1. Introduction
It is well known that successful protein crystallization is typically (as a rule) carried out with the addition of special crystallization agents-precipitants. A number of works (see, for example, [
1,
2,
3,
4]) have shown that the addition of precipitants leads to a change in the interaction between proteins from repulsion to attraction. However, the mechanisms of interaction between proteins and precipitant ions have not been sufficiently studied.
In a series of papers [
5,
6,
7], it has recently been established that the addition of precipitants to protein solutions proteins leads to the formation of precursor clusters representing 3D fragments of the crystal structure. In the case of tetragonal modification of lysozyme, crystal octamers are such clusters that are the units of growth of single crystals. In addition to octamers, dimers are formed in crystallization solutions of lysozyme. However, small-angle X-ray scattering (SAXS) methods allow determining only the size of protein oligomers and their concentrations in solutions.
The paper [
8] shows the possibility of studying the interaction of protein molecules with each other and with precipitant ions by the example of a lysozyme solution with a sodium chloride precipitant using the molecular dynamics (MD) method as it allows investigating the systems with atomic accuracy and obtaining of a large amount of structural data in details. Further, the stability of dimers and octamers in lysozyme solutions with the addition of sodium chloride as a precipitant was calculated by MD methods in [
8]. The approach applied in that work implies MD simulation of oligomers extracted from PDB files of the crystal structures. As a result, the investigation demonstrated the importance of taking into account the positions of precipitant ions incorporated in the crystal structures. However, it considered only solutions with NaCl precipitant. According to earlier work [
9] on lysozyme crystallization with five chlorides of Li
+, Na
+, K
+, Ni
2+ and Cu
2+, the positions of chloride ions and cations were revealed in the lattice.
The main aim of this work was to establish the dependence of the behaviour of lysozyme dimers and octamers on the type of precipitant cation (namely metal) added to the protein solution to initiate crystallization. For this purpose, we simulated the molecular dynamics of lysozyme dimers and octamers in aqueous solutions with different precipitants: LiCl, NaCl, KCl and CuCl
2, and then evaluated the stability, the change of the initial structure and compactness of oligomers, based on root mean square fluctuation (RMSF), root-mean-square deviation (RMSD) and radius of gyration (R
g) plots, respectively. Furthermore, we compared the simulation results with the experimental ones carried out using small angle X-ray scattering [
10].
2. Materials and Methods
Dimer and octamer models were derived using PyMol software (Version 1.5 [
11]) from the structures of lysozyme tetragonal crystals (HEWL) deposited in the Protein Data Bank. The structures used, obtained with LiCl, NaCl, KCl and CuCl
2 precipitants, have PDB ID: 6QX0, 6QWY, 6QWZ and 6QWW, respectively. The precipitant ions bound to the protein in the crystal, which were detected by X-ray crystallography, were retained in the structures (in the pdb files of the oligomers) and the crystalline water was removed.
There are several ways of the extraction of oligomers from the crystal structure, but in our work, we investigated only one type of octamers and dimers, most likely formed in the crystallization solution of lysozyme [
12]. Calculation of the ionization states of amino acid residues at pH 4.5 (according to the experimental conditions) was performed using PROPKA server (Version 2.0.0 [
13]). All residues that were found in crystals with multiple occupancies have the same probability of being in one of two conformations (A or B), as their occupancy factor is 0.5. Since there is no privilege, only conformation A was retained.
All calculations and structure preparation were done in the GROMACS software package version 2021 [
14] and were performed in the same way for different oligomer models. The Amber ff99SB-ILDN field [
15] with new potential parameters for torsion angles was used as the force field. During the equilibration of the system and productive MD simulation, three-dimensional periodic boundary conditions were applied, and the long-range electrostatic interactions were considered by the smooth particle mesh Ewald (PME) summation method [
16] with cubic interpolation and grid spacing in Fourier space of 0.16 nm. Noncovalent interactions were considered only for atoms at less than 1 nm from each other. The bond lengths in the oligomers were constrained using the LINCS algorithm [
17].
