Structural, Non-Covalent Interaction, and Natural Bond Orbital Studies on Bromido-Tricarbonyl Rhenium(I) Complexes Bearing Alkyl-Substituted 1,4-Diazabutadiene (DAB) Ligands


Abstract
:
1. Introduction
2. Experimental
2.1. General Methods
2.2. X-ray Crystallography
2.3. Computational Details
3. Results and Discussion
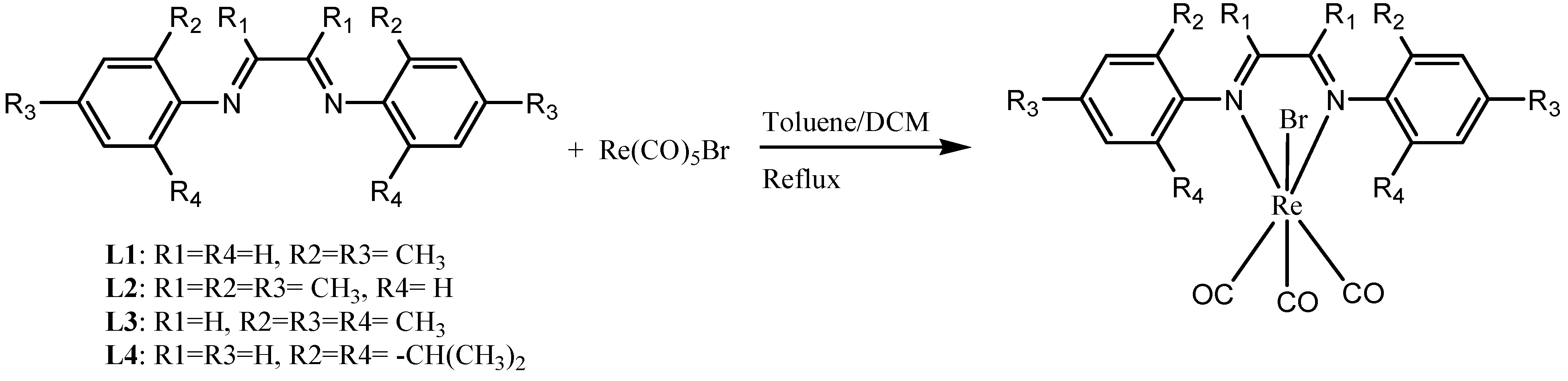
3.1. Synthesis and Characterization
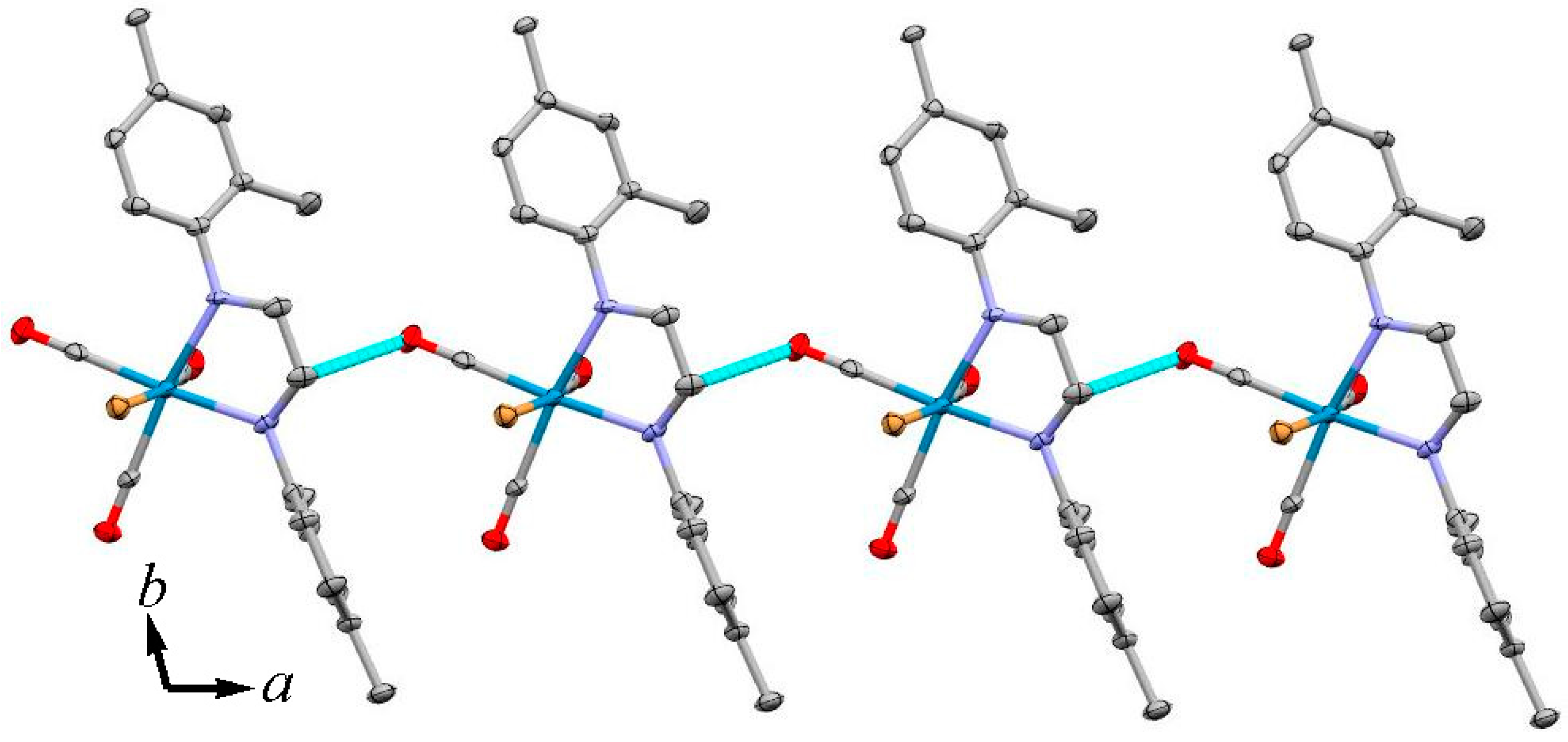
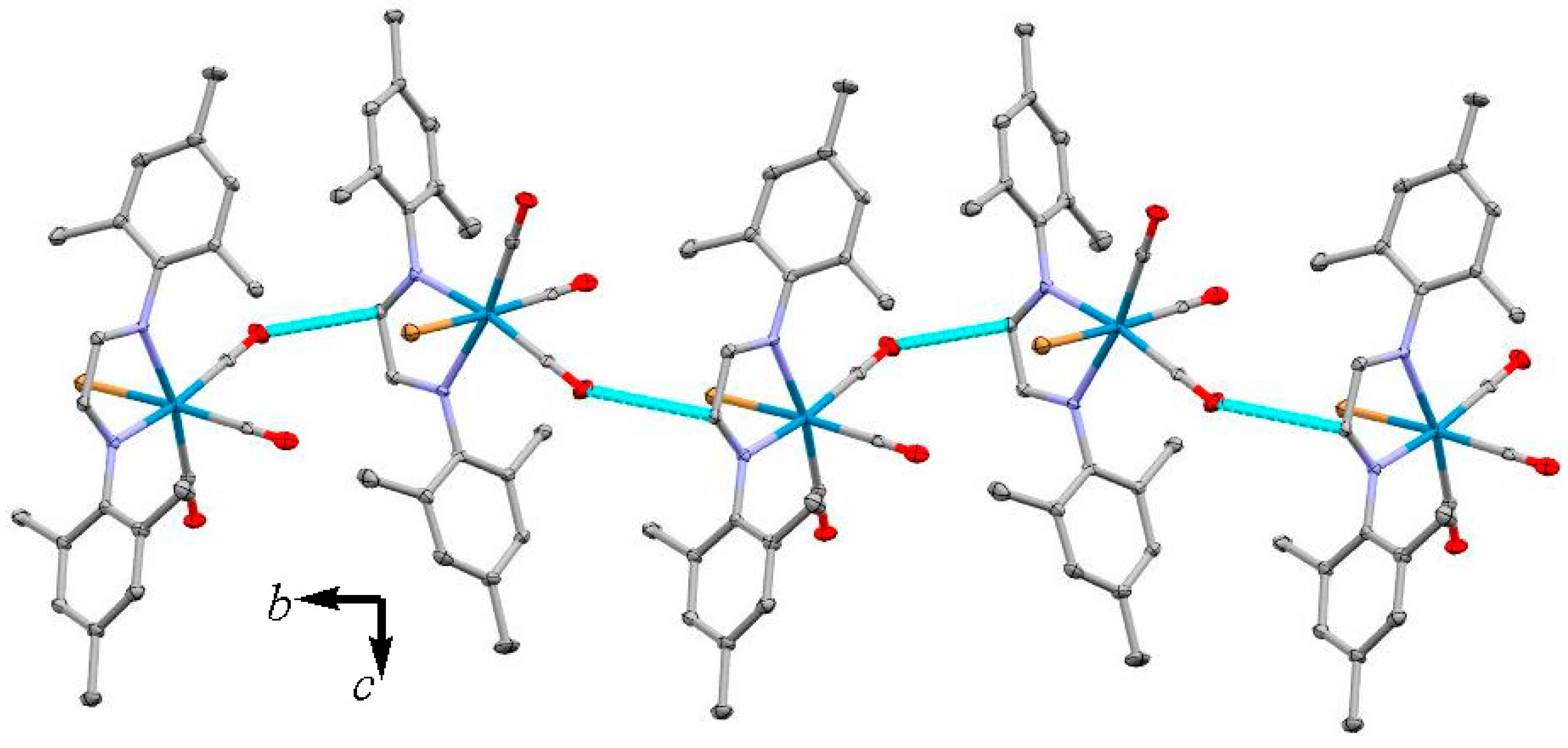
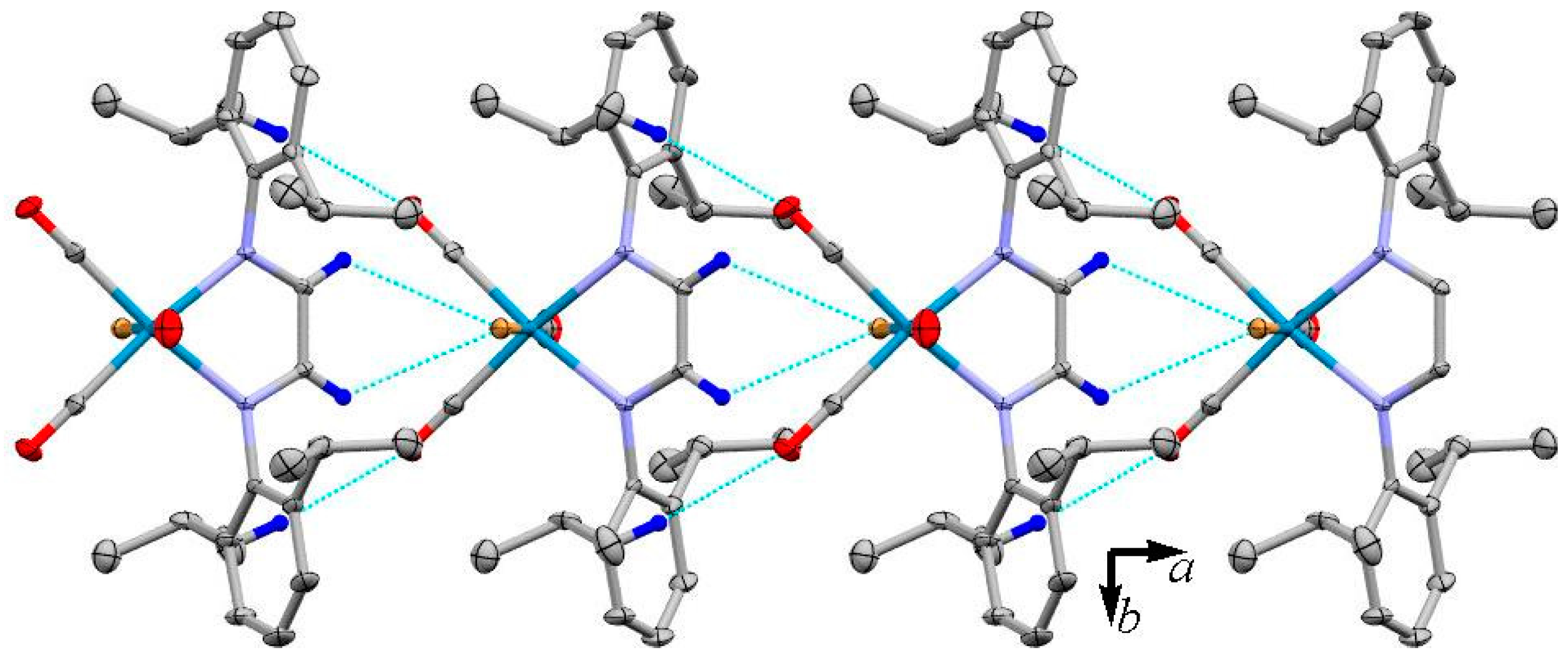
3.2. X-ray Crystal Structures
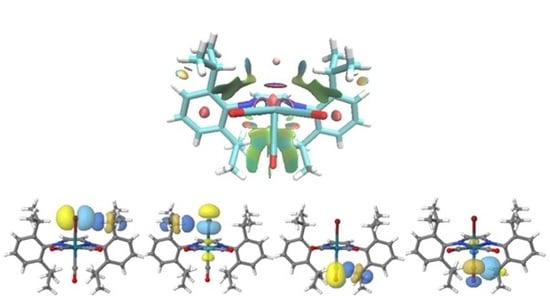
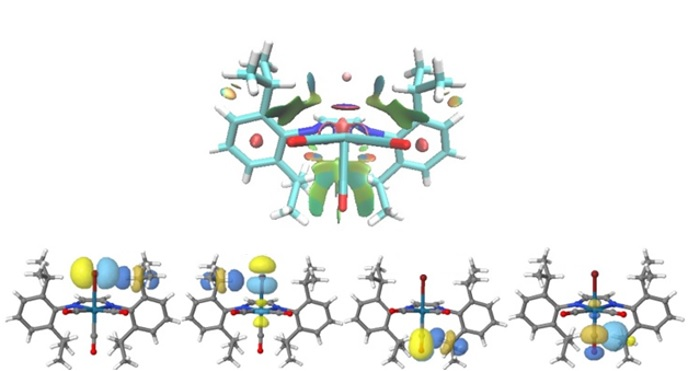
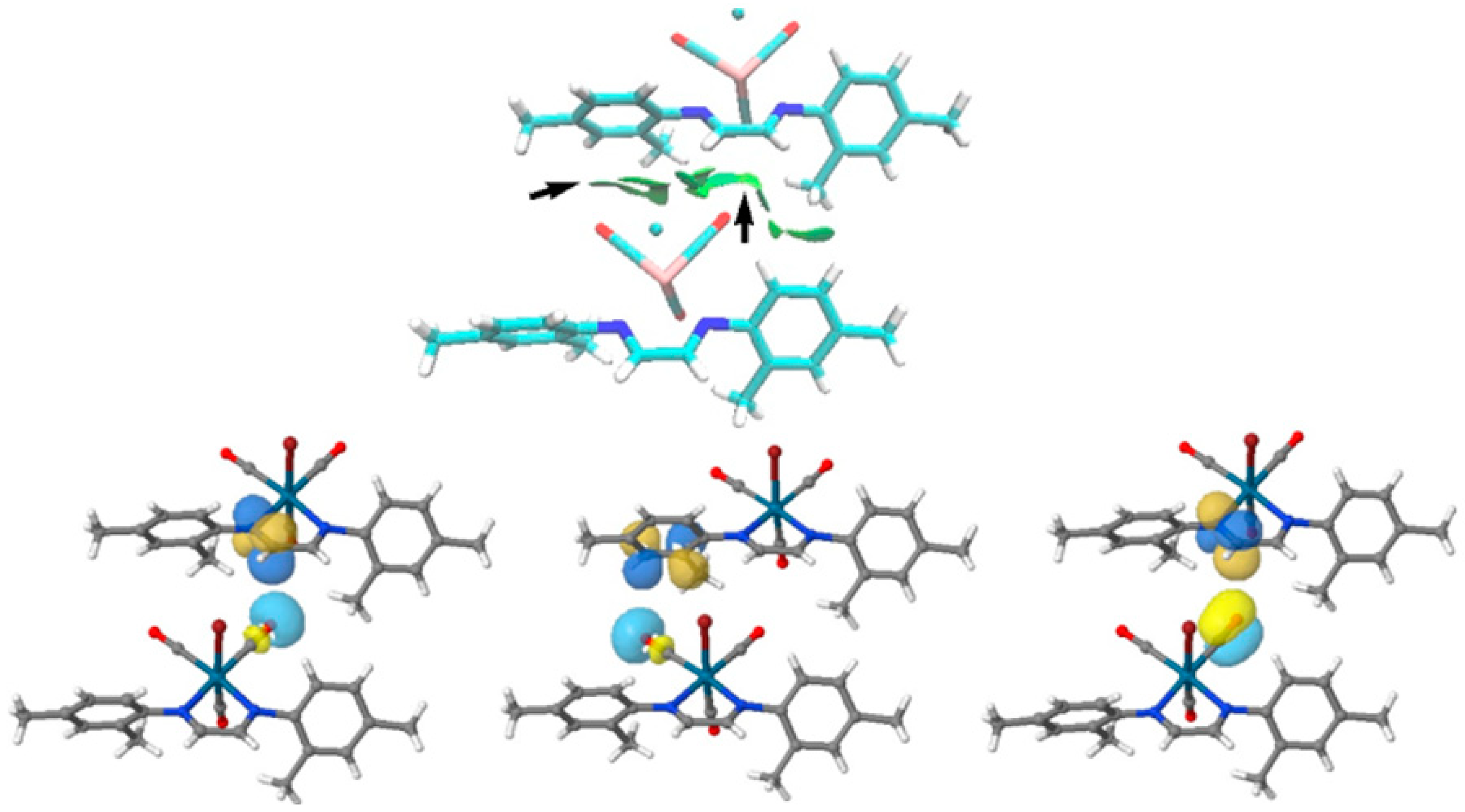
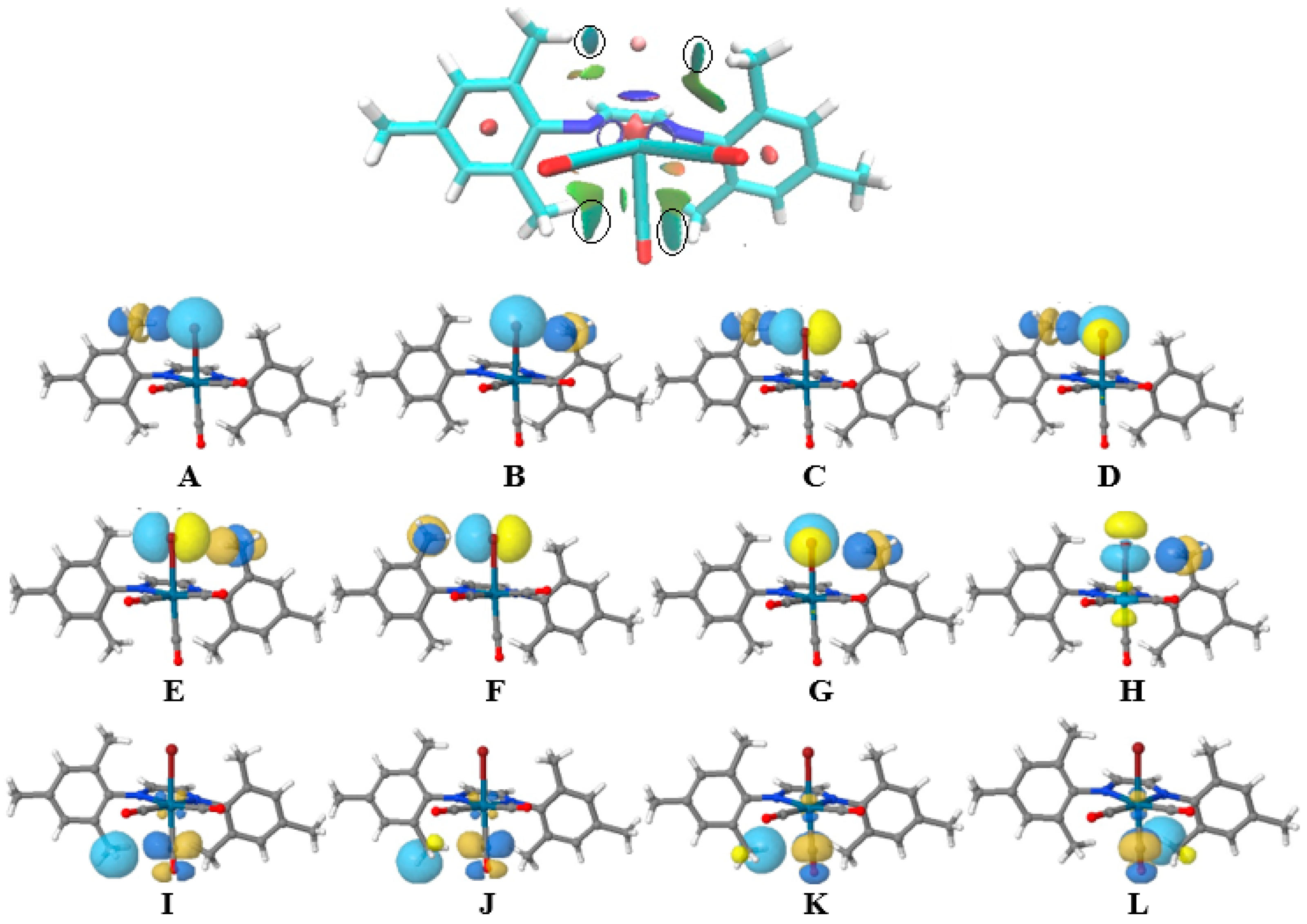
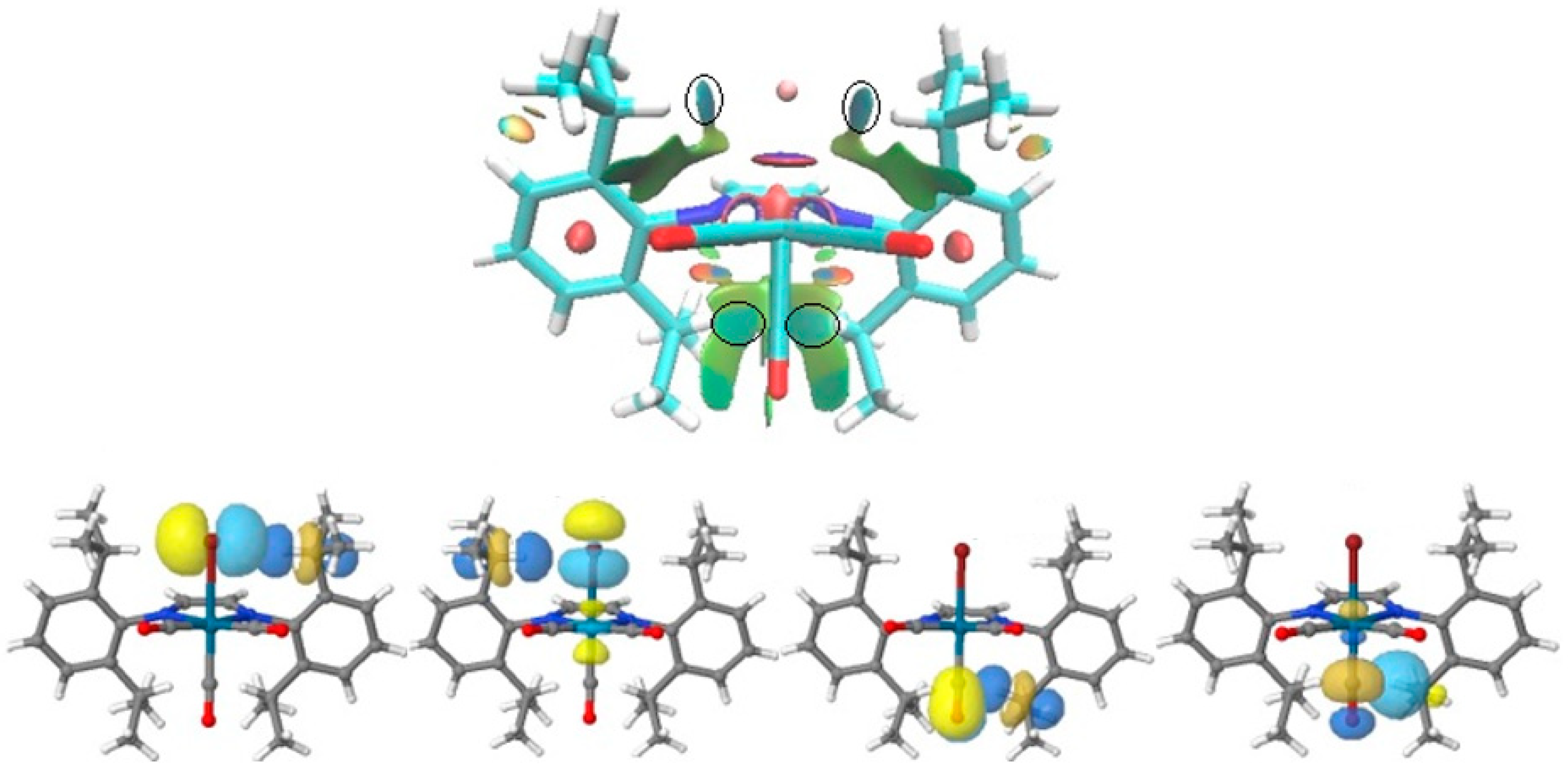
3.3. Non-Covalent Interaction Index and Natural Bond Orbitals (NBOs)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ehlers, A.W.; Dapprich, S.; Vyboishchikov, S.F.; Frenking, G. Structure and Bonding of the Transition-Metal Carbonyl Complexes M(CO)5L (M = Cr, Mo, W) and M(CO)3L (M = Ni, Pd, Pt; L = CO, SiO, CS, N2, NO+, CN-, NC-, HCCH, CCH2, CH2, CF2, H2). Organometallics 1996, 15, 105–117. [Google Scholar] [CrossRef]
- Lee, A.J. Luminescence properties of organometallic complexes. Chem. Rev. 1987, 87, 711–743. [Google Scholar]
- Farrell, I.R.; Vlcek, A., Jr. Mechanisms of ultrafast metal–ligand bond splitting upon MLCT excitation of carbonyl-diimine complexes. Coord. Chem. Rev. 2000, 208, 87–101. [Google Scholar] [CrossRef]
- Striplin, D.R.; Crosby, G.A. Photophysical investigations of rhenium(I)Cl(CO)3(phenanthroline) complexes. Coord. Chem. Rev. 2001, 211, 163–175. [Google Scholar] [CrossRef]
- Collin, J.P.; Sauvage, J.P. Electrochemical reduction of carbon dioxide mediated by molecular catalysts. Coord. Chem. Rev. 1989, 93, 245–268. [Google Scholar] [CrossRef]
- Rossenaar, B.R.; Hartl, F.; Stufkens, D.J. Reduction of [Re(X)(CO)3(R′-DAB)] (X = Otf-, Br-; DAB = Diazabutadiene; R′ = iPr, pTol, pAn) and [Re(R)(CO)3(iPr-DAB)] (R = Me, Et, Bz) Complexes: A Comparative (Spectro)electrochemical Study at Variable Temperatures. Inorg. Chem. 1996, 35, 6194–6203. [Google Scholar] [CrossRef] [Green Version]
