Impact of Surface Roughness on Crystal Nucleation



Abstract
:
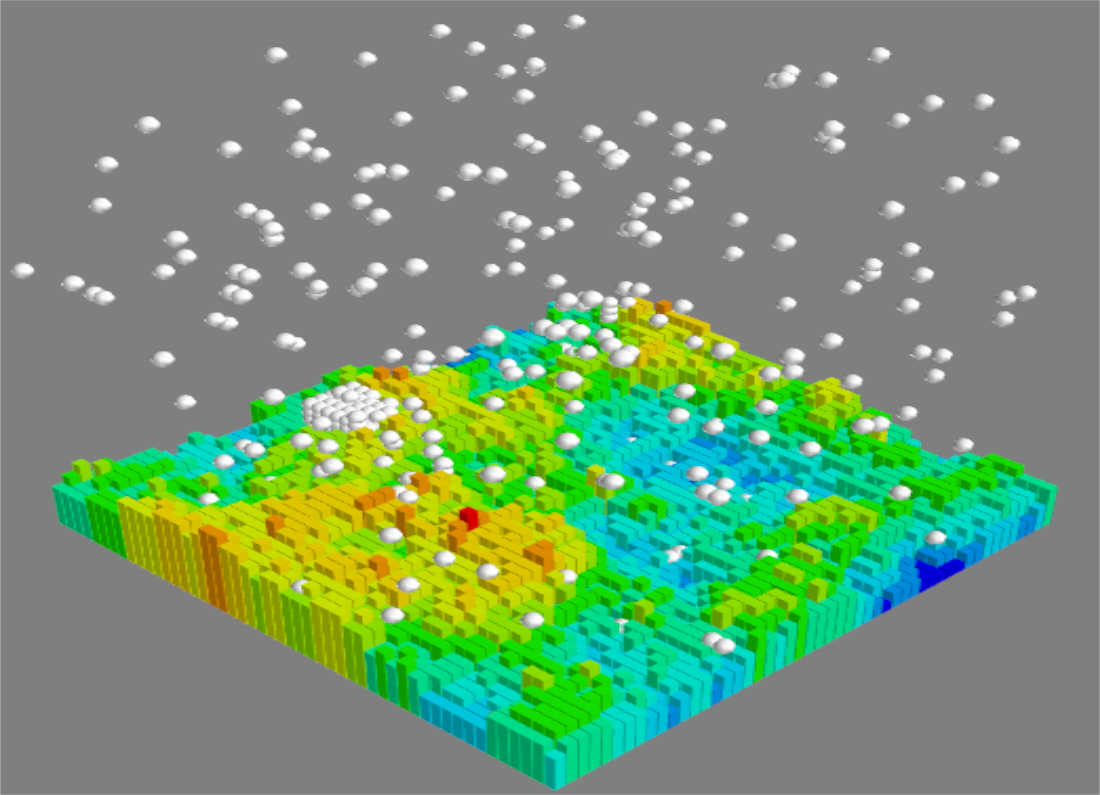{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
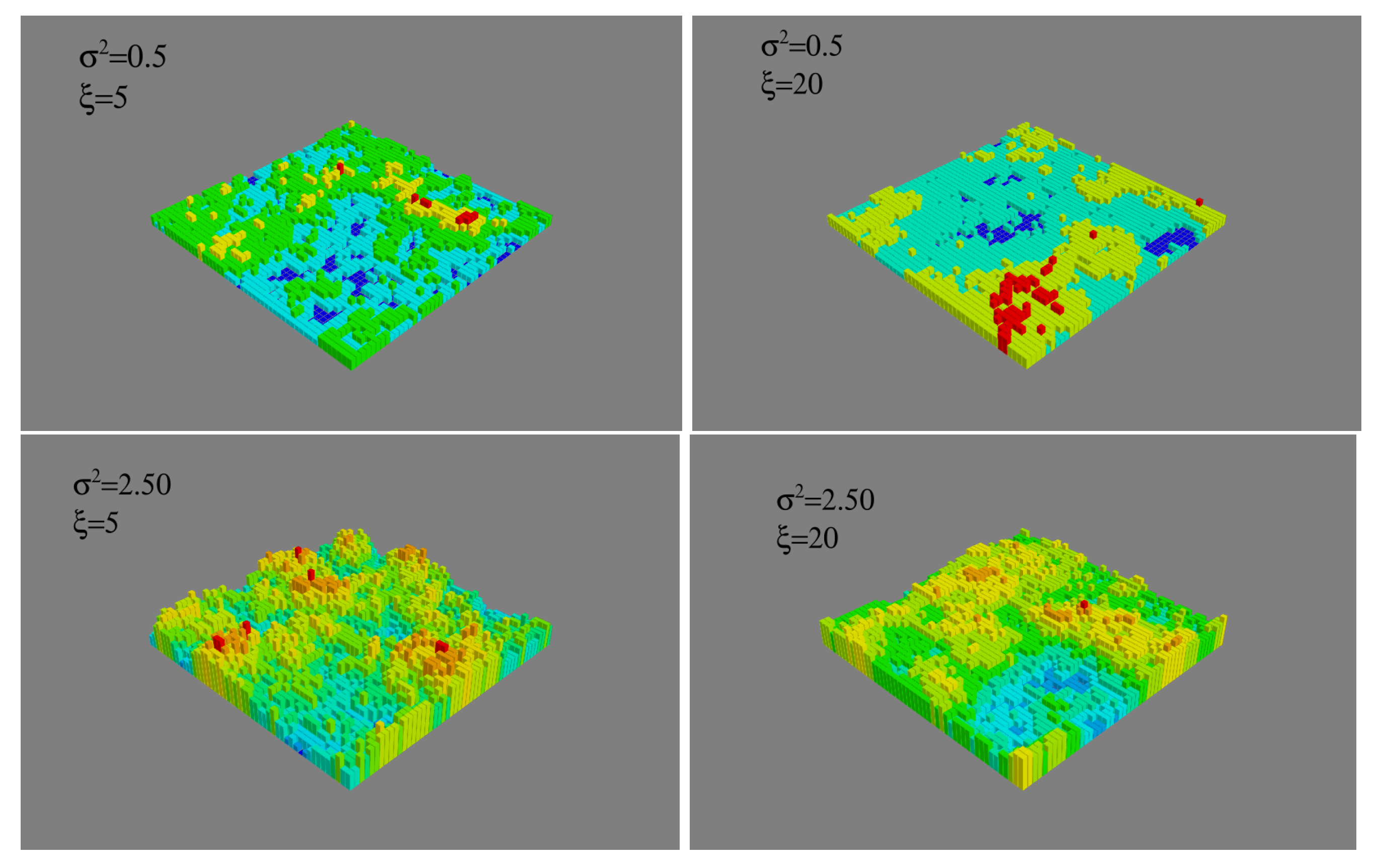
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
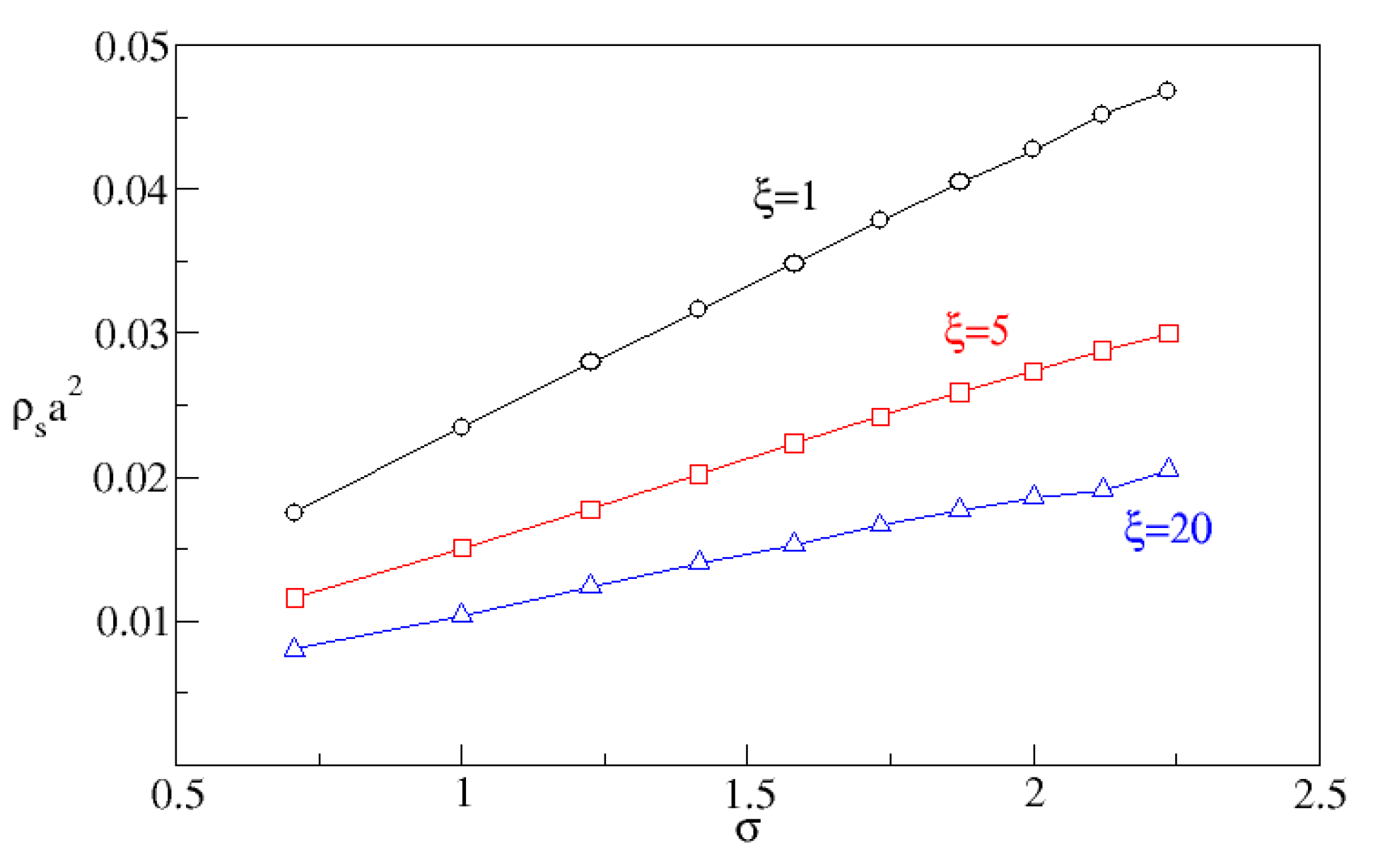
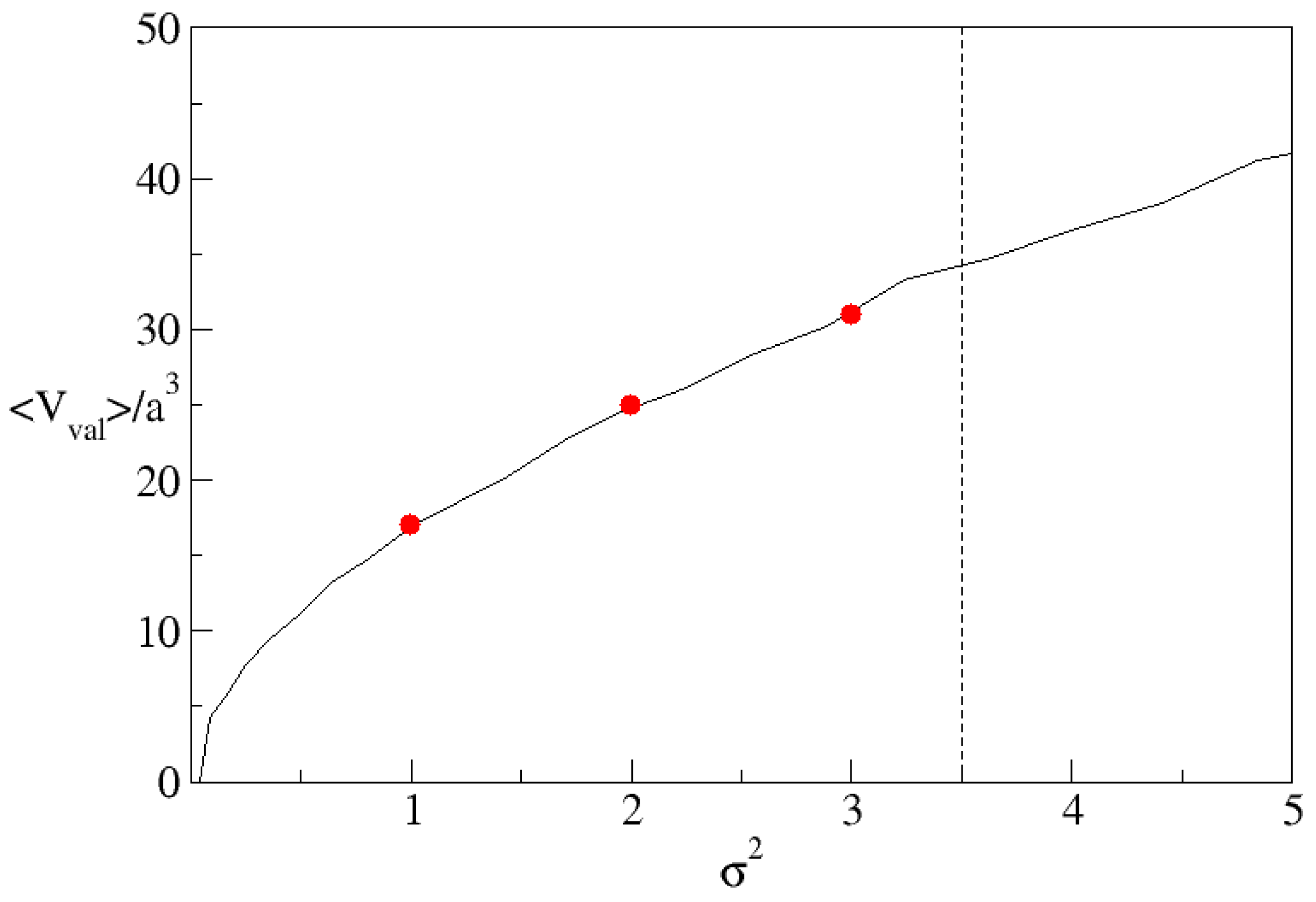{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Methods
2.1. Simulation Model
2.2. Model of Rough Surface
2.3. Rate Calculations
3. Results and Discussion
3.1. For Hard Walls, Roughness Is not Important
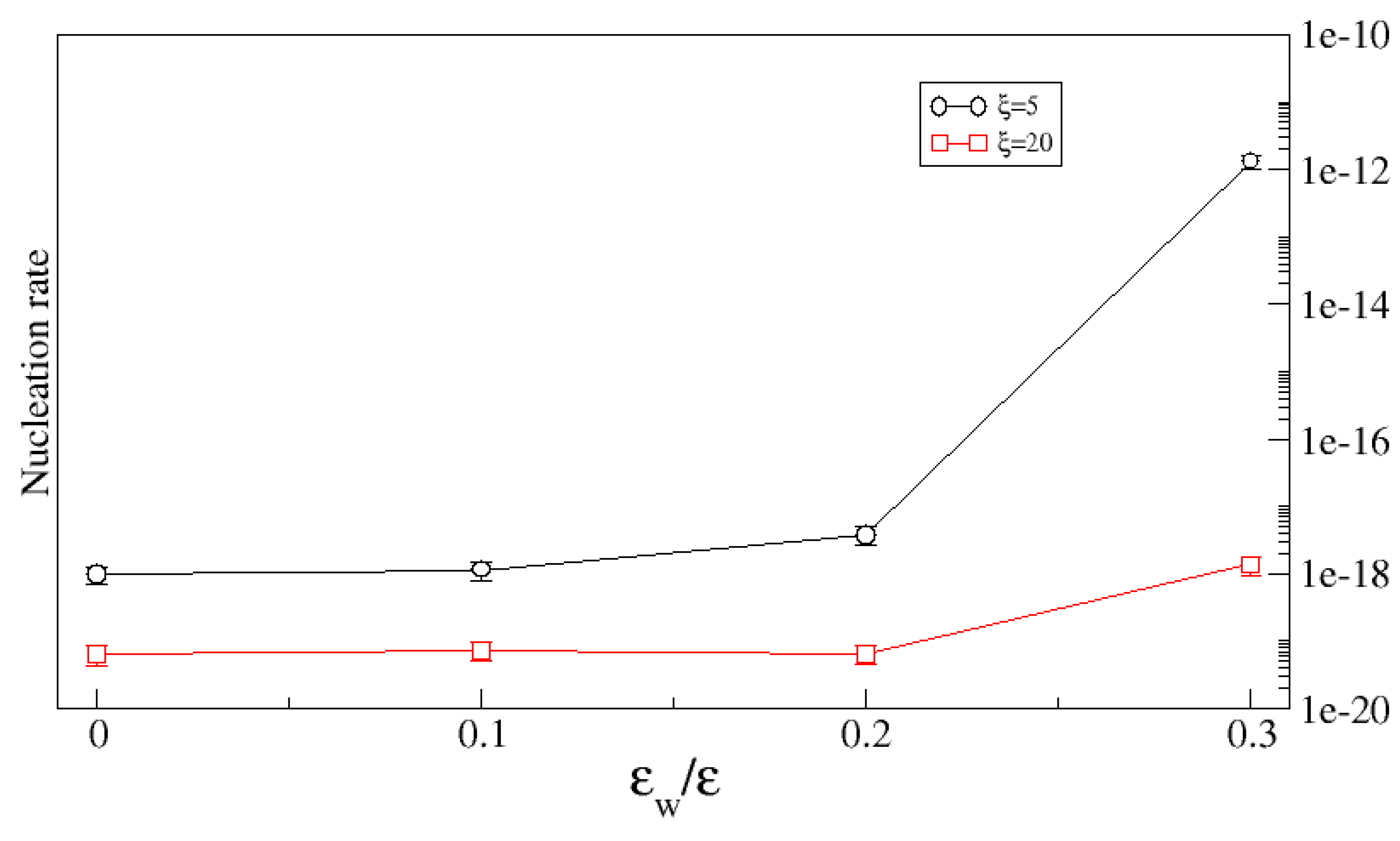
3.2. Roughness of Attractive Walls Increases Absorption
3.3. Correlation Plays an Important Role
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Model of Rough Surface
Appendix A.1. Generating the Surface

Appendix A.2. Characterizing Roughness

References
- Delmas, T.; Roberts, M.M.; Heng, J.Y.Y. Nucleation and Crystallization of Lysozyme: Role of Substrate Surface Chemistry and Topography. J. Adhes. Sci. Technol. 2011, 25, 357–366. [Google Scholar] [CrossRef]