The prepared oligomer structures were placed in the centre of a cubic simulation box with a minimum distance of 1 nm between its edge and the protein molecule. The remaining space of the box was filled with TIP4P-Ew water [
18] for performing MD calculations using the Ewald summation method. To reproduce the conditions of the crystallization experiment water molecules were replaced with one of the cation types (Li
+, Na
+, K
+, Cu
2+) and Cl
− anions so that the precipitant concentration in the box was 0.4 M. Using the PME algorithm for calculating long-range electrostatic interactions formally requires the neutrality of the total charge of the molecular system; therefore, the necessary amount of Cl
− ions was added to each box. Thus, boxes with LiCl, NaCl and KCl contained approximately the same number of cations and anions (chlorine ions were slightly more due to charge neutralisation), whereas in the case of CuCl
2, twice as many chlorine ions were added as copper ions. Since there were no parameters for copper ions in the ff99SB-ILDN force field, the data on them was included in the residue database (aminoacids.rtp) and the ions topology (ions.itp) files.
As a result, each box with a dimer contained 159 ± 1 cations and 175 ± 4 chlorine anions (both dissolved in bulk solution and bound to the protein), except for the case with copper, in which 151 copper cations and 327 chlorine ions were added. Each box with an octamer contained from 628 to 684 cations and from 689 to 774 anions, except for the case with copper, in which 627 copper cations and 1360 chlorine ions were added.
The energy minimization and equilibration of the systems were carried out according to the following protocol. First, to get rid of atomic overlaps in the original crystal structure and those created by the addition of water, the energy of each system was minimized using the steepest descent algorithm (50,000 steps) so that the force acting on any atom did not exceed 1000 kJ∙M
−1nm
−2. The systems were then sequentially equilibrated in NVT- and NPT- ensembles by the modified Berendsen (V-rescale) [
19] and Parrinello-Rahman methods [
20], respectively (for 100 ps each). The integration time step was set at 2 fs, and the temperature and pressure were 283 K and 1 atm. Productive MD was calculated in NPT ensemble (at P = 1 atm, T = 283 K) using modified Berendsen thermostat and Parrinello-Rahman barostat. Integration was carried out by standard leap-frog algorithm [
21]. The duration of each of the calculated lysozyme dimer trajectories was 100 ns. Three independent MD simulations were performed for each precipitant.
Before the analysis of the trajectories, artifacts arising from quasi-infinite periodic boundary conditions were removed by using the gmx trjconv command with the −pbc nojump flag to restore the integrity of the oligomers. RMSF, RMSD and Rg were then plotted by running the commands gmx rmsf, gmx rms and gmx gyrate, respectively. The structural alignment of the trajectories of lysozyme atoms to the initial position in the RMSD calculation was performed by the command gmx trjconv with the flag −fit rot + trans excluding the movement of the protein as a solid body (parallel transfers and rotations around the axis) because it does not affect its conformational changes. For each curve in the RMSF, RMSD and Rg plots, the values were averaged over three independent simulations.
