
Phase Behaviour of Methane Hydrates in Confined Media


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
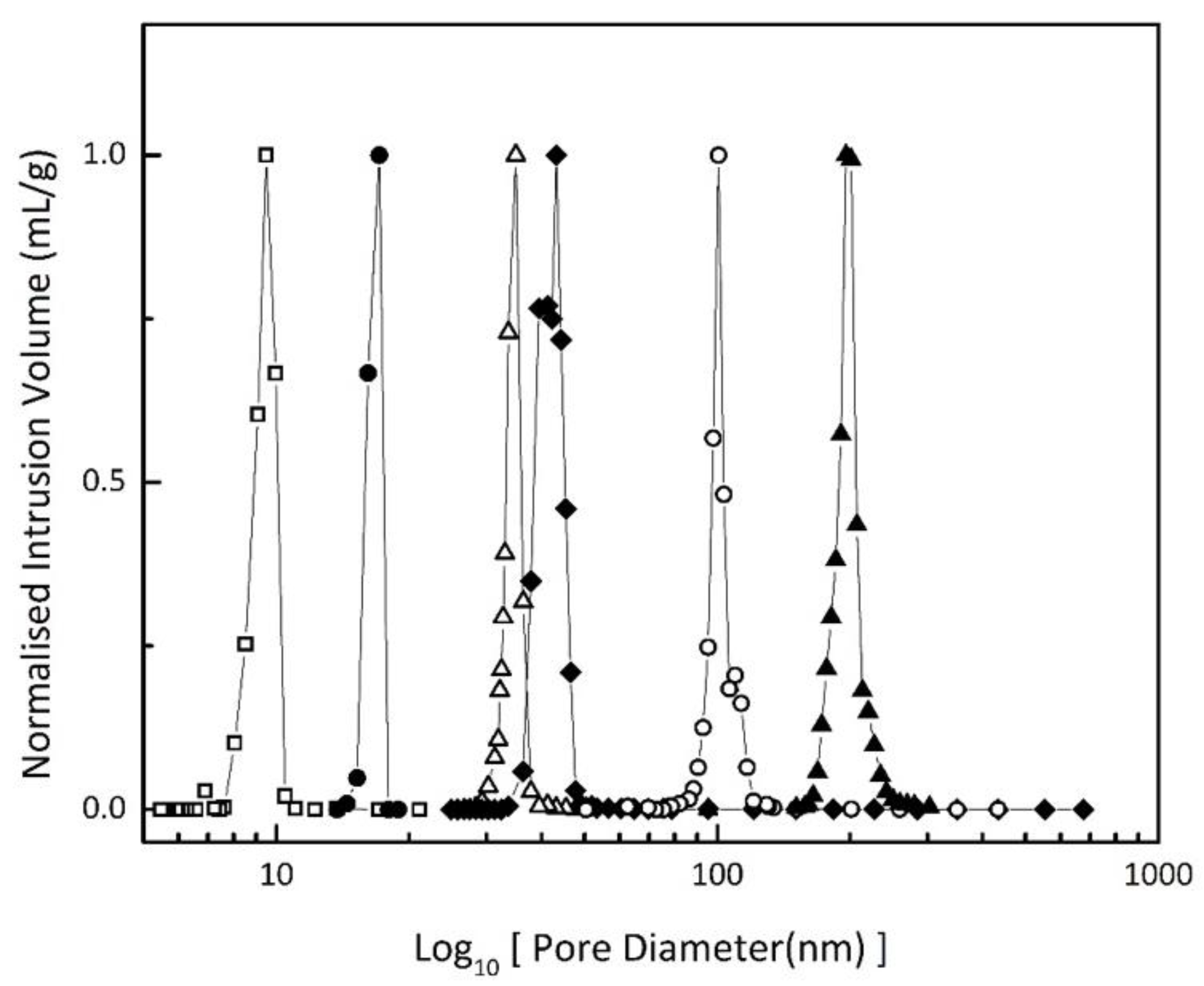
2.2. Characterisation
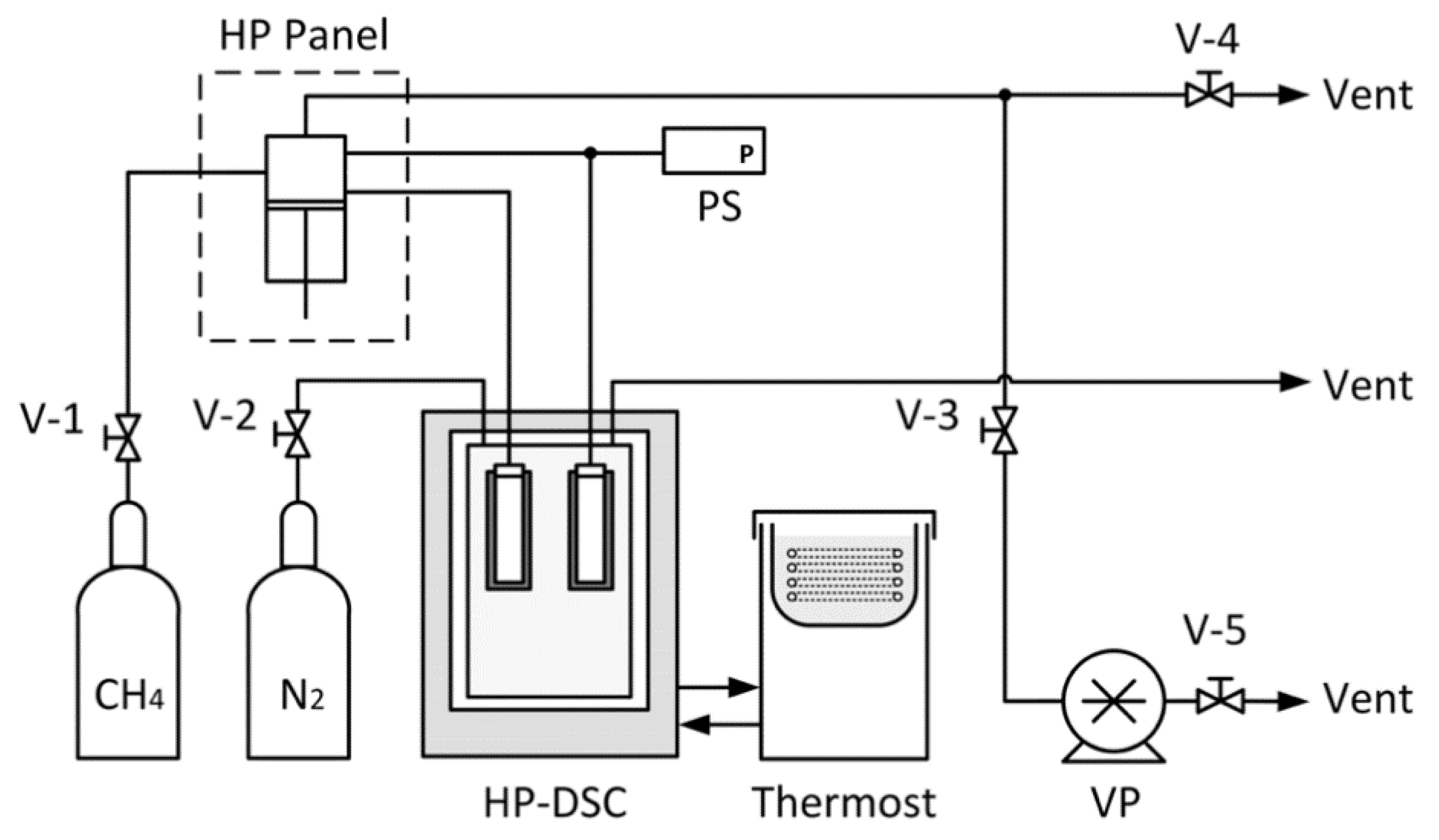
2.3. DSC Measurement
2.3.1. Calibration
2.3.2. Formation of Methane Hydrates in Porous Media
3. Results and Discussion
3.1. Experimental Conditions
3.1.1. Impact of Particle Size
3.1.2. Impact of Sample Mass
3.1.3. Impact of Evacuation Duration
3.1.4. Impact of Formation Durations
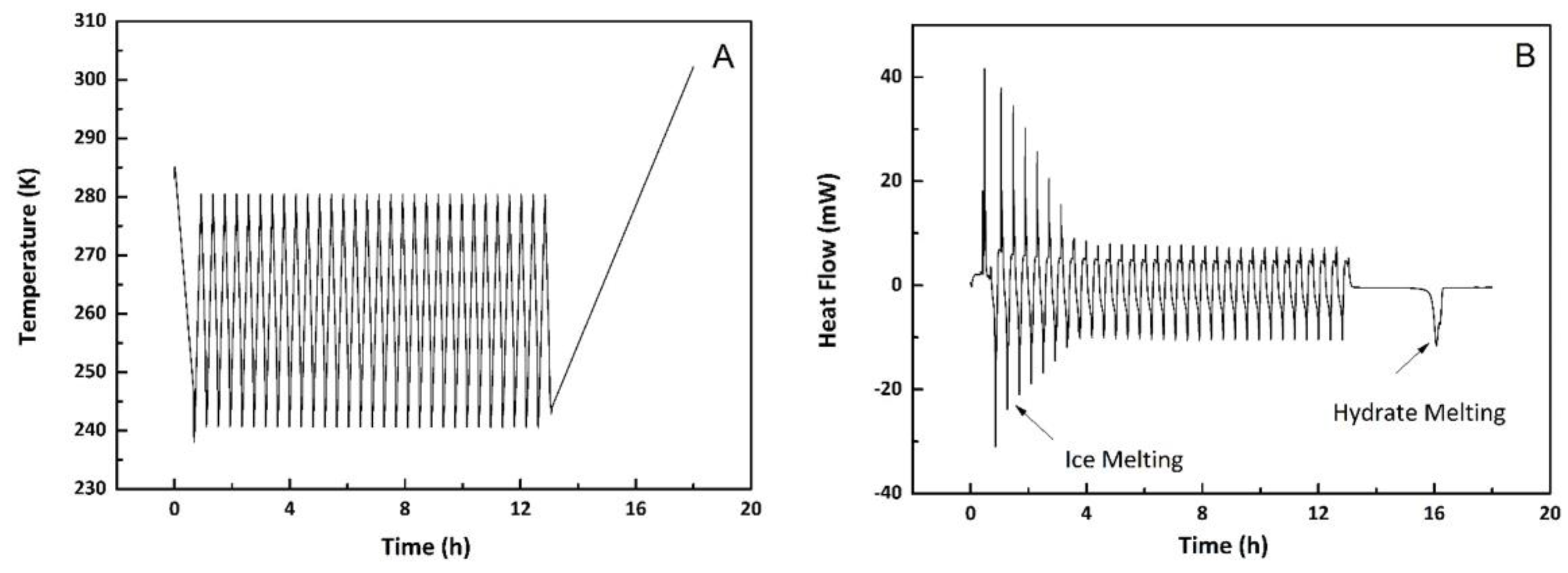
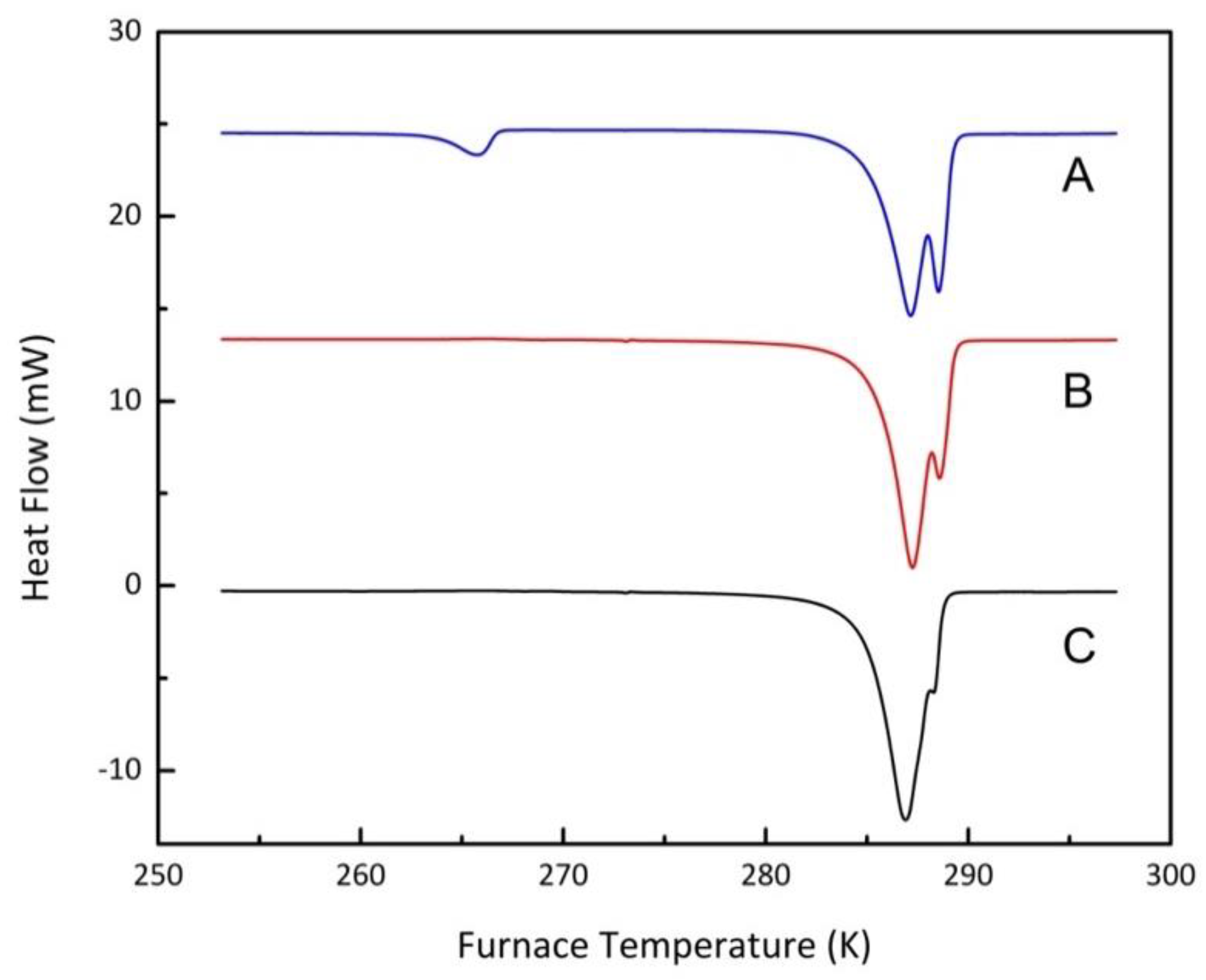
3.1.5. Memory Effects
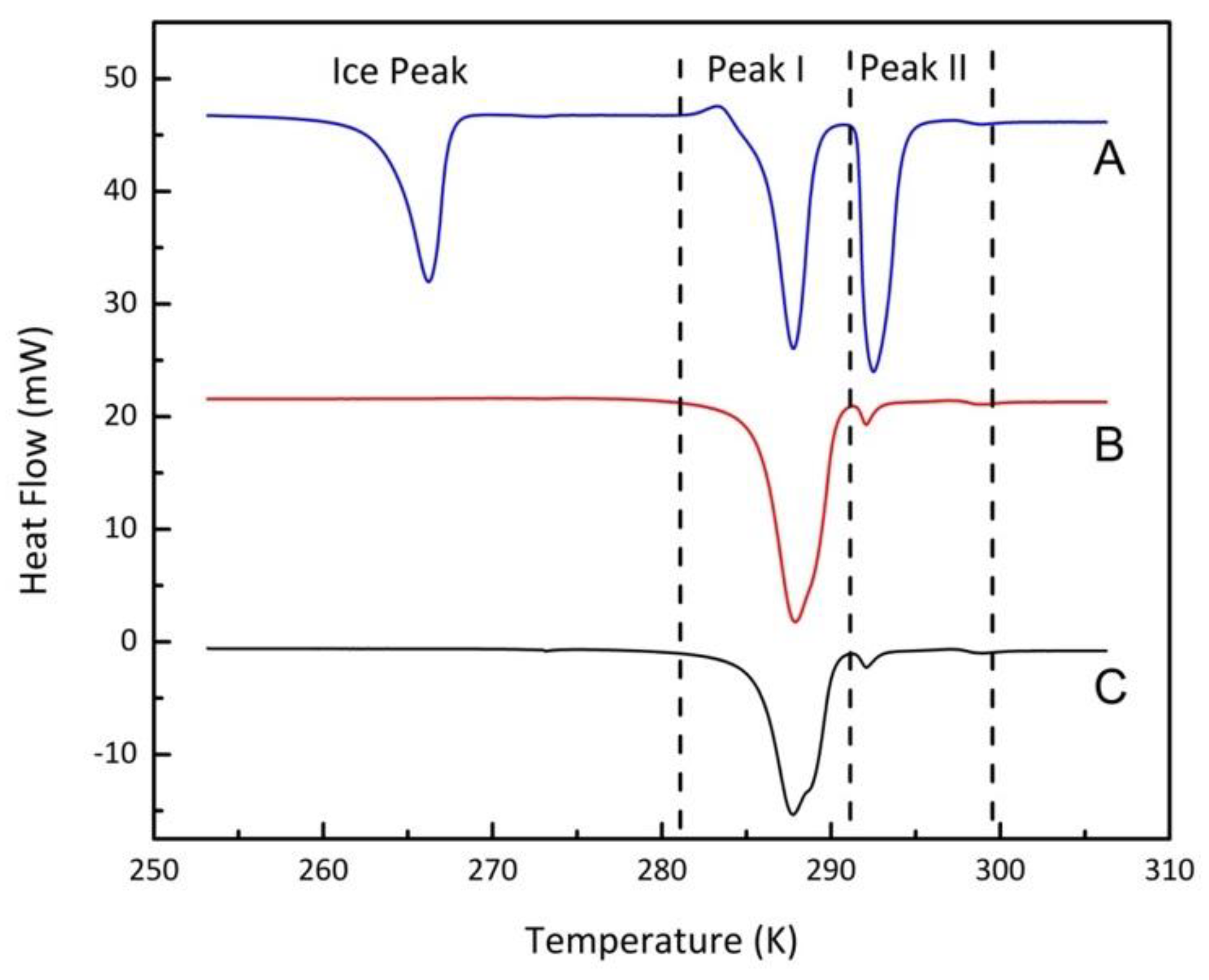
3.2. Experimental Results
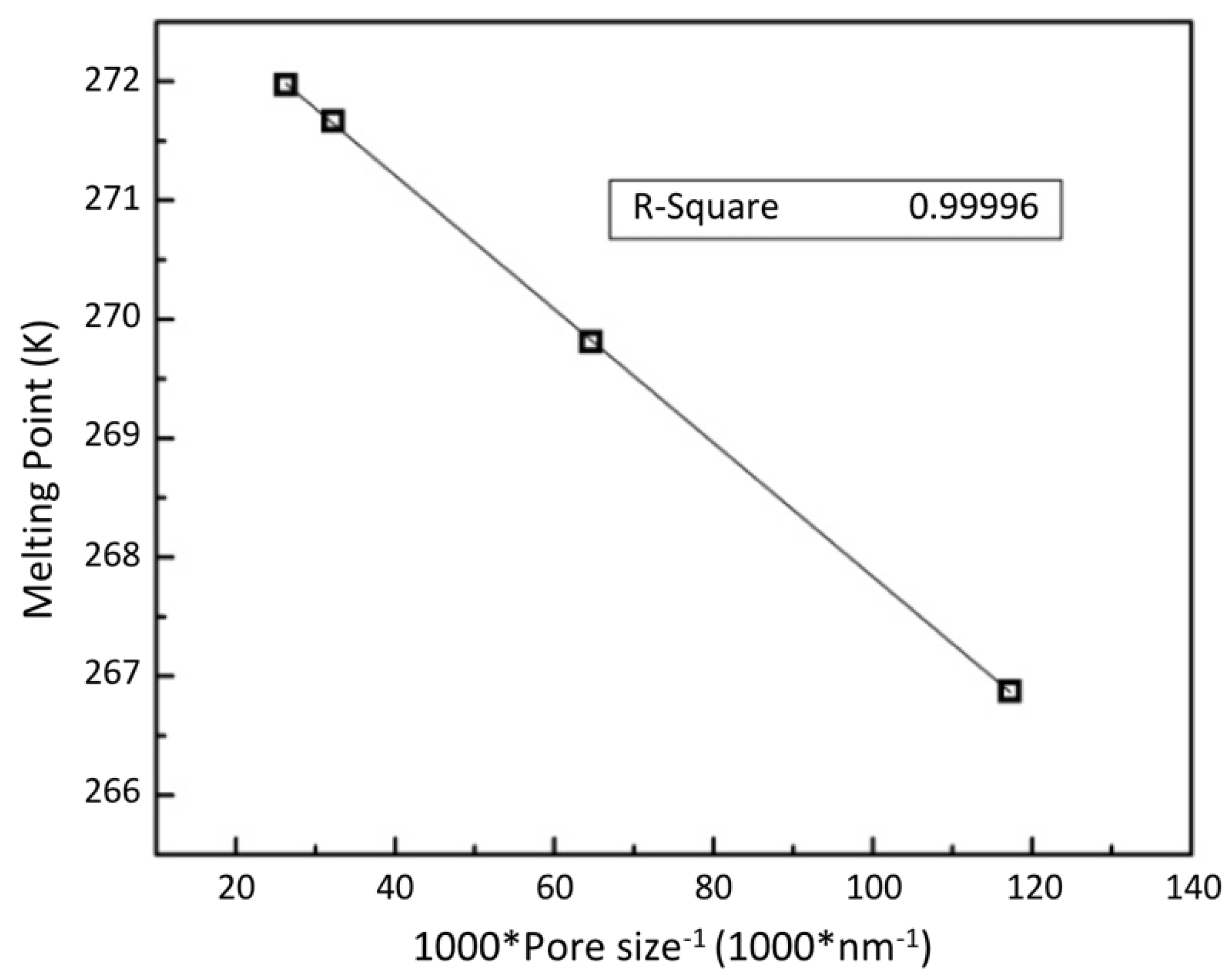
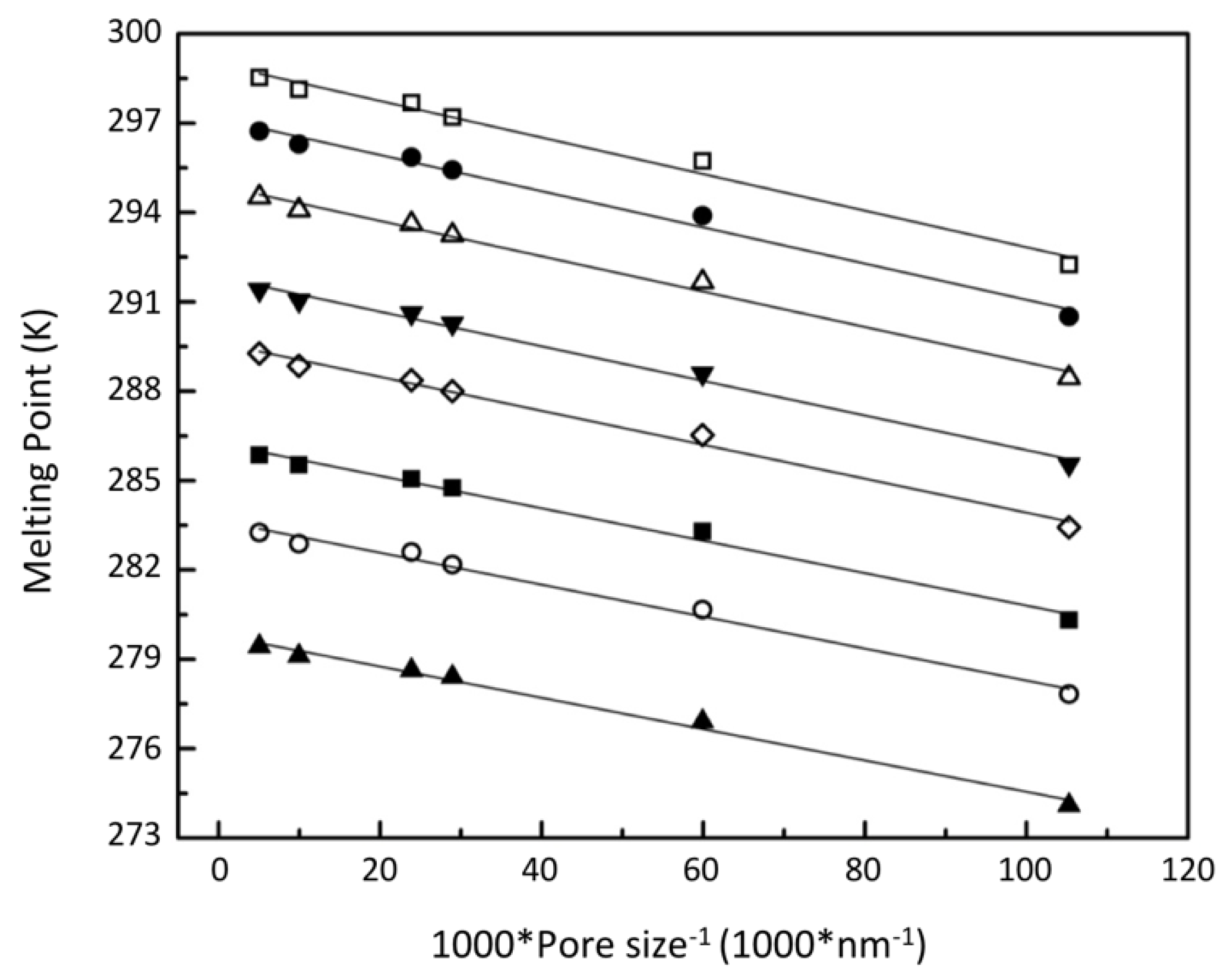
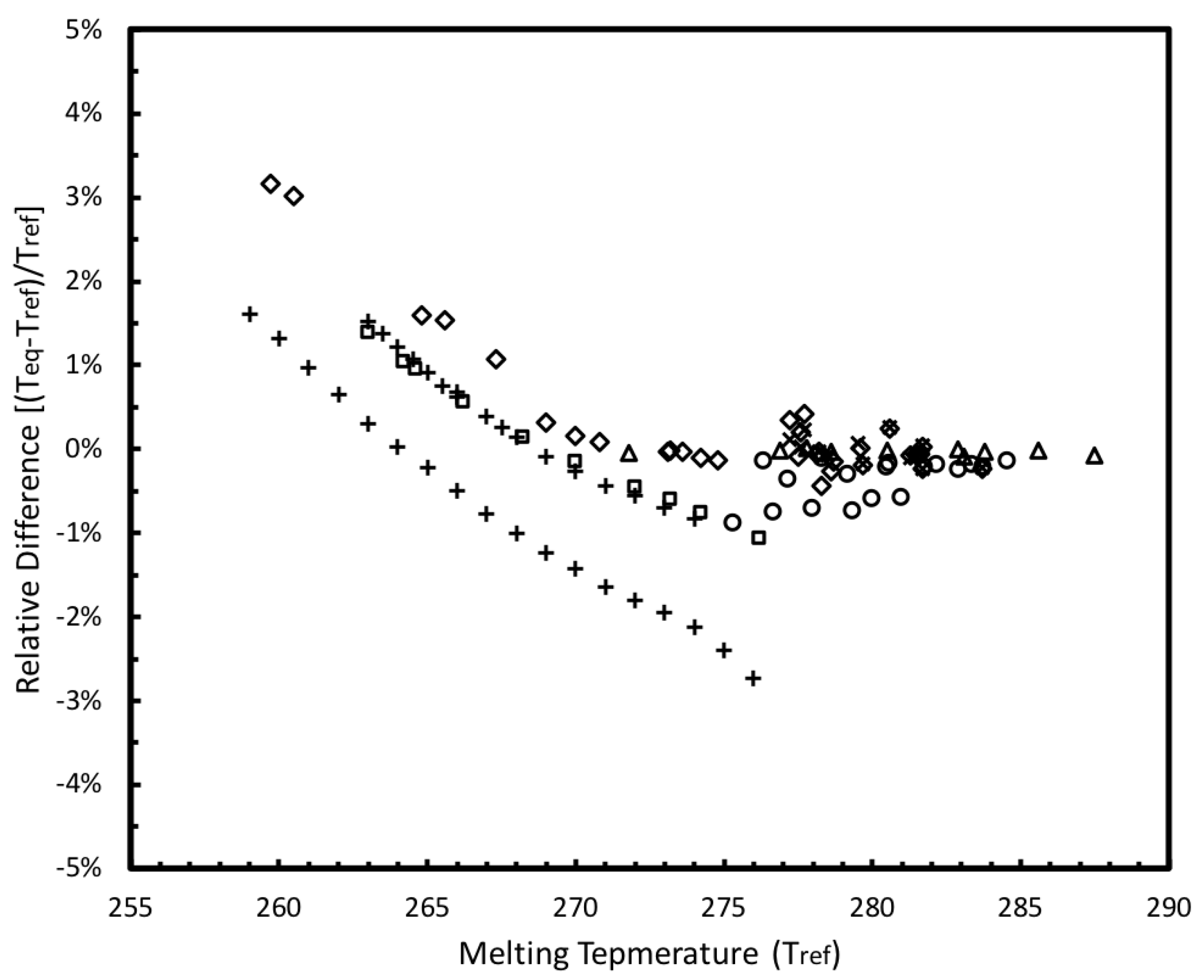
3.2.1. Confined Melting Points of Ice
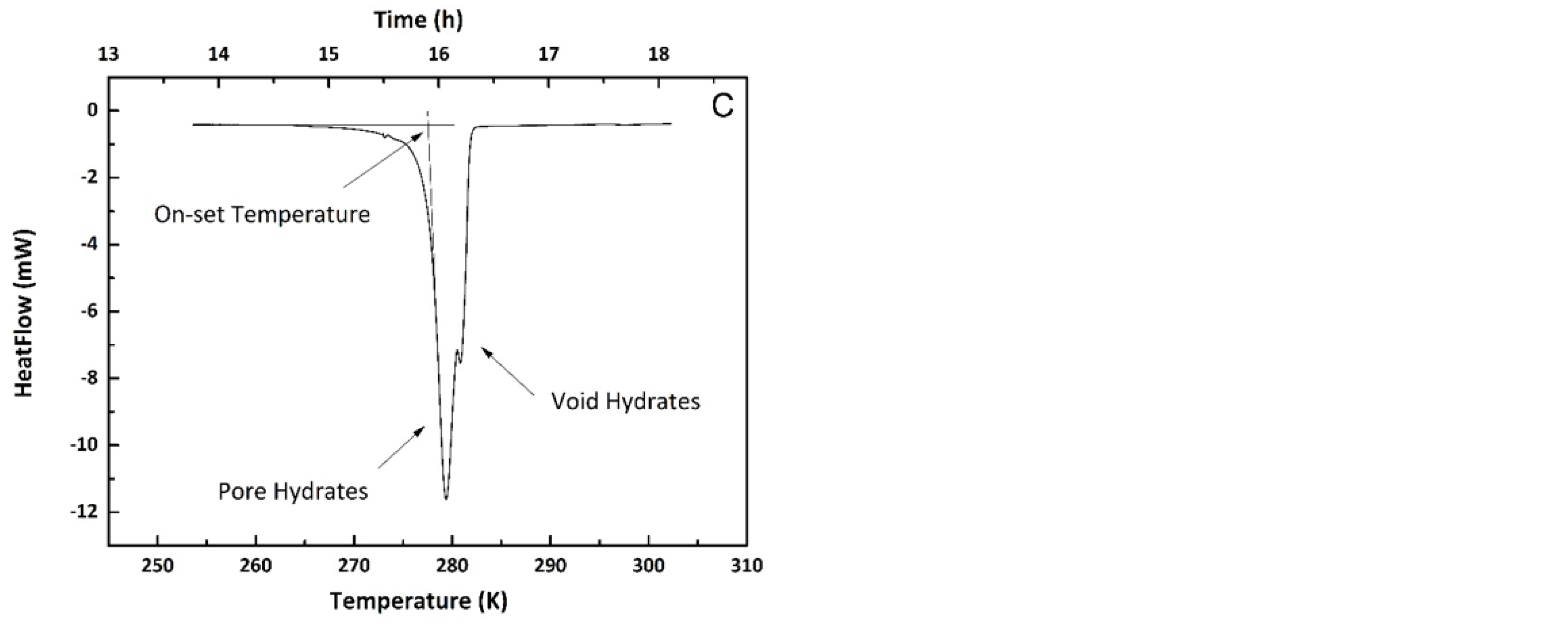
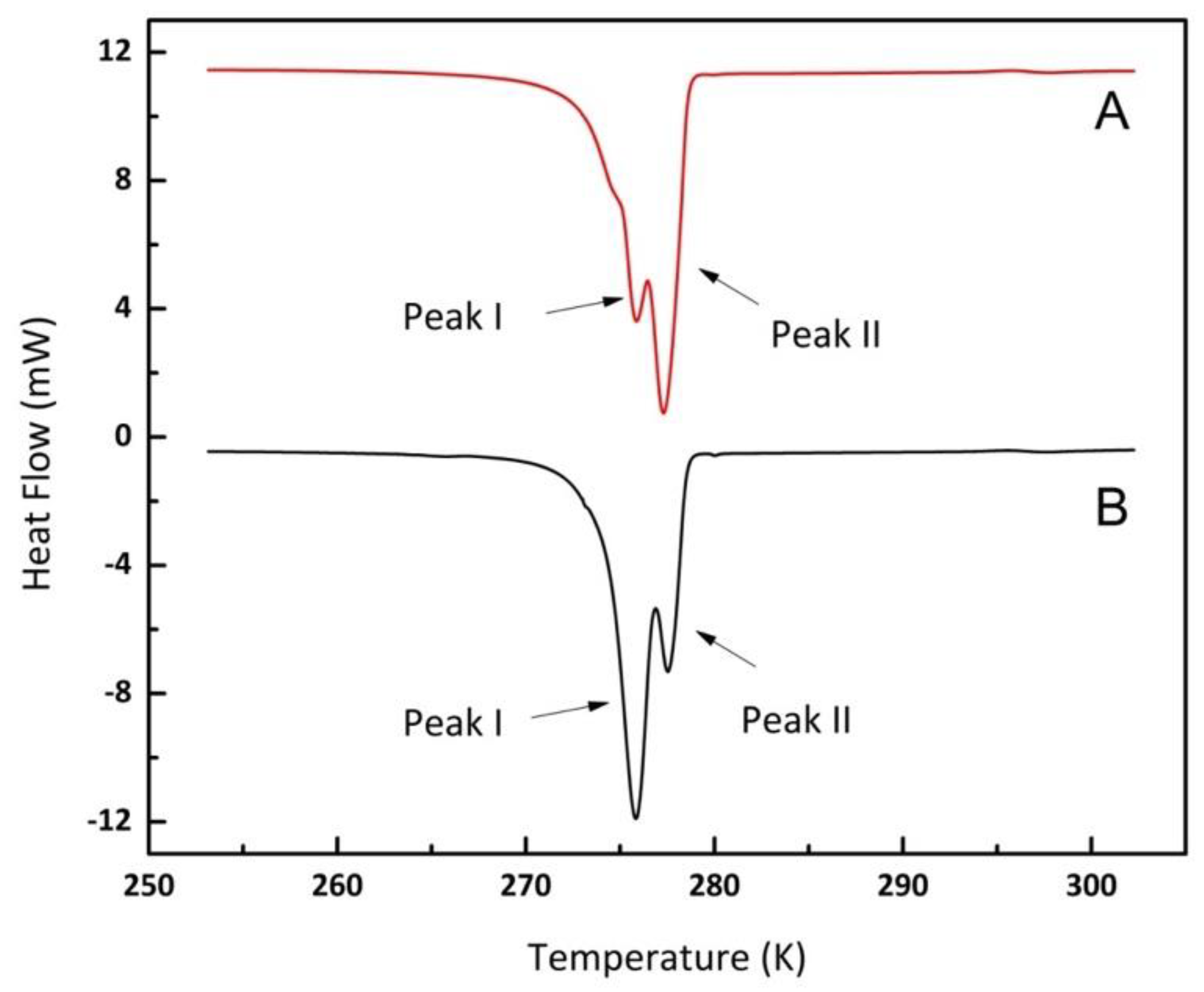
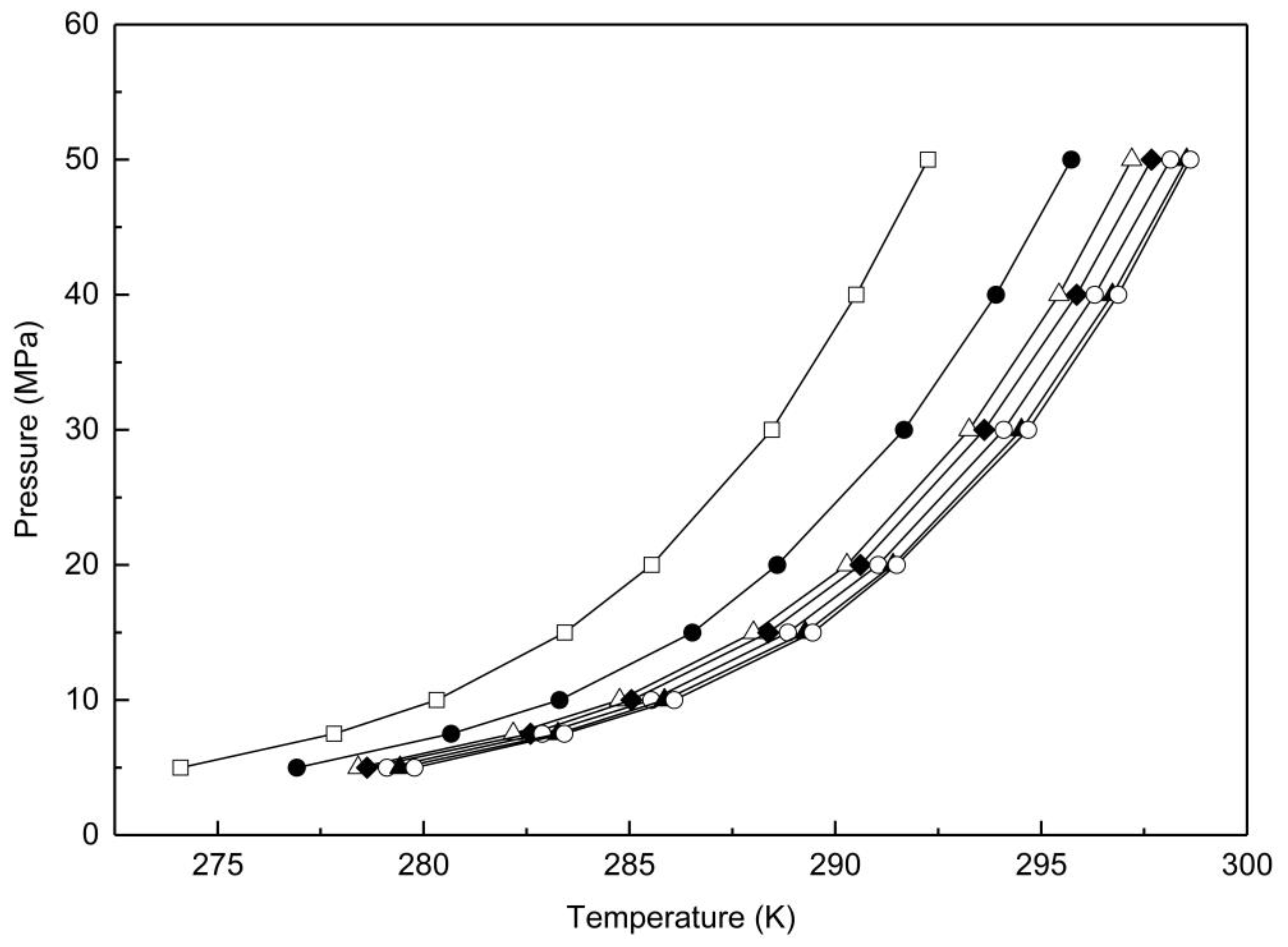
3.2.2. Methane Hydrates in Confined Media
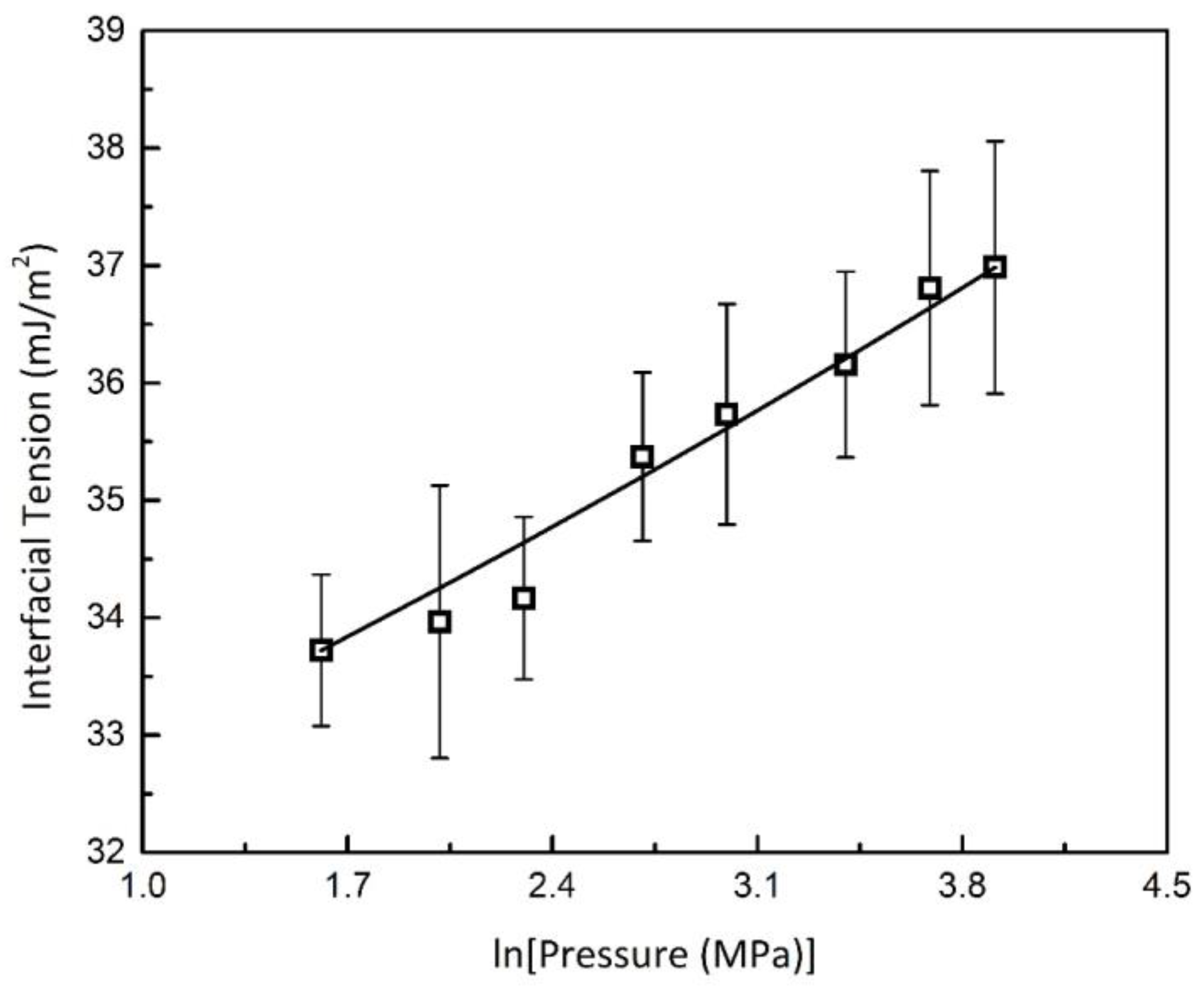
3.2.3. Interfacial Energy and the Gibbs–Thomson Equation
3.2.4. Empirical Correlation
3.2.5. Enthalpy of Fusion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sloan, E.D., Jr.; Koh, C.A. Clathrate Hydrates of Natural Gases, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 1–730. [Google Scholar]