- Artusio, F.; Pisano, R. Surface-induced crystallization of pharmaceuticals and biopharmaceuticals: A review. Int. J. Pharm. 2018, 547, 190–208. [Google Scholar] [CrossRef] [PubMed]
- Curcio, E.; Fontananova, E.; Profio, G.D.; Drioli, E. Influence of the Structural Properties of Poly(vinylidene fluoride) Membranes on the Heterogeneous Nucleation Rate of Protein CrystalsHeterogeneous Nucleation in and out of Pores. J. Phys. Chem. B 2006, 110, 12438–12445. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Sear, R.P. Heterogeneous Nucleation in and out of Pores. Phys. Rev. Lett. 2006, 97, 065701. [Google Scholar] [CrossRef] [Green Version]
- Sear, R.P. Heterogeneous and Homogeneous Nucleation Compared: Rapid Nucleation on Microscopic Impurities. J. Phys. Chem. B 2006, 110, 4985–4989. [Google Scholar] [CrossRef] [PubMed]
- Curcio, E.; Curcio, V.; Profio, G.D.; Fontananova, E.; Drioli, E. Heterogeneous Nucleation in and out of Pores. J. Phys. Chem. B 2010, 114, 13650–13655. [Google Scholar] [CrossRef] [PubMed]
- Chayen, N.E.; Saridakis, E.; Sear, R.P. Experiment and theory for heterogeneous nucleation of protein crystals in a porous medium. Proc. Natl. Acad. Sci. USA 2006, 103, 597–601. [Google Scholar] [CrossRef] [Green Version]
- Saridakis, E.; Chayen, N.E. Towards a ‘universal’ nucleant for protein crystallization. Trends Biotechnol. 2009, 27, 99–106. [Google Scholar] [CrossRef]
- Wang, K.; Zhou, C.; Hong, Y.; Zhang, X. A review of protein adsorption on bioceramics. Interface Focus 2012, 2, 259–277. [Google Scholar] [CrossRef]
- Pinholt, C.; Hartvig, R.A.; Medlicott, N.J.; Jorgensen, L. The importance of interfaces in protein drug delivery—Why is protein adsorption of interest in pharmaceutical formulations? Expert Opin. Drug Deliv. 2011, 8, 949–964. [Google Scholar] [CrossRef]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation; Academic Press, Inc.: Orlando, FL, USA, 2001. [Google Scholar]
