
A New L-Proline Amide Hydrolase with Potential Application within the Amidase Process

 ,
, 
 and
and 
Abstract
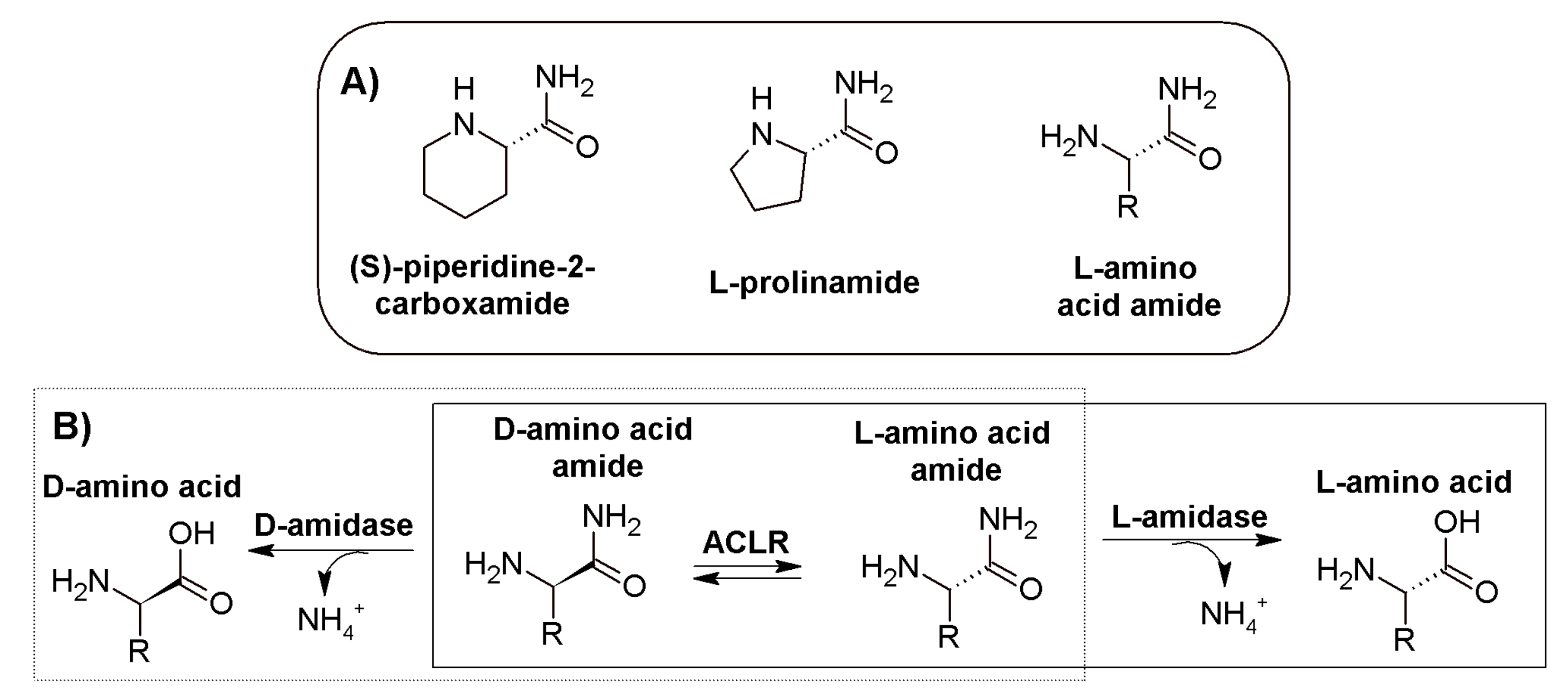
:1. Introduction
2. Materials and Methods
2.1. Cloning, Overexpression, and Purification of PsyPAH
2.2. Activity Measurement
2.3. Size Exclusion Chromatography (SEC-FPLC)
2.4. Dynamic Light Scattering
2.5. Thermal Shift Assays
2.6. Crystallization
2.7. Data Collection and Refinement
2.8. Sequence and Structure Analysis
3. Results and Discussion
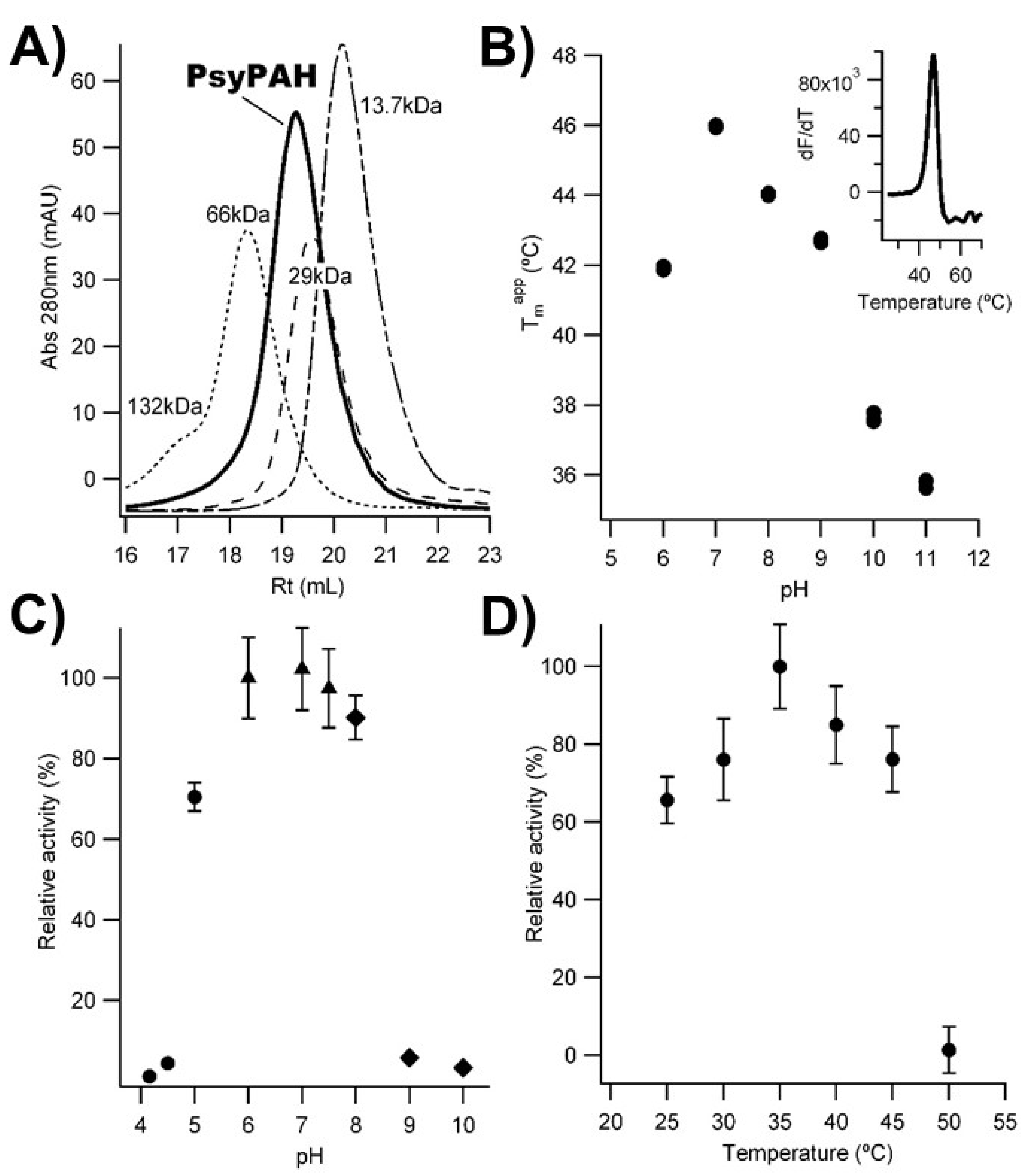
3.1. PsyPAH Characterization
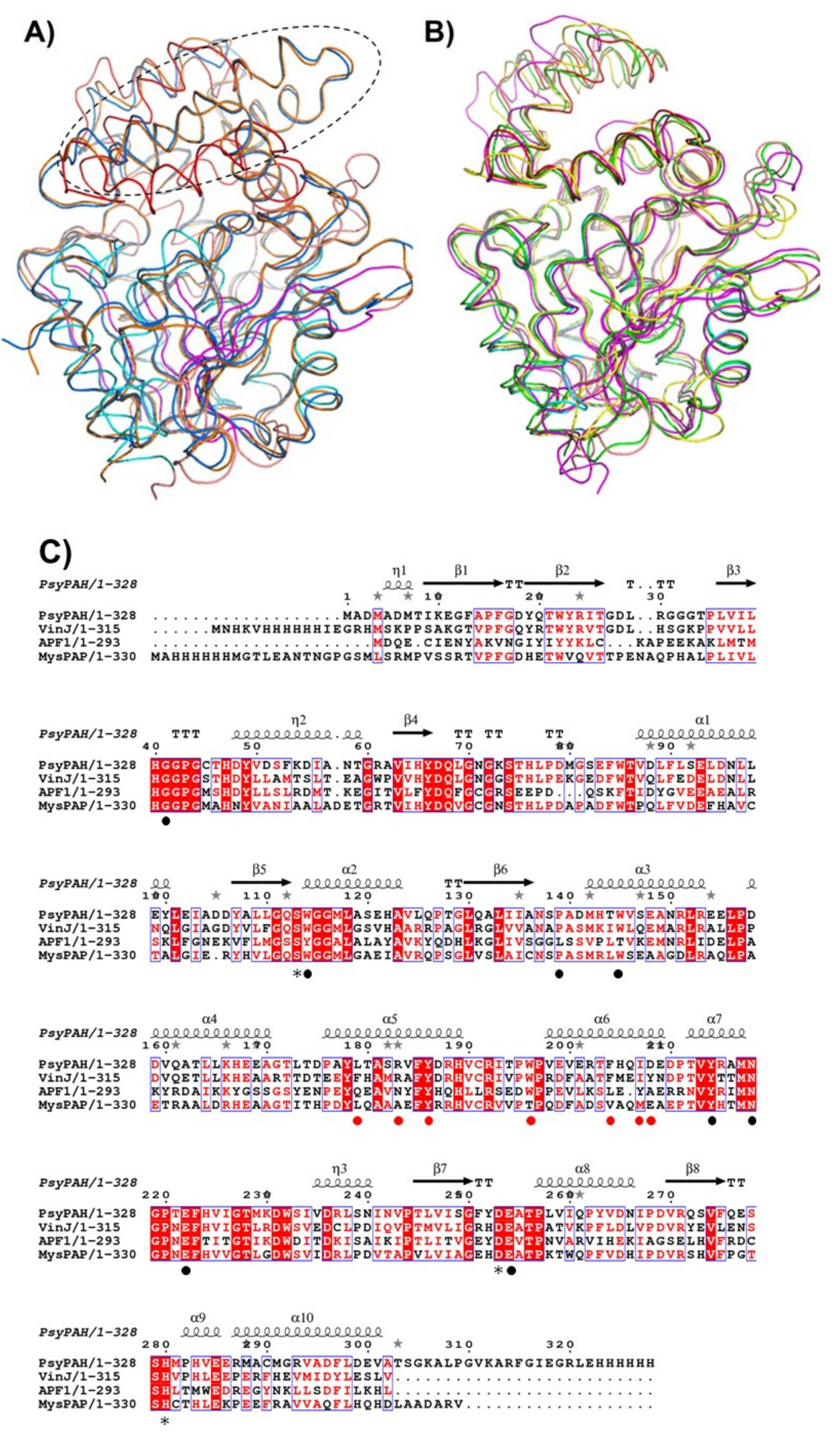