- Jamadagni, S.N.; Godawat, R.; Garde, S. Annual review of chemical and biomolecular engineering. Ann. Rev. Palo Alto 2011, 2, 237–257. [Google Scholar]
- Chong, Z.R.; Yang, S.H.B.; Babu, P.; Linga, P.; Li, X.-S. Review of natural gas hydrates as an energy resource: Prospects and challenges. Appl. Energy 2016, 162, 1633–1652. [Google Scholar] [CrossRef]
- Boswell, R.; Collett, T.S. Current perspectives on gas hydrate resources. Energy Environ. Sci. 2011, 4, 1206–1215. [Google Scholar] [CrossRef]
- Li, X.-S.; Xu, C.-G.; Zhang, Y.; Ruan, X.-K.; Li, G.; Wang, Y. Investigation into gas production from natural gas hydrate: A review. Appl. Energy 2016, 172, 286–322. [Google Scholar] [CrossRef] [Green Version]
- Konno, Y.; Fujii, T.; Sato, A.; Akamine, K.; Naiki, M.; Masuda, Y.; Yamamoto, K.; Nagao, J. Key Findings of the World’s First Offshore Methane Hydrate Production Test off the Coast of Japan: Toward Future Commercial Production. Energy Fuels 2017, 31, 2607–2616. [Google Scholar] [CrossRef]
- Casco, M.E.; Silvestre-Albero, J.; Ramírez-Cuesta, A.J.; Rey, F.; Jordá, J.L.; Bansode, A.; Urakawa, A.; Peral, I.; Martínez-Escandell, M.; Kaneko, K.; et al. Methane hydrate formation in confined nanospace can surpass nature. Nat. Commun. 2015, 6, 6432. [Google Scholar] [CrossRef] [Green Version]
- Klauda, J.B.; Sandler, S.I. Predictions of gas hydrate phase equilibria and amounts in natural sediment porous media. Mar. Pet. Geol. 2003, 20, 459–470. [Google Scholar] [CrossRef]
