. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.Q.; Cheng, Q.D.; Chen, J.J.; Sun, D.S.; Ao, L.B.; Li, D.W.; Lu, Q.Q.; Yin, D.C. An investigation of the effects of varying pH on protein crystallization screening. CrystEngComm 2017, 19, 860–867. [Google Scholar] [CrossRef]
- Smith, G.D.; Pangborn, W.A.; Blessing, R.H. Phase changes in T3R3f human insulin: Temperature or pressure induced? Acta Cryst. D 2001, 57, 1091–1100. [Google Scholar] [CrossRef] [Green Version]
- Bentley, G.; Dodson, E.; Dodson, G.U.Y.; Hodgkin, D.; Mercola, D.A.N. Structure of insulin in 4-zinc insulin. Nature 1976, 261, 166–168. [Google Scholar] [CrossRef]
- Huus, K.; Havelund, S.; Olsen, H.B.; Sigurskjold, B.W.; van de Weert, M.; Frokjaer, S. Ligand Binding and Thermostability of Different Allosteric States of the Insulin Zinc-Hexamer. Biochemistry 2006, 45, 4014–4024. [Google Scholar] [CrossRef]
- Matthews, B.W. Solvent content of protein crystals. J. Mol. Biol. 1968, 33, 491–497. [Google Scholar] [CrossRef]
- McGrath, B.M.; Walsh, G. Directory of Therapeutic Enzymes; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2005. [Google Scholar]
- Yang, M.X.; Shenoy, B.; Disttler, M.; Patel, R.; McGrath, M.; Pechenov, S.; Margolin, A.L. Crystalline monoclonal antibodies for subcutaneous delivery. Proc. Natl. Acad. Sci. USA 2003, 100, 6934–6939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanisevic, I.; McClurg, R.B.; Schields, P.J. Uses of X-ray powder diffraction in the pharmaceutical industry. Pharm. Sci. Encycl. Drug Discov. Dev. Manuf. 2010, 1–42. [Google Scholar] [CrossRef]
- Litteer, B.; Beckers, D. Increasing application of X-ray powder diffraction in the pharmaceutical industry. Am. Lab. 2005, 37, 22–25. [Google Scholar]
- Rathbone, M.J.; Hadgraft, J.; Roberts, M.S.; Lane, M.E. Modified-Release Drug Delivery Technology; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2008. [Google Scholar]