3. Results
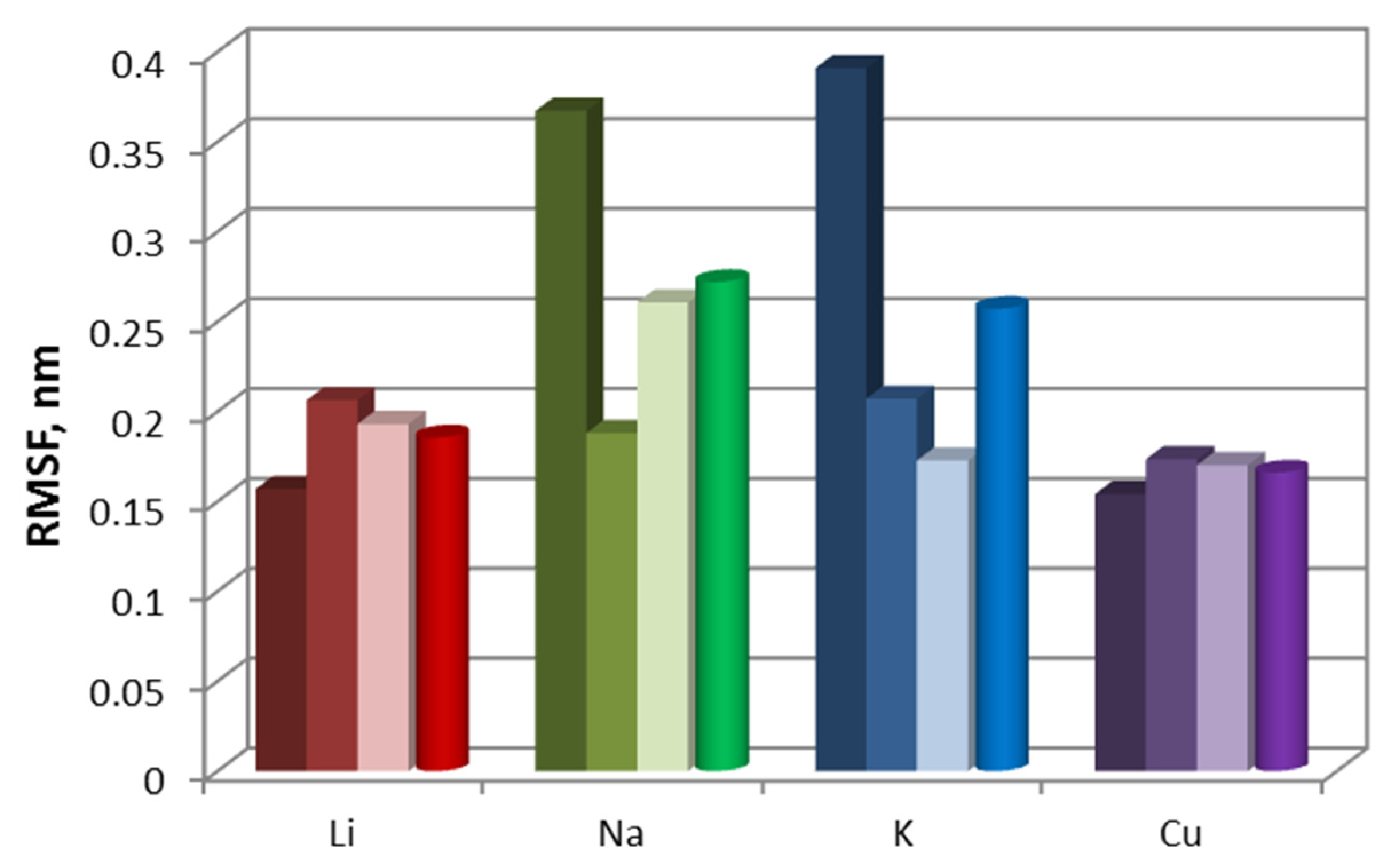
The stability of the oligomers was assessed by RMSF plots of the Cα atoms. RMSF values serve as a measure of the flexibility of the polypeptide chain, as they indicate the degree of deviation of the Cα atoms from their average position. The higher the RMSF value of an atom, the more mobile it is.
From
Figure 1 and
Table 1 it follows that the most stable dimers are those with sodium and lithium ions—their averaged over all atoms RMSF values are 0.116 nm (
Table 1). Dimers with copper (RMSF is 0.137 nm) and potassium (RMSF is 0.138 nm) are more flexible. The region of amino acid residues ARG125, GLY126, CYS127, ARG128 and LEU129 of the polypeptide chain A is mainly destabilized when simulated with potassium cations (green curve in
Figure 1) and chain B—in case of using copper cations (blue curve). It should be noted that these residues are located on the surface of the protein molecule but do not participate in the formation of covalent or hydrogen bonds between monomers. Moreover, they are far from the interface between monomers; residue LEU129 is terminal in the polypeptide chain.
In
Figure 2, the precipitants have different effects on the different polypeptide chains of the octamer. For example, chain “A” was relatively stable only when the simulation was conducted with copper (blue curve), while chains “B” and “C” were mostly destabilized by sodium (yellow curve).
From
Figure 2 and
Table 2 it is noticeable that the most stable octamers are with copper and lithium whereas octamers with potassium and sodium are the least rigid.
It should be noted that the three independent simulations for both octamers with sodium and octamer with potassium are quite different from each other, unlike the MD of octamers with lithium and copper, as demonstrated in
Figure 3.
The change of oligomer structure during molecular dynamics was evaluated based on RMSD plots, i.e., root-mean-square deviation of all Cα atoms from the initial (in our case crystalline) structure. The difference between RMSF and RMSD is that RMSF reflects the fluctuations of each atom around its average position over the entire simulation time, while RMSD reflects the deviation of all atoms as a function of time.
Figure 4 and
Table 1 show that most of all, especially starting from the 60th ns, the dimer with copper is destabilized (RMSD~0.31 nm). The structure of the dimer with lithium (RMSD~0.21 nm) remained the most similar to the initial one (as in the crystal) throughout the simulation. Dimers with sodium and potassium, although they underwent significant transformations in the course of dynamics, have an RMSD of 0.25 nm on average over 100 ns. As can be seen from
Table 1, the dependence of RMSD on the type of cation is almost ideally correlated with the experimental results on the study of precrystallization solutions of lysozyme using SAXS, since the more the dimer is destabilized, the lower its concentration.