- Balzani, V.; Juris, A.; Venturi, M.; Campagna, S.; Serroni, S. Luminescent and Redox-Active Polynuclear Transition Metal Complexes. Chem. Rev. 1996, 96, 759–834. [Google Scholar] [CrossRef]
- Slone, R.V.; Hupp, J.T. Synthesis, Characterization, and Preliminary Host−Guest Binding Studies of Porphyrinic Molecular Squares Featuring fac-Tricarbonylrhenium(I) Chloro Corners. Inorg. Chem. 1997, 36, 5422–5423. [Google Scholar] [CrossRef]
- Nieto, S.; Perez, J.; Riera, L.; Riera, V.; Miguel, D. Non-covalent interactions between anions and a cationic rhenium diamine complex: Structural characterization of the supramolecular adducts. New J. Chem. 2006, 30, 838–843. [Google Scholar] [CrossRef]
- Hevia, E.; Perez, J.; Riera, V.; Miguel, D.; Kassel, S.; Rheingold, A.L. New Synthetic Routes to Cationic Rhenium Tricarbonyl Bipyridine Complexes with Labile Ligands. Inorg. Chem. 2002, 41, 4673–4679. [Google Scholar] [CrossRef]
- Machura, B.; Wolff, M.; Jaworska, M.; Lodowski, P.; Benoist, E.; Carrayon, C.; Saffon, N.; Kruszynski, R.; Mazurak, Z. Rhenium(I) carbonyl complex of 4,7-diphenyl-1,10-phenanthroline–Spectroscopic properties, X-Ray structure, theoretical studies of ground and excited electronic states. J. Organomet. Chem. 2011, 696, 3068–3075. [Google Scholar] [CrossRef]
- Drozdz, A.; Bubrin, M.; Fiedler, J.; Zalis, S.; Kaim, W. (α-Diimine)tricarbonylhalorhenium complexes: The oxidation side. Dalton Trans. 2012, 41, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Grupp, A.; Bubrin, M.; Ehret, F.; Kvapilova, H.; Zalis, S.; Kaim, W. Oxidation and reduction response of α-diimine complexes with tricarbonylrhenium halides and pseudohalides. J. Organomet. Chem. 2014, 751, 678–685. [Google Scholar] [CrossRef]
- Kumar, A.; Sun, S.-S.; Lees, A.J. Photophysics and Photochemistry of Organometallic Rhenium Diimine Complexes. Top. Organomet. Chem. 2010, 29, 1–35. [Google Scholar]
- Kirgan, R.A.; Sullivan, B.P.; Rillema, D.P. Photochemistry and Photophysics of Coordination Compounds: Rhenium. Top. Curr. Chem. 2007, 281, 45–100. [Google Scholar]