- Gozzo, F. The Power of Synchrotron X-Ray Powder Diffraction for the Characterization of Pharmaceuticals. Pharm. Technol. 2018, 42, 18. [Google Scholar]
- Shargel, L.; Andrew, B.C.; Wu-Pong, S. Applied Biopharmaceutics and Pharmacokinetics; Appleton & Lange: Stamford, CT, USA, 1999; pp. 32–35. [Google Scholar]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug. Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.Q. Somatostatin: Likely the most widely effective gastrointestinal hormone in the human body. World J. Gastroenterol. 1997, 3, 201–204. [Google Scholar] [CrossRef]
- Pohl, E.; Heine, A.; Sheldrick, G.M.; Dauter, Z.; Wilson, K.S.; Kallen, J.; Pfaffli, P.J. Structure of octreotide, a somatostatin analogue. Acta Cryst. D 1995, 51, 48–59. [Google Scholar] [CrossRef]
- Brown, M.; Rivier, J.; Vale, W. Somatostatin: Analogs with selected biological activities. Science 1977, 196, 1467–1469. [Google Scholar] [CrossRef]
- Long, R.K.; Cakmak, H. Hypothalamic & Pituitary Hormones. In Basic and Clinical Pharmacology, 14th ed.; Katzung, B.G., Ed.; McGraw Hill Professional: New York, NY, USA, 2018; pp. 667–686. [Google Scholar]
- Fitch, A.N. The high resolution powder diffraction beam line at ESRF. J. Res. Natl. Inst. Stan. 2004, 109, 133–142. [Google Scholar] [CrossRef]
- Willmott, P.R.; Meister, D.; Leake, S.J.; Lange, M.; Bergamaschi, A.; Böge, M.; Calvi, M.; Cancellieri, C.; Casati, N.; Cervellino, A.; et al. The materials science beamline upgrade at the Swiss Light Source. J. Synchrotron Radiat. 2013, 20, 667–682. [Google Scholar] [CrossRef]
- Datta, S.; Grant, D.J.W. Crystal structures of drugs: Advances in determination, prediction and engineering. Nat. Rev. Drug Discov. 2004, 3, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Borghetti, G.S.; Carini, J.P.; Honorato, S.B.; Ayala, A.P.; Moreira, J.C.F.; Bassani, V.L. Physicochemical properties and thermal stability of quercetin hydrates in the solid state. Thermochim. Acta 2012, 539, 109–114. [Google Scholar] [CrossRef]