- Shukla, K.M.; Kumar, P.; Yadav, U.S. Gas hydrate reservoir identification, delineation, and characterization in the Krishna-Godavari basin using subsurface geologic and geophysical data from the national gas hydrate program 02 expedition, offshore India. Mar. Pet. Geol. 2019, 108, 185–205. [Google Scholar] [CrossRef]
- Lei, L.; Seol, Y.; Choi, J.-H.; Kneafsey, T.J. Pore habit of methane hydrate and its evolution in sediment matrix—Laboratory visualization with phase-contrast micro-CT. Mar. Pet. Geol. 2019, 104, 451–467. [Google Scholar] [CrossRef] [Green Version]
- Moridis, G.J.; Seol, Y.; Kneafsey, T.J. Studies of Reaction Kinetics of Methane Hydrate Dissocation in Porous Media. 2005. Available online: https://escholarship.org/uc/item/8q50w5cn (accessed on 28 January 2021).
- Kneafsey, T.J.; Tomutsa, L.; Moridis, G.J.; Seol, Y.; Freifeld, B.; Taylor, C.E.; Gupta, A. Methane hydrate formation and dissociation in a partially saturated sand—measurements and observations. J. Pet. Sci. Eng. 2007, 56, 108–126. [Google Scholar] [CrossRef] [Green Version]
- Nikitin, V.V.; Dugarov, G.A.; Duchkov, A.A.; Fokin, M.I.; Drobchik, A.N.; Shevchenko, P.D.; De Carlo, F.; Mokso, R. Dynamic in-situ imaging of methane hydrate formation and self-preservation in porous media. Mar. Pet. Geol. 2020, 115, 104234. [Google Scholar] [CrossRef]