In
Figure 5, the RMSDs of octamers with sodium and potassium increase with time, i.e., these clusters did not manage to reach their equilibrium states in 100 ns, and their structures changed dramatically. The lithium and copper octamers retained the best similarity to their initial structures (derived from the crystal).
The radius of gyration characterizes the size (compactness) of a protein. It is calculated as the root-mean-square distance of all the atoms in the protein from the centre of mass of the molecule.
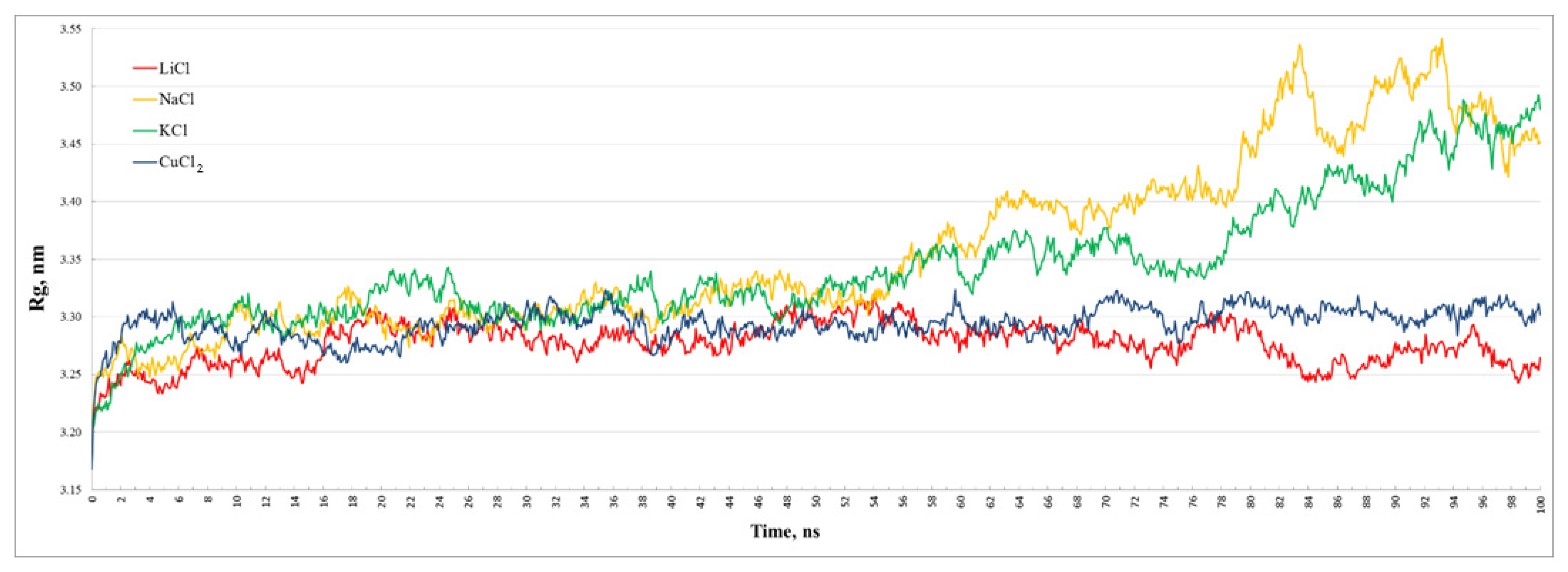
From
Figure 6 and
Table 1, it follows that the dimer with copper remained the least compact during almost the entire dynamics, while the dimer with lithium, on the contrary, thickens slightly with time. The volumes of dimers with sodium and potassium, on average, take intermediate values, despite the fact that they underwent noticeable changes. However, all dimers generally retain their sizes, and the radii of gyration vary within 0.15 nm (from 1.96 to 2.11 nm).
Table 1 and comparison of the plots of the radius of gyration and RMSD of the dimers show that they correlate: with an increase in RMSD, R
g also grows, while the root-mean-square deviations of dimers with sodium and potassium, as well as their compactness, are approximately the same.
In
Figure 7, the sizes of the sodium and potassium octamers increase the most, their radii of gyration varying in the range of 0.3 nm. Lithium octamer practically does not change its compactness throughout the whole simulation; its radius of gyration fluctuates within 0.1 nm. The volume of octamer with copper remains the most constant throughout the 100 ns dynamics, although it noticeably increases already in 3 ns, after which its R
g vary in the range of 0.06 nm.