- Khatchaturyan, A.G. Phase transformation in crystals composed of macromolecules. Zh. Eksp. Teor. Fiz. 1977, 72, 1149–1155. [Google Scholar]
- Morozov, V.N.; Morozova, T.Y. Viscoelastic properties of protein crystals: Triclinic crystals of hen egg white lysozyme in different conditions. Biopolymers 1981, 20, 451–467. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A. Protein crystallization in the structural genomics era. J. Struct. Funct. Genom. 2004, 5, 3–12. [Google Scholar] [CrossRef]
- Naschberger, A.; Juyoux, P.; von Velsen, J.; Rupp, B.; Bowler, M.W. Controlled dehydration, structural flexibility, and Gadolinium MRI contrast compound binding in human plasma glycoprotein afamin. bioRxiv 2019, 673376. [Google Scholar] [CrossRef]
- Crowfoot, D.; Riley, D. Crystal Structures of the Proteins An X-Ray Study of Palmar’s Lactoglobulin. Nature 1938, 141, 521–522. [Google Scholar] [CrossRef] [Green Version]
- Perutz, M.F. The composition and swelling properties of haemoglobin crystals. Trans. Faraday Soc. B 1946, 42, 187–194. [Google Scholar] [CrossRef]
- Huxley, H.E.; Kendrew, J.C. Discontinuous lattice changes in haemoglobin crystals. Acta Cryst. 1953, 6, 76–80. [Google Scholar] [CrossRef]
- Nagendra, H.G.; Sukumar, N.; Vijayan, M. Role of water in plasticity, stability, and action of proteins: The crystal structures of lysozyme at very low levels of hydration. Proteins 1998, 32, 229–240. [Google Scholar] [CrossRef]
- Salunke, D.M.; Veerapandian, B.; Kodandapani, R.; Vijayan, M. Water-mediated transformations in protein crystals. Acta Cryst. B 1985, 41, 431–436. [Google Scholar] [CrossRef]
- Morozov, V.N.; Morozova, T.Y.; Kachalova, G.S.; Myachin, G.S. Interpretation of water desorption isotherms of lysozyme. Int. J. Biol. Macromol. 1988, 10, 329–336. [Google Scholar] [CrossRef]
- Kodandapani, R.; Suresh, C.G.; Vijayan, M. Crystal structure of low humidity tetragonal lysozyme at 2.1-A resolution. Variability in hydration shell and its structural consequences. J. Biol. Chem. 1990, 265, 16126–16131. [Google Scholar] [PubMed]