- Handa, Y.P.; Stupin, D.Y. Thermodynamic properties and dissociation characteristics of methane and propane hydrates in 70-.ANG.-radius silica gel pores. J. Phys. Chem. 1992, 96, 8599–8603. [Google Scholar] [CrossRef]
- Uchida, T.; Ebinuma, T.; Ishizaki, T. Dissociation Condition Measurements of Methane Hydrate in Confined Small Pores of Porous Glass. J. Phys. Chem. B 1999, 103, 3659–3662. [Google Scholar] [CrossRef]
- Uchida, T.; Ebinuma, T.; Takeya, S.; Nagao, J.; Narita, H. Effects of pore sizes on dissociation temperatures and pressures of methane, carbon dioxide, and propane hydrates in porous media. J. Phys. Chem. B 2002, 106, 820–826. [Google Scholar] [CrossRef]
- Smith, D.H.; Wilder, J.W.; Seshadri, K. Methane Hydrate Equilibria in Silica Gels with Broad Pore-Size Distributions. Aiche J. 2002, 48, 393–400. [Google Scholar] [CrossRef]
- Seo, Y.; Lee, H.; Uchida, T. Methane and Carbon Dioxide Hydrate Phase Behavior in Small Porous Silica Gels: Three-Phase Equilibrium Determination and Thermodynamic Modeling. Langmuir 2002, 18, 9164–9170. [Google Scholar] [CrossRef]
- Anderson, R.; Llamedo, M.; Tohidi, B.; Burgass, R.W. Experimental Measurement of Methane and Carbon Dioxide Clathrate Hydrate Equilibria in Mesoporous Silica. J. Phys. Chem. B 2003, 107, 3507–3514. [Google Scholar] [CrossRef]