- Allen, R.J.; Frenkel, D.; ten Wolde, P.R. Simulating rare events in equilibrium or nonequilibrium stochastic systems. J. Chem. Phys. 2006, 124, 024102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, R.J.; Valeriani, C.; ten Wolde, P.R. Forward flux sampling for rare event simulations. J. Phys. Condens. Matter 2009, 21, 463102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutsko, J.F. How crystals form: A theory of nucleation pathways. Sci. Adv. 2019, 5, eaav7399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutsko, J.F.; Lam, J. Long-wavelength density fluctuations as nucleation precursors. Phys. Rev. E 2020, 101, 052122. [Google Scholar] [CrossRef]
- Lord, M.S.; Foss, M.; Besenbacher, F. Influence of nanoscale surface topography on protein adsorption and cellular response. Nano Today 2010, 5, 66–78. [Google Scholar] [CrossRef]
- Chervanyov, A.I.; Heinrich, G. What really enhances the adsorption of polymers onto chemically nonuniform surfaces: Surface randomness or its heterogeneity? J. Chem. Phys. 2006, 125, 084703. [Google Scholar] [CrossRef]
- Wu, J.J. Simulation of non-Gaussian surfaces with FFT. Tribol. Int. 2004, 37, 339–346. [Google Scholar] [CrossRef]