- Bernal, J.D.; Crowfoot, D. X-ray photographs of crystalline pepsin. Nature 1934, 133, 794–795. [Google Scholar] [CrossRef]
- Perutz, M.F. The structure of haemoglobin III. Direct determination of the molecular transform. Proc. R. Soc. Lond. Ser. A 1954, 225, 264–286. [Google Scholar]
- Einstein, J.R. Humidity control device for the Buerger precession camera. J. Sci. Instrum. 1961, 38, 449–451. [Google Scholar] [CrossRef]
- Pickford, M.G.; Garman, E.F.; Jones, E.Y.; Stuart, D.I. A design of crystal mounting cell that allows the controlled variation of humidity at the protein crystal during X-ray diffraction. J. Appl. Cryst. 1993, 26, 465–466. [Google Scholar] [CrossRef]
- Heras, B.; Edeling, M.A.; Byriel, K.A.; Jones, A.; Raina, S.; Martin, J.L. Dehydration converts DsbG crystal diffraction from low to high resolution. Structure 2003, 11, 139–145. [Google Scholar] [CrossRef]
- Abergel, C. Spectacular improvement of X-ray diffraction through fast desiccation of protein crystals. Acta Cryst. D 2004, 60, 1413–1416. [Google Scholar] [CrossRef]
- Hofmeister, F. Zur lehre von der wirkung der salze. Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol. 1888, 24, 247–260. [Google Scholar] [CrossRef] [Green Version]
- Zellnitz, S.; Narygina, O.; Resch, C.; Schroettner, H.; Urbanetz, N.A. Crystallization speed of salbutamol as a function of relative humidity and temperature. Int. J. Pharm. 2015, 489, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Boultif, A.; Louër, D. Indexing of powder diffraction patterns for low-symmetry lattices by the successive dichotomy method. J. Appl. Cryst. 1991, 24, 987–993. [Google Scholar] [CrossRef]
- Degen, T.; Sadki, M.; Bron, E.; König, U.; Nénert, G. The highscore suite. Powder Diffr. 2014, 29, S13–S18. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.; Biswal, B.K.; Vijayan, M. The effect of stabilizing additives on the structure and hydration of proteins: A study involving tetragonal lysozyme. Acta Cryst. D 2001, 57, 1614–1620. [Google Scholar] [CrossRef] [Green Version]