- Kinghat, R.; Khatyr, A.; Knorr, M.; Kubicki, M.M.; Vigier, E.; Villafañe, F. Mono- and di-nuclear 2,3-diazabutadiene and 2-azabutadiene complexes of Rhenium(I): Syntheses, luminescence spectra and X-ray structures. Inorg. Chem. Commun. 2008, 11, 1060. [Google Scholar] [CrossRef]
- Zou, W.; Chen, C. Influence of Backbone Substituents on the Ethylene (Co)polymerization Properties of a-diimine Pd(II) and Ni(II) Catalysts. Organometallics 2016, 35, 1794–1801. [Google Scholar] [CrossRef]
- Kong, S.; Song, K.; Liang, T.; Guo, C.-Y.; Sun, W.-H.; Redshaw, C. 18 Methylene-bridged bimetallic α-diimino nickel(II) complexes: Synthesis and high efficiency in ethylene polymerization. Dalton Trans. 2013, 42, 9176–9178. [Google Scholar] [CrossRef]
- Abakumov, G.A.; Druzhkov, N.O.; Egorova, E.N.; Kocherova, T.N.; Shavyrin, A.S.; Cherkasov, A.V. Intramolecular cyclization–decyclization of new sterically hindered diiminophenol. Synthesis and coordination abilities. RSC Adv. 2014, 4, 14495–14500. [Google Scholar] [CrossRef]
- Egorova, E.N.; Druzhkov, N.O.; Shavyrin, A.S.; Cherkasov, A.V.; Abakumova, G.A.; Fedorov, A.Y. The group 13 metal complexes of stericallyhindered substituted iminophenol: Synthesis and structure. RSC Adv. 2015, 5, 19362–19367. [Google Scholar] [CrossRef]
- Echeverría, J. The n → π* interaction in metal complexes. Chem. Commun. 2018, 54, 3061–3064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgi, H.B.; Dunitz, J.D.; Lehn, J.M.; Wipff, G. Stereochemistry of reaction paths at carbonyl centres. Tetrahedron 1974, 30, 1563–1572. [Google Scholar] [CrossRef]
- Burgi, H.B.; Dunitz, J.D.; Shefter, E. Geometrical reaction coordinates. II. Nucleophilic addition to a carbonyl group. J. Am. Chem. Soc. 1973, 95, 5065–5067. [Google Scholar] [CrossRef]
- Mooibroek, T.J.; Gamez, P.; Reedijk, J. Lone pair–π interactions: A new supramolecular bond? CrystEngComm 2008, 10, 1501–1515. [Google Scholar] [CrossRef]
- Jakobsche, C.E.; Choudhary, A.; Miller, S.J.; Raines, R.T. n → π* Interaction and n)(π Pauli Repulsion Are Antagonistic for Protein Stability. J. Am. Chem. Soc. 2010, 132, 6651–6653. [Google Scholar] [CrossRef] [Green Version]