- Kang, S.P.; Ryu, H.J.; Seo, Y. Phase Behavior of CO2 and CH4 Hydrate in porous media. Int. J. Appl. Sci. Eng. Technol. 2007, 4, 189–194. [Google Scholar]
- Kang, S.P.; Lee, J.W.; Ryu, H.J. Phase behavior of methane and carbon dioxide hydrates in meso-and macro-sized porous media. Fluid Phase Equilibria 2008, 274, 68–72. [Google Scholar] [CrossRef]
- Kluijtmans, S.G.J.M.; Dhont, J.K.G.; Philipse, A.P. A Light-Scattering Contrast-Variation Study of Bicontinuous Porous Glass Media. Langmuir 1997, 13, 4976–4981. [Google Scholar] [CrossRef]
- Sing, K.S. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Allen, T. Particle Size Measurement; Springer: Dordrecht, The Netherlands, 1997; Volume 1, ISBN 978-94-009-0417-0. [Google Scholar]
- Lee, S.; Lee, Y.; Lee, J.; Lee, H.; Seo, Y. Experimental verification of methane–carbon dioxide replacement in natural gas hydrates using a differential scanning calorimeter. Environ. Sci. Technol. 2013, 47, 13184–13190. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Lachance, J.; Sloan Jr, E.D.; Koh, C.A. Measurements of methane hydrate heat of dissociation using high pressure differential scanning calorimetry. Chem. Eng. Sci. 2008, 63, 5848–5853. [Google Scholar] [CrossRef]
- Takeya, S.; Hori, A.; Hondoh, T.; Uchida, T. Freezing-memory effect of water on nucleation of CO2 hydrate crystals. J. Phys. Chem. B 2000, 104, 4164–4168. [Google Scholar] [CrossRef]