- Di, L.; Kerns, E.H. Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization; Academic Press, Elsevier: Oxford, UK, 2015. [Google Scholar]
- Dejoie, C.; Coduri, M.; Petitdemange, S.; Giacobbe, C.; Covacci, E.; Grimaldi, O.; Autran, P.O.; Mogodi, M.W.; Jung, D.Š.; Fitch, A.N. Combining a nine-crystal multi-analyser stage with a two-dimensional detector for high-resolution powder X-ray diffraction. J. Appl. Cryst. 2018, 51, 1721–1733. [Google Scholar] [CrossRef]
- Spence, J.C.H. XFELs for structure and dynamics in biology. IUCrJ 2017, 4, 322–339. [Google Scholar] [CrossRef]
- Coe, J.; Fromme, P. Serial femtosecond crystallography opens new avenues for Structural Biology. Protein Pept. Lett. 2016, 23, 255–272. [Google Scholar] [CrossRef] [Green Version]
- Hunter, M.S.; Fromme, P. Toward structure determination using membrane-protein nanocrystals and microcrystals. Methods 2011, 55, 387–404. [Google Scholar] [CrossRef] [Green Version]
- Chapman, H.N.; Caleman, C.; Timneanu, N. Diffraction before destruction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130313. [Google Scholar] [CrossRef] [Green Version]
- Doerr, A. The new XFELs. Nat. Methods 2018, 15, 33. [Google Scholar] [CrossRef]
- Chapman, H.N.; Fromme, P.; Barty, A.; White, T.A.; Kirian, R.A.; Aquila, A.; Doak, R.B. Femtosecond X-ray protein nanocrystallography. Nature 2011, 470, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Nogales, E.; Scheres, S.H. Cryo-EM: A unique tool for the visualization of macromolecular complexity. Mol. Cell 2015, 5, 677–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egelman, E.H. The current revolution in cryo-EM. Biophys. J. 2016, 110, 1008–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
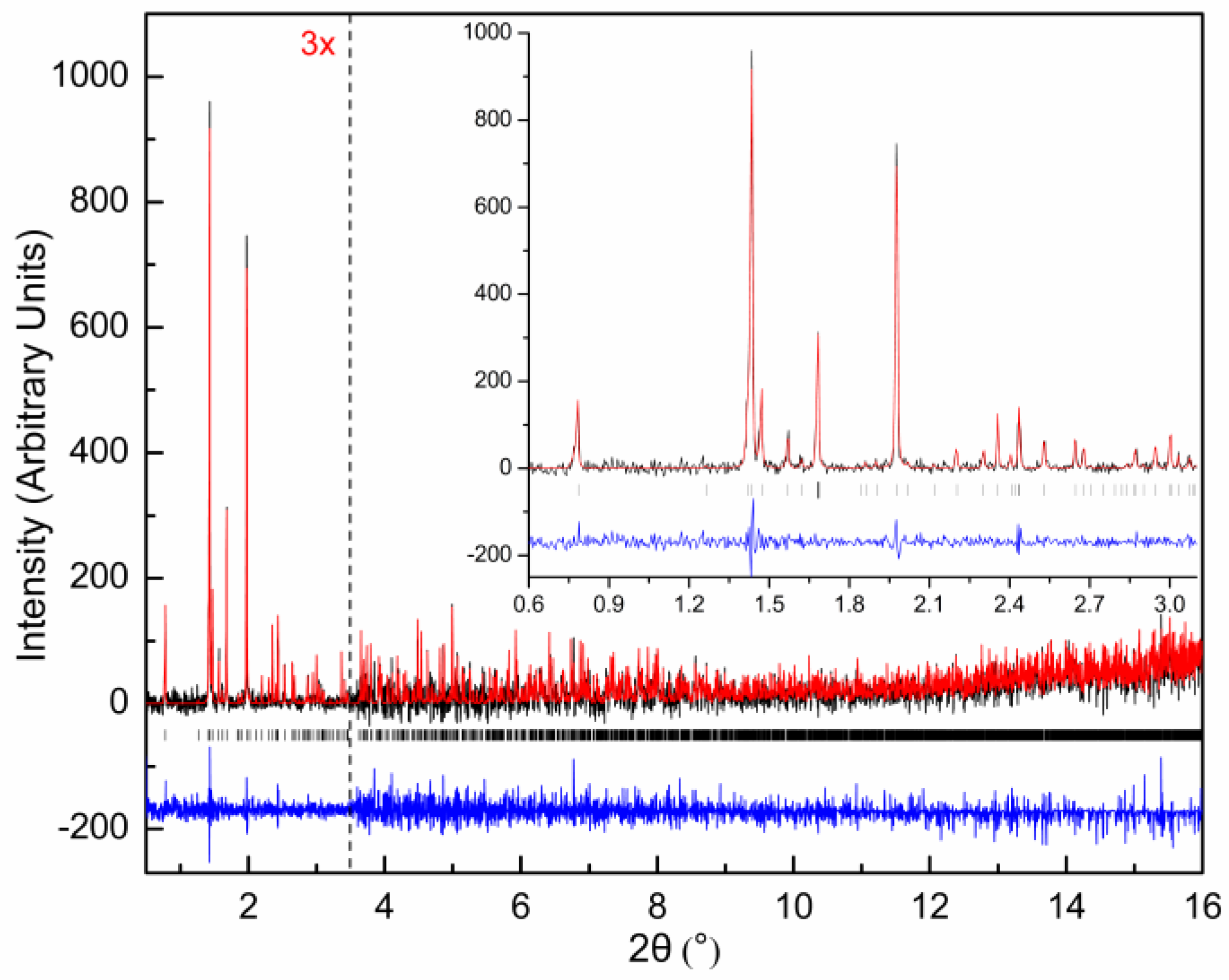{kind=link}
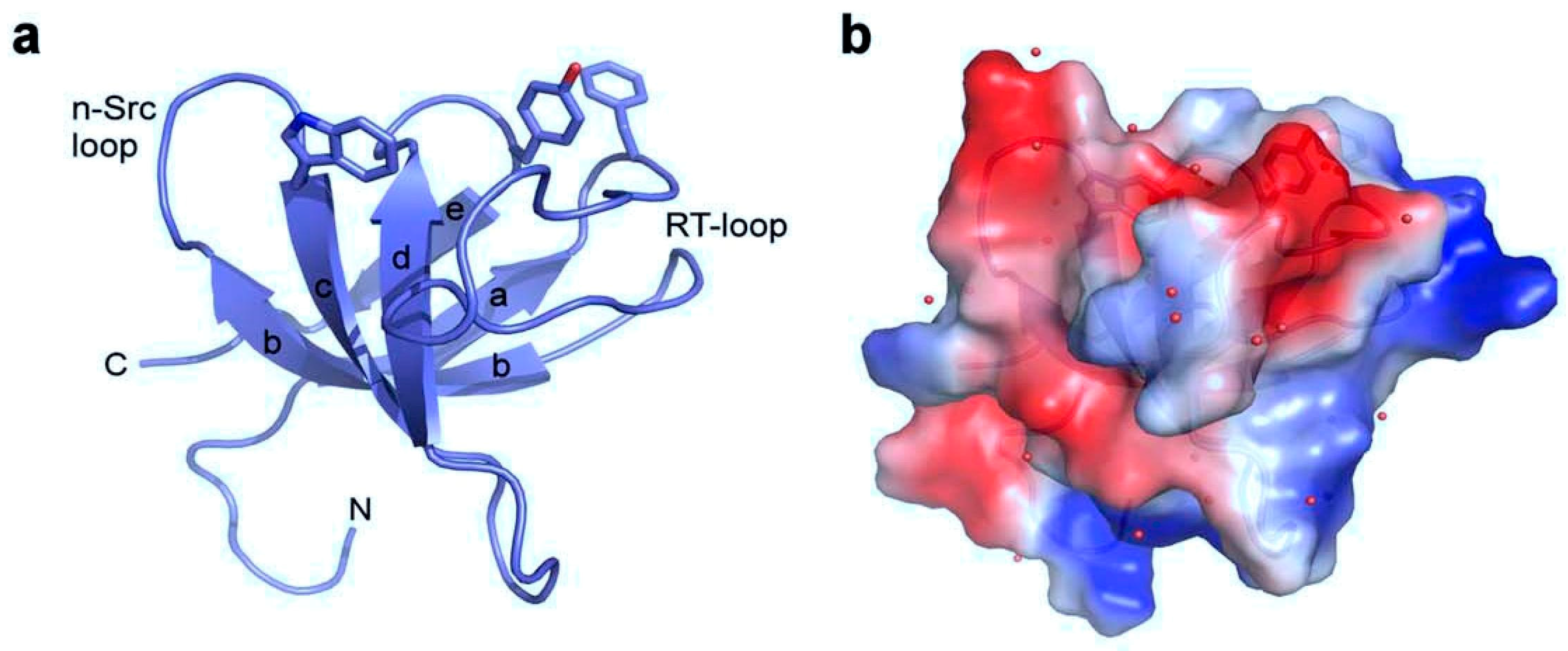{kind=link}