- Kramer, K.J.; Choudhary, A.; Raines, R.T. Intimate Interactions with Carbonyl Groups: Dipole–Dipole or n→π*? J. Org. Chem. 2013, 78, 2099–2103. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, G.J.; Newberry, R.W.; Van Veller, B.; Raines, R.T.; Woolfson, D.N. Interplay of Hydrogen Bonds and n → π* Interactions in Proteins. J. Am. Chem. Soc. 2013, 135, 18682–18688. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Das, A. The n → π* interaction: A rapidly emerging non-covalent interaction. Phys. Chem. Chem. Phys. 2015, 17, 9596–9612. [Google Scholar] [CrossRef] [Green Version]
- Velásquez, J.D.; Echeverría, J.; Alvarez, S. Effect of the Substituents on the Nature and Strength of Lone-Pair–Carbonyl Interactions in Acyl Halides. Cryst. Growth Des. 2019, 11, 6511–6518. [Google Scholar] [CrossRef]
- Newberry, R.W.; Raines, R.T. The n→π* Interaction. Acc. Chem. Res. 2017, 50, 1838–1846. [Google Scholar] [CrossRef] [Green Version]
- Doppert, M.T.; van Overeem, H.; Mooibroek, T.J. Intermolecular π-hole/n→π* interactions with carbon monoxide ligands in crystal structures. Chem. Commun. 2018, 54, 12049–12052. [Google Scholar] [CrossRef] [PubMed]
- van der Werve Ad, R.; van Dijk, Y.R.; Mooibroek, T.J. π-Hole/n→π* interactions with acetonitrile in crystal structures. Chem. Commun. 2018, 54, 10742–10745. [Google Scholar] [CrossRef] [PubMed]
- Zukerman-Schpector, J.; Haiduc, I.; Tiekink, E.R.T. The metal–carbonyl⋯π(aryl) interaction as a supramolecular synthon for the stabilisation of transition metal carbonyl crystal structures. Chem. Commun. 2011, 47, 12682–12684. [Google Scholar] [CrossRef] [PubMed]
- Zukerman-Schpector, J.; Haidu, I.; Tiekink, E.R.T. Supramolecular Self-assembly of Transition Metal Carbonyl Molecules Through M–CO(Lone Pair)…π(Arene) Interactions. Adv. Organomet. Chem. 2012, 60, 49–92. [Google Scholar]
- Wan, C.-Q.; Chen, X.-D.; Mak, T.C.W. Supramolecular frameworks assembled via intermolecular lone pair-aromatic interaction between carbonyl and pyridyl groups. CrystEngComm 2008, 10, 475–478. [Google Scholar] [CrossRef]