- Schmidt, R.; Hansen, E.W.; Stoecker, M.; Akporiaye, D.; Ellestad, O.H. Pore size determination of MCM-51 mesoporous materials by means of 1H NMR spectroscopy, N2 adsorption, and HREM. A Preliminary Study. J. Am. Chem. Soc. 1995, 117, 4049–4056. [Google Scholar] [CrossRef]
- Liu, E.; Dore, J.C.; Webber, J.B.W.; Khushalani, D.; Jähnert, S.; Findenegg, G.; Hansen, T. Neutron diffraction and NMR relaxation studies of structural variation and phase transformations for water/ice in SBA-15 silica: I. The over-filled case. J. Phys. Condens. Matter 2006, 18, 10009. [Google Scholar]
- Jackson, C.L.; McKenna, G.B. The melting behavior of organic materials confined in porous solids. J. Chem. Phys. 1990, 93, 9002–9011. [Google Scholar] [CrossRef]
- Hillig, W.B. Measurement of interfacial free energy for ice/water system. J. Cryst. Growth 1998, 183, 463–468. [Google Scholar] [CrossRef]
- Overloop, K.; Vangerven, L. Freezing phenomena in adsorbed water as studied by NMR. J. Magn. Reson. Ser. A 1993, 101, 179–187. [Google Scholar] [CrossRef]
- Schreiber, A.; Ketelsen, I.; Findenegg, G.H. Melting and freezing of water in ordered mesoporous silica materials. Phys. Chem. Chem. Phys. 2001, 3, 1185–1195. [Google Scholar] [CrossRef]