- Wenzel, R. Crystallization seed favour crystallization only during initial growth. Ind. Eng. Chem. 1936, 28, 988–994. [Google Scholar] [CrossRef]
- Quéré, D. Wetting and roughness. Annu. Rev. Mater. Res. 2008, 38, 71–99. [Google Scholar] [CrossRef]
- de Gennes, P. Wetting: Statics and dynamics. Rev. Mod. Phys. 1985, 57, 827–863. [Google Scholar] [CrossRef]
- Israelachvili, J. Intermolecular and Surface Forces, 3rd ed.; Elsevier Academic Press Inc.: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Chow, T. Wetting of rough surfaces. J. Phys. Condens. Matter 1998, 10, L445. [Google Scholar] [CrossRef]
- Allahyarov, E.; Sandomirski, K.; Egelhaaf, S.U.; Lowen, H. Crystallization seed favour crystallization only during initial growth. Nat. Commun. 2015, 6, 7110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, P. Some aspects of surface roughness measurement. Wear 1973, 26, 165–174. [Google Scholar] [CrossRef]
- Bracewell, R. The Fourier Transform and Its Applications; McGraw-Hill Series in Electrical and Computer Engineering. Circuits and Systems; McGraw-Hill: New York, NY, USA, 2000. [Google Scholar]
- Greenwood, J. A unified theory of surface roughness. Proc. R. Soc. Lond. A 1984, 393, 133–157. [Google Scholar]
- Herminghaus, S. Wetting, spreading, and adsorption on randomly rough surfaces. Eur. Phys. J. E 2012, 35, 43. [Google Scholar] [CrossRef] [Green Version]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grosfils, P.; Lutsko, J.F. Impact of Surface Roughness on Crystal Nucleation. Crystals 2021, 11, 4. https://doi.org/10.3390/cryst11010004
Grosfils P, Lutsko JF. Impact of Surface Roughness on Crystal Nucleation. Crystals. 2021; 11(1):4. https://doi.org/10.3390/cryst11010004
Chicago/Turabian StyleGrosfils, Patrick, and James F. Lutsko. 2021. "Impact of Surface Roughness on Crystal Nucleation" Crystals 11, no. 1: 4. https://doi.org/10.3390/cryst11010004
APA StyleGrosfils, P., & Lutsko, J. F. (2021). Impact of Surface Roughness on Crystal Nucleation. Crystals, 11(1), 4. https://doi.org/10.3390/cryst11010004