- Echeverría, J. Intermolecular Carbonyl Carbonyl Interactions in Transition-Metal Complexes. Inorg. Chem. 2018, 57, 5429–5437. [Google Scholar] [CrossRef]
- Murcia-García, C.; Bauzá, A.; Schnakenburg, G.; Frontera, A.; Streubel, R. Surprising behaviour of M–CO(lone pair)⋯π(arene) interactions in the solid state of fluorinated oxaphosphirane complexes. CrystEngComm 2015, 17, 1769–1772. [Google Scholar] [CrossRef]
- Mark-Lee, W.F.; Chong, Y.Y.; Kassim, M.B. Supramolecular structures of rhenium (I) complexes mediated by ligand planarity via the interplay of substituents. Acta Crystallogr. Sect. C Struct. Chem. 2018, 74, 997–1006. [Google Scholar]
- Kia, R.; Hosseini, M.; Abdolrahimi, A.; Mahmoudi, M. Intermolecular C–H⋯O and n→π* and short intramolecular σ → π* interactions in the molybdenum(0) tetracarbonyl complex of a very twisted 14-membered tetraazaannulene macrocyclic ligand: Structural and computational studies. CrystEngComm 2019, 21, 5222–5226. [Google Scholar] [CrossRef]
- Kia, R.; Mahmoudi, S.; Raithby, P.R. New rhenium-tricarbonyl complexes bearing halogen-substituted bidentate ligands: Structural, computational and Hirshfeld surfaces studies. CrystEngComm 2019, 21, 77–93. [Google Scholar] [CrossRef] [Green Version]
- Villegas, J.M.; Stoyanov, S.R.; Huang, W.; Rillema, D.P. A spectroscopic and computational study on the effects of methyl and phenyl substituted phenanthroline ligands on the electronic structure of Re(I) tricarbonyl complexes containing 2,6-dimethylphenylisocyanide. Dalton Trans. 2005, 34, 1042–1051. [Google Scholar] [CrossRef] [PubMed]
- Kliegman, J.M.; Barnes, R.K. Glyoxal derivatives-I: Conjugated aliphatic diimines from glyoxal and aliphatic primary amines. Tetrahedron 1970, 26, 2555–2560. [Google Scholar] [CrossRef]