Finally,
Figure 8 and
Figure 9 represent a comparison of the temperature B-factor with the RMSF obtained from the MD simulation. It is noticeable that the B-factors of dimers significantly correlate with RMSFs (
Figure 8). In the case of octamers, the correlation is worse, which can be explained by the movement of monomers included in octamers relative to each other during MD (
Figure 9).
4. Discussion
Comparison of concentration and plots of RMSF, RMSD and radius of gyration of dimers (
Table 1) shows that concentration strongly correlates with RMSD and radius of gyration while the RMSF results do not correspond to the experimental ones (SAXS) to this extent (so well), though the agreement is still satisfactory. The fact that RMSF data do not correlate with concentration ones, as well as RMSD and radius of gyration dependences, may, firstly, indicate that the structures of dimers appearing in solutions before lysozyme crystallization are very close to crystalline. Secondly, the RMSD and radius of gyration results seem to be more reliable than RMSF data during the investigation of pre-crystallization oligomers by molecular dynamics. Overall, the dependence of dimers’ behaviour in solutions before lysozyme crystallization on precipitant cations is confirmed.
From a comparison of RMSF, RMSD and radius of gyration of octamers on
Figure 2 and
Table 2, it follows that they correlate—the most rigid molecules are with copper and lithium (their average RMSFs are 0.166 and 0.185 nm, respectively), whereas the clusters modelled with sodium and potassium are the least stable. However, the experimentally observable concentration of octamers is the lowest in the case of lithium and copper and the highest when modelling with sodium and potassium.
In
Figure 3, for the potassium and sodium octamers, the largest contribution to the average RMSF is made by the first simulation, during which octamers began to decompose into smaller oligomers as visual inspection of the trajectories revealed. Their average RMSFs are 0.257 and 0.272 nm, respectively. However, even if these simulations are accidental, the results are reliable as RMSFs averaged over two other simulations equal to 0.19 nm for potassium and 0.224 nm for sodium; therefore, octamers with these precipitants are still the most unstable. Nevertheless, even considering the data excluding simulations when octamers dissociate, the results are in a disagreement with the experimental ones as the more the octamer structure changes during the dynamics, the higher its concentration. Moreover, taking into account all three independent simulations for each type of precipitant leads to an absence of correlation.
The reason for the correspondence of simulation results to the experimental ones only for dimers and not for octamers can be in the fact that dimers and octamers both were modelled in solutions with the precipitant concentration of 0.4 M, whereas octamers probably form after dimers when precipitant concentration is higher than 0.4 M.
While in the work [
2] there was no significant effect of cation nature of precipitant on the ability to switch protein interactions to attractive ones found, but a number of chlorine anions incorporated in crystal (and oligomers) structures undoubtedly depend on cation type of the precipitant (
Table 3 and
Figure 10). Nevertheless, the positions of Cl
− did not change with varying NiCl
2 concentrations, whereas it affected a number of Ni
2+ [
22]. The monomer extracted from the lysozyme crystal grown from the LiCl solution has the least number (three) of chlorine ions bound to it, whereas structures corresponding to the CuCl
2 solution have the most number of them (six per monomer). NaCl and KCl lysozyme monomers incorporate four and five chlorine ions, respectively. It should be noted that lithium and potassium cations are absent in the lysozyme structures. Lithium ions may not be detected by X-ray structural analysis, whereas potassium ions might be too large to incorporate in the protein but they force more chlorine anions (five per monomer) to associate with lysozyme (
Table 3 and
Figure 10).
From [
3], it is known that a majority of anion sites are observed in all lysozyme crystals, whatever their form or precipitant nature is, while there is no site that is common to all structures. However, analysis of structures of oligomers considered above showed that chorine ions are always present in three constant sites (SER-24, GLY-26, GLN-121; THR-69, ARG-68, GLY-67, SER-72, ASN-65; LYS-33) that may be the most important for lysozyme oligomers forming before tetragonal crystal growth. Additionally, other sites seem to slightly destabilize dimers, since the lower the concentration of dimers is, the more sites are involved. Moreover, a site with chlorine ion bound to residues (TYR-23 and ASN-113) of two different chains might be responsible for octamers sustainability: firstly, this is the only site when chlorine ion is associated with two chains simultaneously. Secondly, this site is found only in octamers extracted from lysozyme crystals grown from NaCl and KCl solutions, whereas octamers’ concentration is the highest in the case of these precipitants. It should be noted that our inferences concern only the relative efficiency of different chlorine sites to influence the stability of lysozyme oligomers considered here as their number and position strongly vary depending on precipitant concentration; see, for example, [
23].













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}