{kind=link}
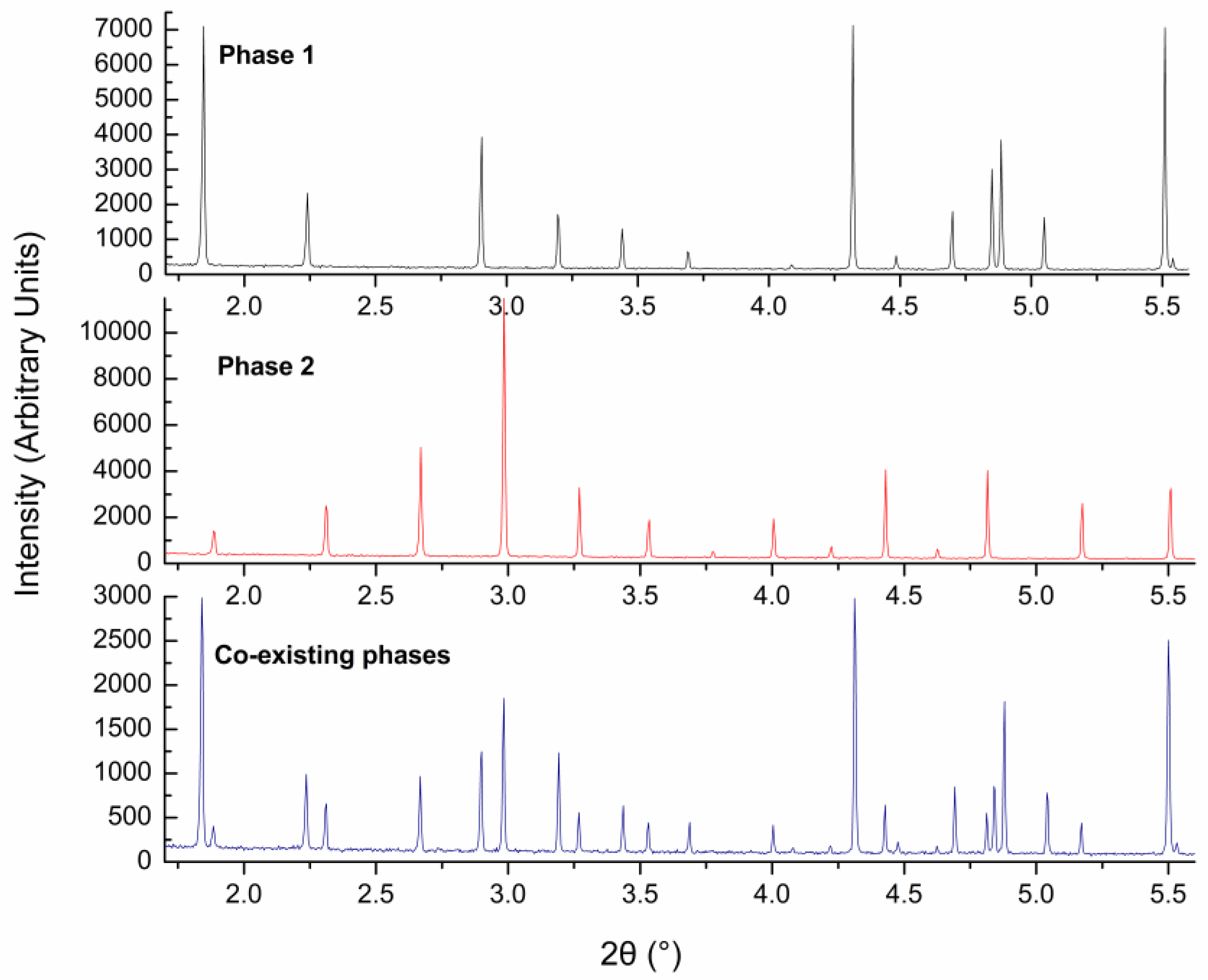{kind=link}
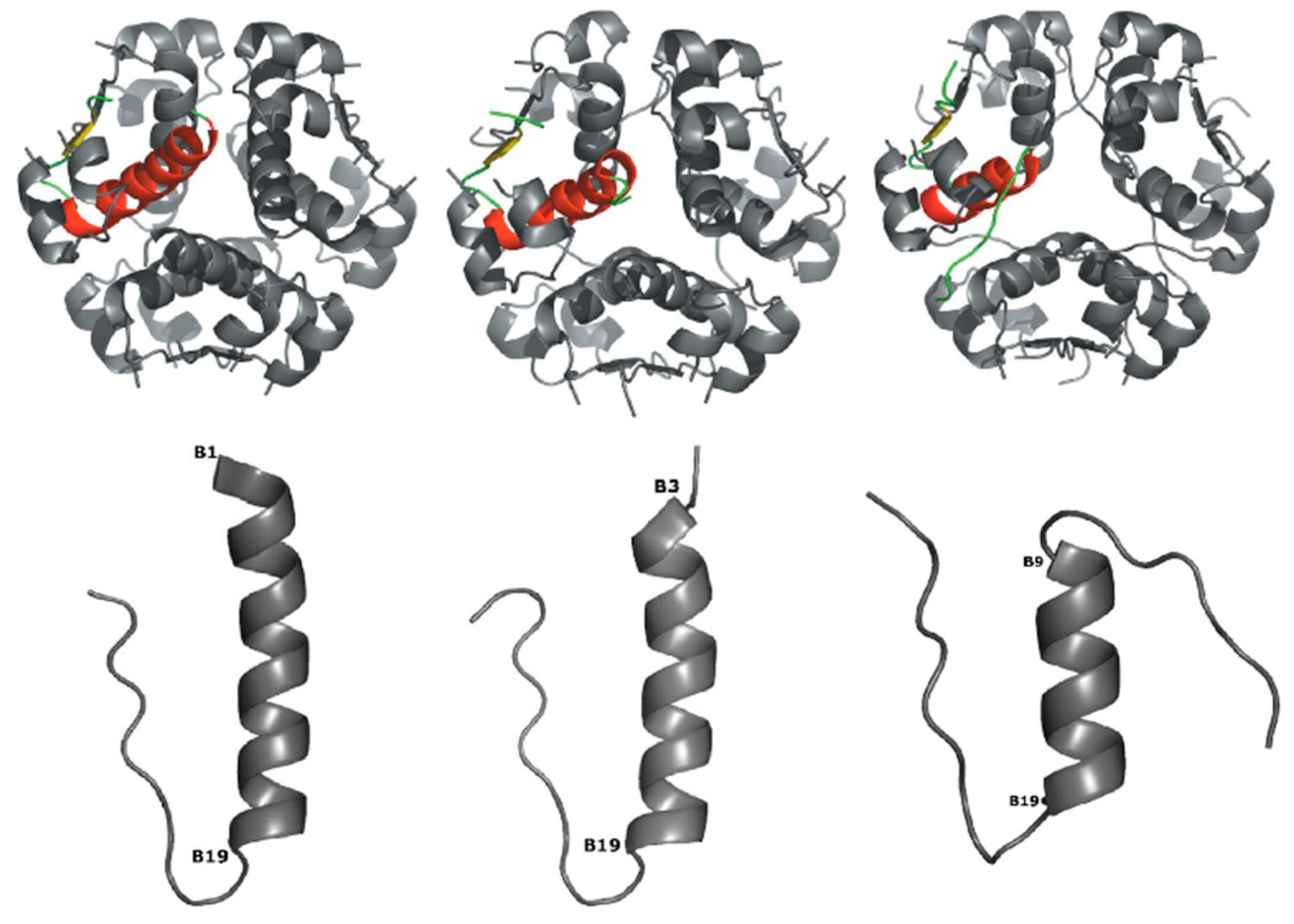{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Formulation | Insulin Formulation | Start Action | Maximum Action | Duration of Action |
|---|---|---|---|---|
| Rapid-acting analogues | Insulin lispro | 5–15 min | 30–90 min | 3–5 h |
| Insulin aspart | 5–15 min | 30–90 min | 3–5 h | |
| Insulin glusine | 5–15 min | 30–90 min | 3–5 h | |
| Quick-acting analogues | Regular | 30–60 min | 2–3 h | 5–8 h |
| Intermediate-acting analogues | NPH | 30–60 min | 4–10 h | 10–16 h |
| Lente | 30–60 min | 4–12 h | 12–18 h | |
| Semilente | 1–3 h | 2–8 h | 12–16 h | |
| Long-acting analogues | Ultralente | 6–10 h | 10–16 h | 18–24 h |
| Insulin glargine | 2–4 h | Peakless | 20–24 h | |
| Insulin detemir | 2–4 h | 6–14 h | 16–20 h | |
| Insulin mixtures (multiple action) | 75/25 lispro analogue mix(75% intermediate, 25% lispro) | 5–15 min | Peakless | 10–16 h |
| 70/30 aspart analogue mix(70% intermediate, 30% aspart) | 5–15 min | Peakless | 10–16 h | |
| 50/50 lispro analogue mix(50% intermediate, 50% lispro) | 5–15 min | Peakless | 10–16 h | |
| 70/30 human mix (75% NPH, 30% regular) | 30–60 min | Peakless | 10–16 h | |
| 50/50 human mix (50% NPH, 50% regular) | 30–60 min | Peakless | 10–16 h |
| Crystal Polymorph | Ligand | pH Range | Unit-Cell Parameters | |||
|---|---|---|---|---|---|---|
| a (Å) | b (Å) | c (Å) | β (°) | |||
| P21(α) | phenol | 5.47–5.70 | 114.682 (6) | 337.63 (2) | 49.270 (4) | 101.555 (6) |
| rescorsinol | 5.29–5.46 | 114.0228 (8) | 335.43 (3) | 49.211 (6) | 101.531 (8) | |
| 4-ethylresorcinol | 2.64-5.80 | 114.130 (7) | 336.086 (3) | 48.987 (5) | 101.935 (8) | |
| P21(β) | phenol | 7.01–8.25 | 61.0920 (4) | 61.8279 (4) | 47.9302 (4) | 110.6253 (7) |
| rescorsinol | 7.53–8.22 | 61.0008 (4) | 62.0040 (3) | 47.8823 (3) | 110.0465 (5) | |
| 4-ethylresorcinol | 6.70–8.10 | 62.8231 (7) | 62.1078 (5) | 47.8362 (6) | 111.6913 (9) | |