3.2. PsyPAH Sequence Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acronym | Protein Family | Source | Sequence | Reference |
|---|---|---|---|---|
| PsyPAH | Serine peptidase S33 | Pseudomonas psyringae | A0A0Q0CYJ4 | This work |
| LaaAPa | Serine peptidase S33 | Pseudomonas azotoformans | BAD15092.1 | [1] |
| LaaAOa | Acetamidase/formamidase | Ochrobactrum anthropi | AAV87210 | [7] |
| LaaAEc | Acetamidase/formamidase | Enterobacter cloacae | AAR56843 | [43] |
| LaaATs | Acetamidase/formamidase | Thermus sp. | BAL49703.1 | [44] |
| XfAmid | Acetamidase/formamidase | Xantobacter flavus | BAE02548.1 | [45,46] |
| LaaABd | Leucine aminopeptidase (M17) | Brevundimonas Diminuta | BAE91931 | [2] |
| ppLAP | Leucine aminopeptidase (M17) | Pseudomonas putida | CAA09054.1 | [9] |
| apLAP | Leucyl-aminopeptidase (M28) | Aeromonas proteolytica | Q01693 | [38] |
| LaaAMn | - | Mycobacterium neoaurum | n.a. | [10] |
| - | - * | Hog kidney | - * | [5] |
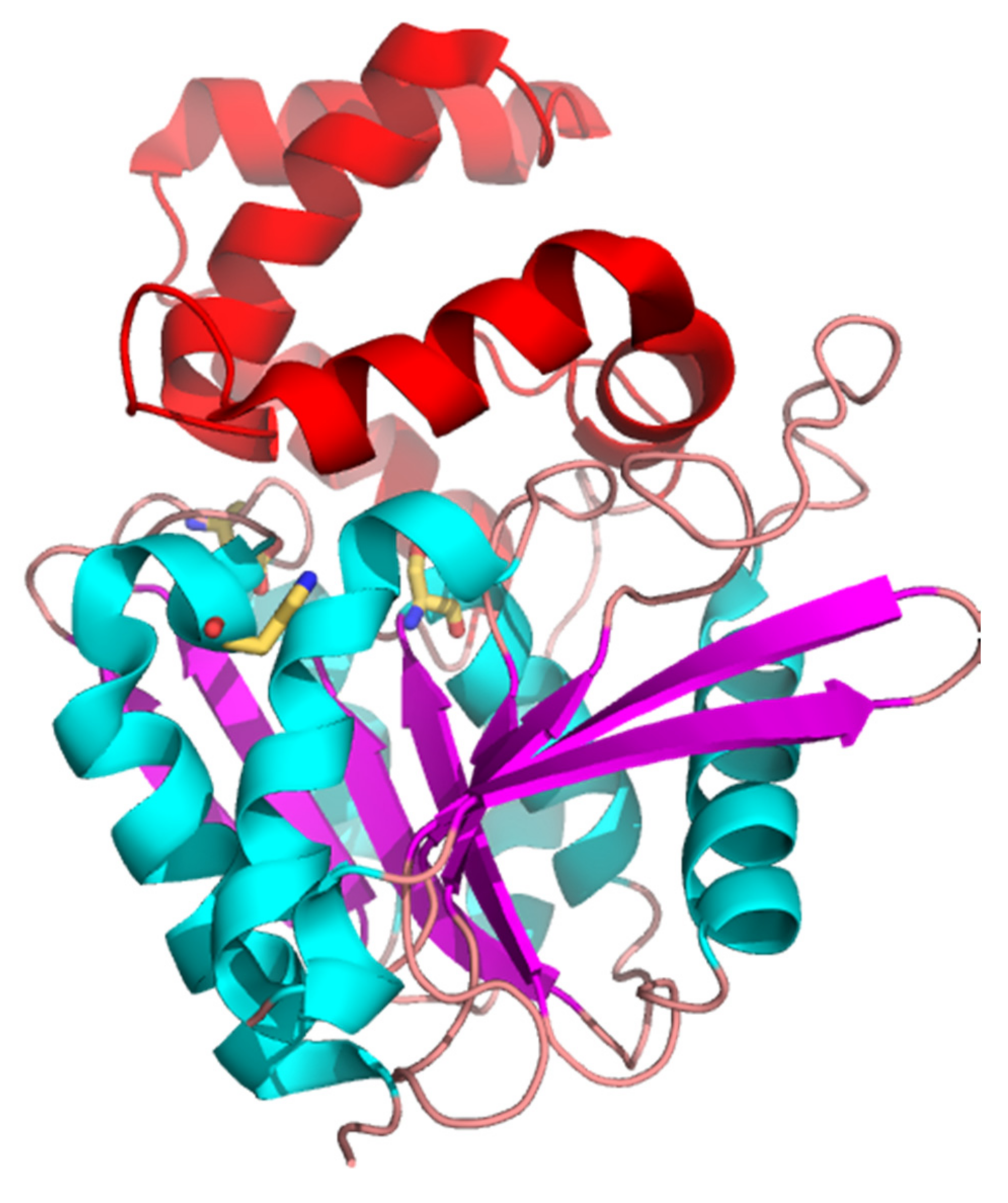
3.3. Overall Structure of PsyPAH
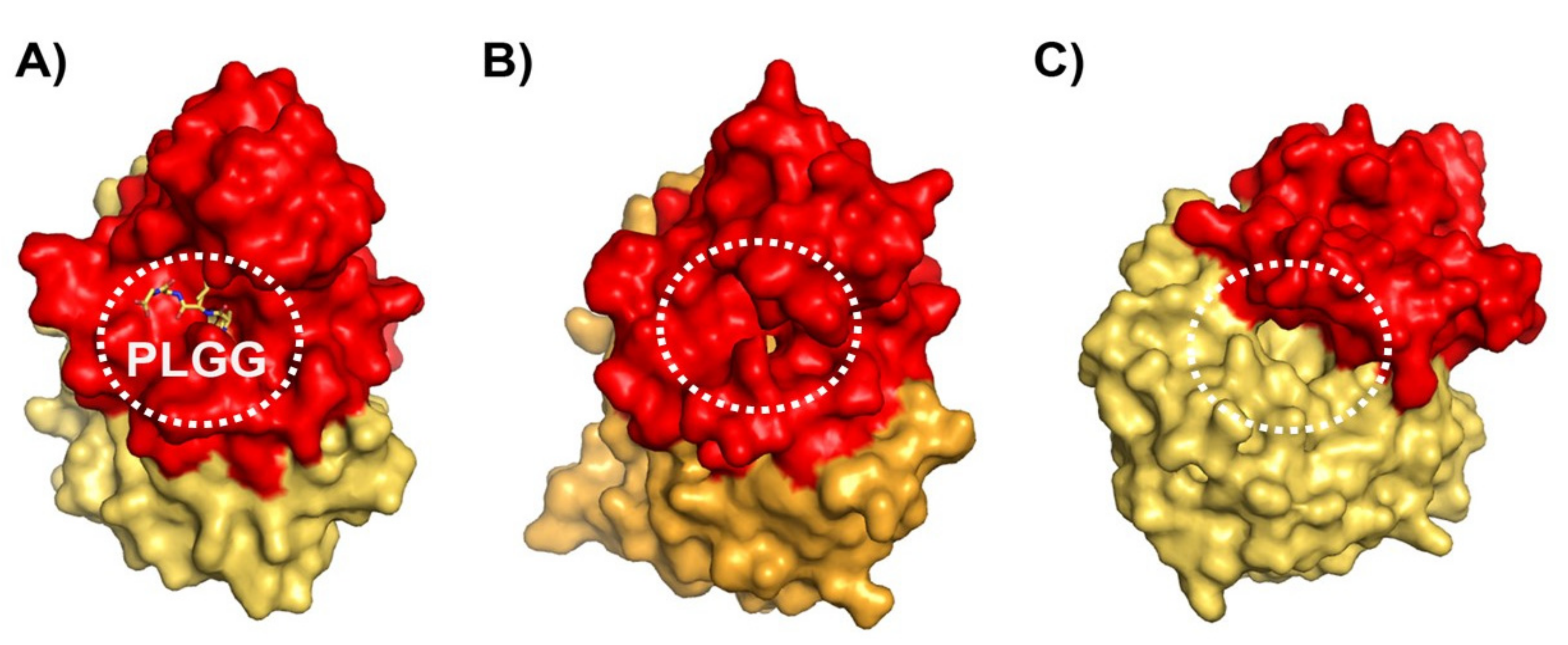
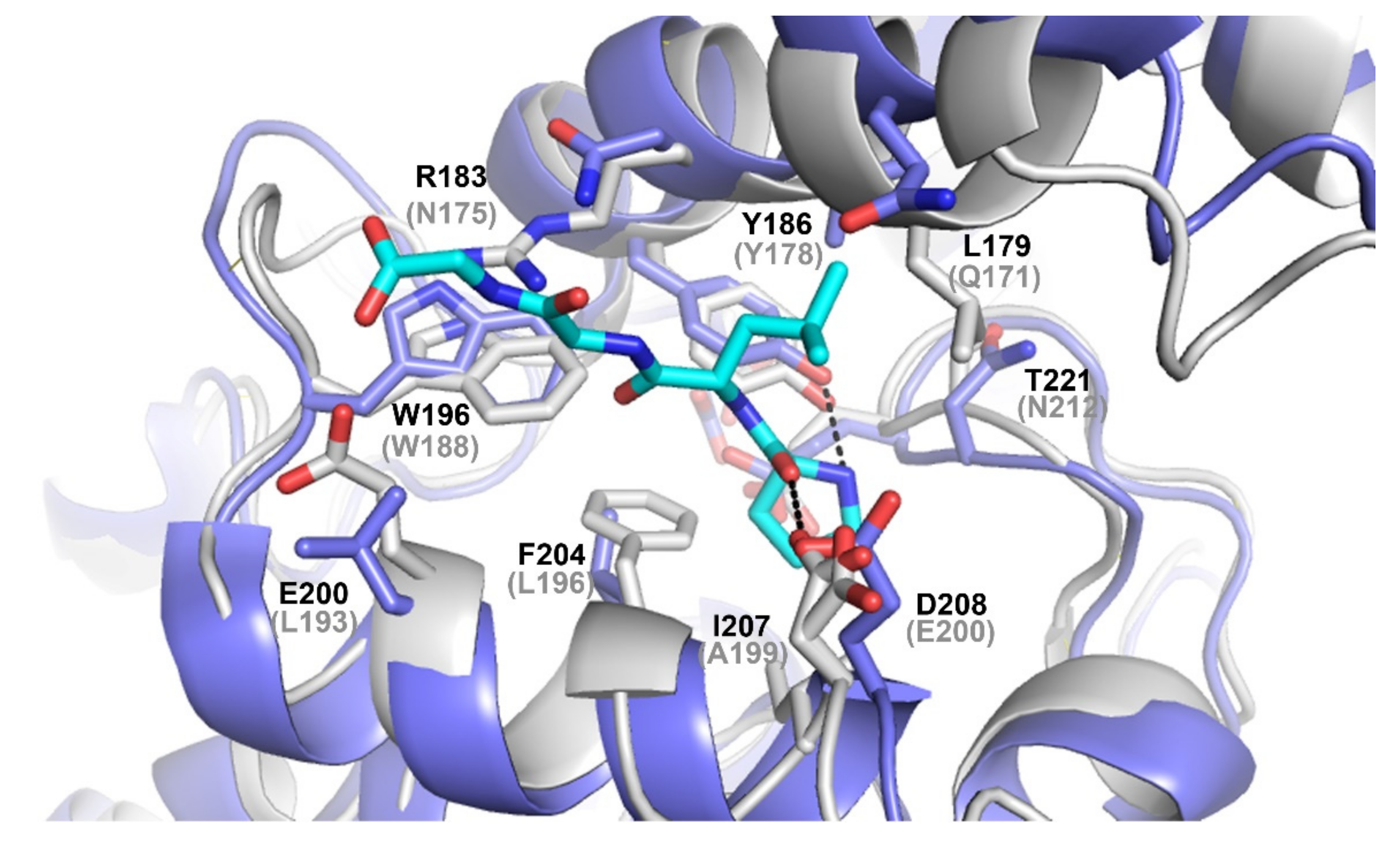
3.4. Differences on the Substrate Binding Groove (SBG) Seem to Account for the Substrate Scope of PsyPAH
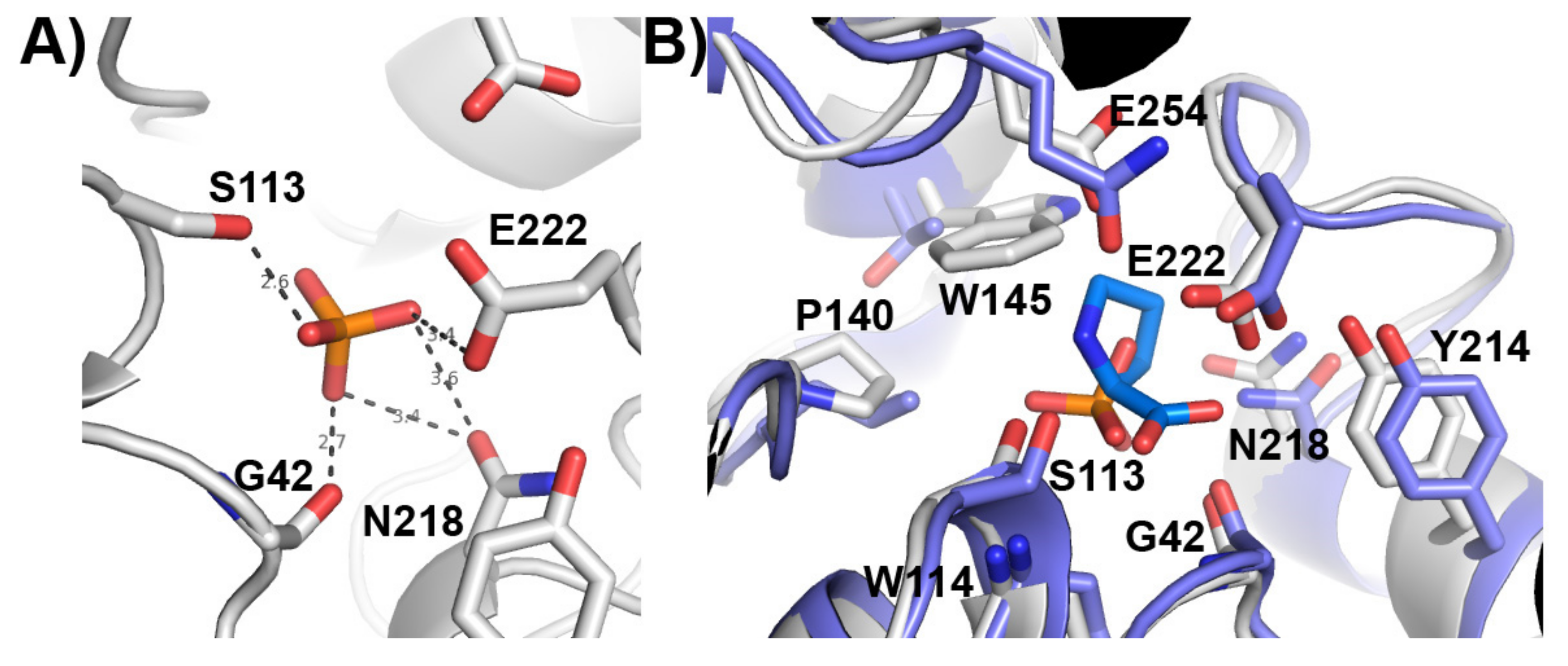
3.5. Putative Catalytic Centre of PsyPAH