- Clark, R.C.; Reid, J.S. The analytical calculation of absorption in multifaceted crystals. Acta Cryst. 1995, A51, 887–897. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. 2009, D65, 148–155. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B. Gaussian 09, Revision A.02; ScienceOpen: Berlin, Germany, 2014. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Cotton, F.A.; Karihanzel, C.S. Vibrational Spectra and Bonding in Metal Carbonyls. I. Infrared Spectra of Phosphine-substituted Group VI Carbonyls in the CO Stretching Region. J. Am. Chem. Soc. 1962, 84, 4432–4438. [Google Scholar] [CrossRef]
- Klein, A.; Vogler, C.; Kaim, W. The δ in 18 + δ Electron Complexes: Importance of the Metal/Ligand Interface for the Substitutional Reactivity of “Re(0)” Complexes (α-diimine)ReI(CO)3(X). Organometallics 1996, 15, 236–244. [Google Scholar] [CrossRef]
- Staal, L.H.; Oskam, A.; Vrieze, K. The syntheses and coordination properties of M(CO)3X(DAB) (M = Mn, Re; X = Cl, Br, I.; DAB = 1,4-diazabutadiene). J. Organomet. Chem. 1979, 170, 235–245. [Google Scholar] [CrossRef]
- Vollmer, M.V.; Machan, C.W.; Clark, M.L.; Antholine, W.E.; Agarwal, J.; Schaefer III, H.F.; Kubiak, C.P.; Walensky, J.R. Synthesis, Spectroscopy, and Electrochemistry of (α-Diimine)M(CO)3Br, M = Mn, Re, Complexes: Ligands Isoelectronic to Bipyridyl Show Differences in CO2 Reduction. Organometallics 2015, 34, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramov, P.A.; Dmitriev, A.A.; Kholin, K.V.; Gritsan, N.P.; Kadirov, M.K.; Gushchin, A.L.; Sokolov, M.N. Mechanistic study of the [(dpp-bian)Re(CO)3Br] electrochemical reduction using in situ EPR spectroscopy and computational chemistry. Electrochim. Acta 2018, 270, 526–534. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting non-covalent interaction regions. J. Chem. Theory Comput. 2010, 7, 625–632. [Google Scholar] [CrossRef]
- Usoltsev, A.N.; Adonin, S.A.; Novikov, A.S.; Samsonenko, D.G.; Sokolova, M.N.; Fedin, V.P. One-dimensional polymeric polybromotellurates (IV): Structural and theoretical insights into halogen···halogen contacts. CrystEngComm 2017, 19, 5934–5939. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. WIREs Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Empirical Formula Formula Mass Crystal Size (mm) Colour Crystal System Space Group θmax (°) a (Å) b (Å) c (Å) α (°) β (°) γ (°) V (Å3) Z Dcalc (Mg/m3) μ (mm−1) F (000) Index Ranges No. of Measured Reflns. No. of independent reflns./Rint No. of observed reflns. I > 2σ(I) No. of parameters Goodness-of-fit (GOF) R1 (observed data) wR2 (all data) | C21H20BrN2O3Re 614.50 0.10 × 0.15 × 0.25 Dark-brown Monoclinic P21/n 26 7.3182(2) 21.8786(9) 13.0930(5) 90 95.658(3) 90 2086.13(13) 4 1.957 7.764 1176 −9 ≤ h ≤8 −26 ≤ k ≤26 −17 ≤ l ≤16 16734 4084/0.046 3477 257 1.07 0.0261 0.0574 | C23H24BrN2O3Re 642.55 0.04 × 0.08 × 0.15 Dark-brown Triclinic P-1 30.4 8.1105(3) 8.3373(4) 18.3651(11) 93.857(4) 97.380(4) 104.588(4) 1185.41(10) 2 1.800 6.836 620 −11 ≤ h ≤ 11 −11 ≤ k ≤ 11 −25 ≤ l ≤ 25 23113 6361/0.067 5247 282 1.18 0.0783 0.1694 | C23H24N2O3Re 642.55 0.10 × 0.18 × 0.35 Dark-brown Orthorhombic Pbca 72.9 14.0730(2) 13.8361(2) 23.2653(3) 90 90 90 4530.11(11) 8 1.884 12.775 2480 −17 ≤ h ≤11 −14 ≤ k ≤17 −28 ≤ l ≤28 21739 4448/0.03 4228 277 1.13 0.0288 0.0726 | C29H36BrN2O3Re 726.71 0.10 × 0.18 × 0.35 Dark-red Orthorhombic Pnma 29.4 13.3103(5) 21.7931(9) 10.3165(5) 90 90 90 2992.5(2) 4 1.613 5.462 1432 −18 ≤ h ≤15 −27 ≤ k ≤18 −13 ≤ l ≤8 8882 3587/0.047 2868 173 1.02 0.0377 0.0722 |
| Bond Lengths (Ǻ) | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Re(1)–N(1) Re(1)–N(2) Re(1)–C(1) Re(1)–C(2) Re(1)–C(3) C(1)–O(1) C(2)–O(2) C(3)–O(3) Re(1)–Br(1) Bond Angles (°) N(1)–Re(1)–N(2) C(1)–Re(1)–N(1) C(2)–Re(1)–N(2) N(1)–Re(1)–C(3) N(2)–Re(1)–C(3) C(1)–Re(1)–C(3) C(2)–Re(1)–C(3) C(1)–Re(1)–Br(1) | 2.180(3) [2.175] a 2.150(4) [2.188] 2.6114(6) [1.916] 1.933(4) [1.935] 1.923(4) [1.936] 1.141(5) [1.161] 1.142(5) [1.156] 1.141(5) [1.157] 2.6114(6) [2.655] 74.59(13) [74.03] 99.20(14) [92.51] 169.99(15) [170.0] 171.97(16) [170.22] 99.45(15) [96.54] 86.35(17) [91.24] 90.09(16) [90.51] | 2.169(9) 2.173] 2.159(9) [2.173] 1.870(14) [1.911] 1.922(16) [1.936] 1.921(13) [1.936] 1.167(19) [1.163] 1.15(2) [1.157] 1.135(17) [1.157] 2.6166(18) [2.668] 73.3(4) [72.98] 95.5(5) [95.11] 171.2(6) [170.04] 168.9(5) [170.05] 95.4(8) [98.08] 89.6(6) [89.98] 89.1(6) [90.44] 177.7(4) [179.19] | 2.182(3) [2.209] 2.179(3) [2.182] 