| 4-chlororesorcinol 1 | 6.60–8.10 | 62.413 (1) | 61.872 (1) | 47.786 (1) | 111.978 (2) | |
| 4-bromoresorcinol 1 | 5.90–8.10 | 62.032 (3) | 62.186 (2) | 47.876 (2) | 113.809 (4) | |
| P21(γ) | m-cresol | 4.50–6.70 | 87.132 (3) | 70.294 (2) | 48.064 (2) | 106.259 (3) |
| 4-nitrophenol | 4.95–5.60 | 87.118 (1) | 70.9493 (9) | 48.4967 (9) | 106.653 (1) | |
| 4-ethylresorcinol | 5.10–6.30 | 87.132 (3) | 70.294 (2) | 48.064 (2) | 106.259 (3) | |
| 4-chlororesorcinol 1 | 4.55–5.43 | 87.731 (1) | 69.9553 (8) | 47.9564 (8) | 106.754 (2) | |
| 4-bromoresorcinol 1 | 4.60–5.60 | 87.065 (4) | 70.191 (2) | 47.822 (3) | 106.539 (4) | |
| P21(δ) | 4-chlororesorcinol 1 | 5.59–5.64 | 48.4206 (9) | 59.663 (1) | 47.7644 (6) | 94.060 (2) |
| 4-bromoresorcinol 1 | 5.88–6.27 | 48.833 (1) | 60.146 (1) | 47.6372 (7) | 93.848 (2) | |
| P21(ε) | m-nitrophenol 1 | 5.60–6.60 | 72.951 (1) | 64.1465 (8) | 59.7727 (8) | 92.091 (1) |
| P21(ζ) | p-coumaric acid | 5.82–6.79 | 48.2712 (8) | 68.513 (1) | 41.6667 (8) | 95.030 (1) |
| resveratrol | 5.46–5.81 | 48.211 (2) | 68.305 (2) | 41.770 (2) | 95.108 (3) | |
| P21(η) | p-coumaric acid | 5.44–5.82 | 77.4210 (1) | 46.7125 (7) | 82.8445 (1) | 111.063 (2) |
| resveratrol | 5.06–5.46 | 77.4454 (1) | 46.7230 (7) | 82.864 (1) | 111.068 (2) | |
| C2 | phenol | 6.70–6.75 | 103.0115 (5) | 61.3213 (2) | 63.5783 (4) | 117.2244 (5) |
| 4-ethylresorcinol | 5.93–6.25 | 103.0848 (4) | 61.6636 (2) | 63.5006 (4) | 117.417 (5) | |
| 4-chlororesorcinol 1 | 5.98–6.50 | 102.947 (2) | 61.502 (1) | 63.372 (2) | 117.221 (3) | |
| C2221 | phenol | 5.93–6.54 | 60.287 (1) | 221.797 (6) | 228.812 (5) | 90 |
| resorcinol | 5.93–7.45 | 60.5579 (7) | 220.907 (3) | 228.320 (3) | 90 | |
| Phase | Unit-Cell Parameters | Volume (Å3) | Matthews Coefficient | Hexamers per Unit-Cell | Solvent (%) | Molecular Packing Efficiency (%) | |||
|---|---|---|---|---|---|---|---|---|---|
| a (Å) | b (Å) | c (Å) | β (°) | ||||||