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Komeda, H.; Harada, H.; Washika, S.; Sakamoto, T.; Ueda, M.; Asano, Y. S-stereoselective piperazine-2-tert-butylcarboxamide hydrolase from Pseudomonas azotoformans IAM 1603 is a novel L-amino acid amidase. Eur. J. Biochem. 2004, 271, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Komeda, H.; Hariyama, N.; Asano, Y. L-Stereoselective amino acid amidase with broad substrate specificity from Brevundimonas diminuta: Characterization of a new member of the leucine aminopeptidase family. Appl. Microbiol. Biotechnol. 2006, 70, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Komeda, H.; Asano, Y. New enzymatic method of chiral amino acid synthesis by dynamic kinetic resolution of amino acid amides: Use of stereoselective amino acid amidases in the presence of alpha-amino-epsilon-caprolactam racemase. Appl. Environ. Microbiol. 2007, 73, 5370–5373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Rodríguez, S.; Torres, J.M.; Sánchez, P.; Ortega, E. Overview on Multienzymatic Cascades for the Production of Non-canonical α-Amino Acids. Front. Bioeng. Biotechnol. 2020, 8, 887. [Google Scholar] [CrossRef]
- Hamer, D.; Greenstein, J.P. An enzymatic resolution of proline. J. Biol. Chem. 1951, 193, 81–89. [Google Scholar] [CrossRef]