1.994(4) [1.914] 1.934(4) [1.938] 1.924(4) [1.932] 1.002(6) [1.162] 1.142(5) [1.156] 1.145(5) [1.157] 2.6161(5) [2.664] 74.08(11) [74.11] 99.86(14) [98.94] 171.19(15) [171.16] 169.73(14) [168.91] 99.32(15) [97.47] 88.05(17) [88.38] 89.49(18) [91.17] 173.55(12) [174.76] | 2.176(3) [2.223] - 1.893(7) [1.927] 1.918(4) [1.951] - 1.166(9) [1.165] 1.148(5) [1.159] - 2.6122(7) [2.712] 75.62(12) [73.92] 96.53(18) [97.10] - - - - - 176.1(2) [170.9] |
| D−H···A | H···A (Å) | D···A (Å) | D−H···A (°) | |
|---|---|---|---|---|
| Complex 1 | C(11)–H(11)…Br(1)i | 2.75 | 3.629(4) | 158 |
| Complex 2 | C(13)–H(16)…Br(1)ii | 2.92 | 3.841(12) | 168 |
| Complex 3 | C(11)–H(11)…O(1)iii | 2.45 | 3.375(6) | 162 |
| C(20)–H(20B)…Br(1) | 2.65 | 3.572(5) | 162 | |
| C(21)–H(21A)…O(1) | 2.51 | 3.303(6) | 140 | |
| C(23)–H(23C)…Br(1) | 2.78 | 3.681(5) | 156 | |
| Complex 4 | C(6)–H(6)…O(2)iv | 2.52 | 3.451(6) | 177 |
| C(10)–H(10)…Br(1) | 2.62 | 3.564(4) | 163 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kia, R.; Kalaghchi, A. Structural, Non-Covalent Interaction, and Natural Bond Orbital Studies on Bromido-Tricarbonyl Rhenium(I) Complexes Bearing Alkyl-Substituted 1,4-Diazabutadiene (DAB) Ligands. Crystals 2020, 10, 267. https://doi.org/10.3390/cryst10040267
Kia R, Kalaghchi A. Structural, Non-Covalent Interaction, and Natural Bond Orbital Studies on Bromido-Tricarbonyl Rhenium(I) Complexes Bearing Alkyl-Substituted 1,4-Diazabutadiene (DAB) Ligands. Crystals. 2020; 10(4):267. https://doi.org/10.3390/cryst10040267
Chicago/Turabian StyleKia, Reza, and Azadeh Kalaghchi. 2020. "Structural, Non-Covalent Interaction, and Natural Bond Orbital Studies on Bromido-Tricarbonyl Rhenium(I) Complexes Bearing Alkyl-Substituted 1,4-Diazabutadiene (DAB) Ligands" Crystals 10, no. 4: 267. https://doi.org/10.3390/cryst10040267
APA StyleKia, R., & Kalaghchi, A. (2020). Structural, Non-Covalent Interaction, and Natural Bond Orbital Studies on Bromido-Tricarbonyl Rhenium(I) Complexes Bearing Alkyl-Substituted 1,4-Diazabutadiene (DAB) Ligands. Crystals, 10(4), 267. https://doi.org/10.3390/cryst10040267