| P21(α)* | 114.0228 (8) | 335.43 (3) | 49.211 (6) | 101.531 (8) | 1,844,168.62 | 2.6457 | 20 | 53.51 | 46.49 |
| 2.4052 | 22 | 48.86 | 51.14 | ||||||
| 2.2047 | 24 | 44.21 | 55.79 | ||||||
| P21(β) | 61.0008 (4) | 62.0040 (3) | 47.8823 (3) | 110.0465 (5) | 170,132.63 | 2.4414 | 2 | 49.62 | 50.38 |
| P21(γ) | 87.5506 (2) | 70.4772 (1) | 48.3231(1) | 107.0332 (2) | 285,089.91 | 2.0452 | 4 | 39.86 | 60.14 |
| P21(δ) | 48.9730 (4) | 60.1422 (5) | 47.7529 (4) | 95.7345 (5) | 139,944.85 | 2.0075 | 2 | 38.73 | 61.27 |
| P21(ε) | 72.951 (1) | 64.1465 (8) | 59.7727 (8) | 92.091 (1) | 279,523.17 | 2.0049 | 4 | 38.65 | 61.35 |
| P21(ζ) | 48.2712 (8) | 68.513 (1) | 41.6667 (8) | 95.030 (1) | 137,269.06 | 1.9689 | 2 | 37.53 | 62.47 |
| P21(η) | 77.41 (3) | 46.728 (2) | 82.96 (3) | 111.148 (6) | 279,873.93 | 2.0082 | 4 | 38.75 | 61.25 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spiliopoulou, M.; Valmas, A.; Triandafillidis, D.-P.; Kosinas, C.; Fitch, A.; Karavassili, F.; Margiolaki, I. Applications of X-ray Powder Diffraction in Protein Crystallography and Drug Screening. Crystals 2020, 10, 54. https://doi.org/10.3390/cryst10020054
Spiliopoulou M, Valmas A, Triandafillidis D-P, Kosinas C, Fitch A, Karavassili F, Margiolaki I. Applications of X-ray Powder Diffraction in Protein Crystallography and Drug Screening. Crystals. 2020; 10(2):54. https://doi.org/10.3390/cryst10020054
Chicago/Turabian StyleSpiliopoulou, Maria, Alexandros Valmas, Dimitris-Panagiotis Triandafillidis, Christos Kosinas, Andrew Fitch, Fotini Karavassili, and Irene Margiolaki. 2020. "Applications of X-ray Powder Diffraction in Protein Crystallography and Drug Screening" Crystals 10, no. 2: 54. https://doi.org/10.3390/cryst10020054
APA StyleSpiliopoulou, M., Valmas, A., Triandafillidis, D. -P., Kosinas, C., Fitch, A., Karavassili, F., & Margiolaki, I. (2020). Applications of X-ray Powder Diffraction in Protein Crystallography and Drug Screening. Crystals, 10(2), 54. https://doi.org/10.3390/cryst10020054