- Jähnert, S.; Chávez, F.V.; Schaumann, G.; Schreiber, A.; Schönhoff, M.; Findenegg, G. Melting and freezing of water in cylindrical silica nanopores. Phys. Chem. Chem. Phys. 2008, 10, 6039–6051. [Google Scholar] [CrossRef] [Green Version]
- Helgerud, M.; Waite, W.F.; Kirby, S.; Nur, A. Elastic wave speeds and moduli in polycrystalline ice Ih, sI methane hydrate, and sII methane-ethane hydrate. J. Geophys. Res. Solid Earth 2009, 114. [Google Scholar] [CrossRef]
- Anderson, G.K. Enthalpy of dissociation and hydration number of methane hydrate from the Clapeyron equation. J. Chem. Thermodyn. 2004, 36, 1119–1127. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pressure (MPa) | Temperature (K) | Pore Size (nm) | |
|---|---|---|---|
| Handa and Stupin [14] | 2.6–5.19 | 263–276.2 | 7 |
| Uchida et al. [15] | 4.80–8.50 | 277.2–283.7 | 10, 30, 50 |
| Uchida et al. [16] | 1.65–9.86 | 259.7–283.7 | 4, 6, 10, 30, 100 |
| Smith et al. [17] | 1.60–4.26 | 244.0–276.0 | 2, 3, 5, 7.5 |
| Seo et al. [18] | 4.01–10.50 | 275.3–284.5 | 6, 15, 30 |
| Anderson et al. [19] | 3.69–14.07 | 271.8–287.5 | 9.2, 15.8, 30.6 |
| Kang et al. [20,21] | 2.38–9.94 | 269.7–284.3 | 6, 30, 100 |
| Sample | Pore Size (nm) | Pore Volume (mL/g) | Surface Area (m2/g) | Particle Size * (µm) |
|---|---|---|---|---|
| Vycor 8 | 8.5 | 0.40 | 162.42 | 74–125 |
| Vycor 17 | 17.1 | 0.40 | 118.62 | 37–74 |
| Vycor 35 | 34.8 | 0.72 | 59.23 | 74–125 |
| Vycor 40 | 40.3 | 0.98 | 57.52 | 74–125 |
| Vycor 95 | 95.4 | 0.79 | 25.96 | 74–125 |
| Vycor 196 | 195.7 | 0.93 | 11.11 | 37–74 |
| Hydrate Density | kg/m3 | 914 |
| Hydrate Heat of Fusion | kJ/mol | 53.2 |
| Hydration ratio | - | 6.0 |
| Interfacial Tension at Pers 5 MPa | mJ/m2 | 33.72 |
| a | - | 1.002612 |
| b | - | −0.031533 |
| a1 | 294.738839 |
| a2 | 0.134310 |
| a3 | −137.059913 |
| a4 | 3.752531 |
| a5 | −48.159863 |
| a6 | −0.186206 |
| a7 | −0.116622 |
| Pressure (MPa) | Enthalpy (J/g) |
|---|---|
| 5 | 494.6 ± 2.0 |
| 10 | 500.5 ± 3.9 |
| 40 | 503.6 ± 4.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bian, H.; Ai, L.; Hellgardt, K.; Maitland, G.C.; Heng, J.Y.Y. Phase Behaviour of Methane Hydrates in Confined Media. Crystals 2021, 11, 201. https://doi.org/10.3390/cryst11020201
Bian H, Ai L, Hellgardt K, Maitland GC, Heng JYY. Phase Behaviour of Methane Hydrates in Confined Media. Crystals. 2021; 11(2):201. https://doi.org/10.3390/cryst11020201
Chicago/Turabian StyleBian, Hao, Lu Ai, Klaus Hellgardt, Geoffrey C. Maitland, and Jerry Y. Y. Heng. 2021. "Phase Behaviour of Methane Hydrates in Confined Media" Crystals 11, no. 2: 201. https://doi.org/10.3390/cryst11020201
APA StyleBian, H., Ai, L., Hellgardt, K., Maitland, G. C., & Heng, J. Y. Y. (2021). Phase Behaviour of Methane Hydrates in Confined Media. Crystals, 11(2), 201. https://doi.org/10.3390/cryst11020201