- Work, E.; Birnbaum, S.; Winitz, M.; Greenstein, J. Separation of the Three Isomeric Components of Synthetic α, ε-Diaminopimelic Acid1. J. Am. Chem. Soc. 1955, 77, 1916–1918. [Google Scholar] [CrossRef]
- Sonke, T.; Ernste, S.; Tandler, R.F.; Kaptein, B.; Peeters, W.P.; van Assema, F.B.; Wubbolts, M.G.; Schoemaker, H.E. L-selective amidase with extremely broad substrate specificity from Ochrobactrum anthropi NCIMB 40321. Appl. Environ. Microbiol. 2005, 71, 7961–7973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Tweel, W.J.J.; van Dooren, T.J.G.M.; de Jonge, P.H.; Kaptein, B.; Duchateau, A.L.L.; Kamphuis, K. Ochrobactrum anthropi NCIMB 40321: A new biocatalyst with broad-spectrum l-specific amidase activity. Appl. Microbiol. Biotechnol. 1993, 39, 296–300. [Google Scholar] [CrossRef]
- Hermes, H.F.; Sonke, T.; Peters, P.J.; van Balken, J.A.; Kamphuis, J.; Dijkhuizen, L.; Meijer, E.M. Purification and Characterization of an l-Aminopeptidase from Pseudomonas putida ATCC 12633. Appl. Environ. Microbiol. 1993, 59, 4330–4334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermes, H.F.; Tandler, R.F.; Sonke, T.; Dijkhuizen, L.; Meijer, E.M. Purification and Characterization of an l-Amino Amidase from Mycobacterium neoaurum ATCC 25795. Appl. Environ. Microbiol. 1994, 60, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Kamphuis, J.; Meijer, E.M.; Boesten, W.H.J.; Broxterman, Q.B.; Kaptein, B.; Hermes, H.F.M.; Schoemaker, H.E. Production of natural and synthetic L- and D-amino acids by aminopeptidases and amino amidases. In Biocatalytic Production of Amino Acids and Derivatives; Rozzell, J.D., Wagner, F., Eds.; Wiley: New York, NY, USA, 1992; pp. 178–206. [Google Scholar]
- Maestracci, M.; Bui, K.; Thiéry, A.; Arnaud, A.; Galzy, P. The amidases from a Brevibacterium strain: Study and applications. Adv. Biochem. Eng. Biotechnol. 1988, 36, 67–115. [Google Scholar] [CrossRef] [PubMed]
- Youshko, M.I.; Luuk, M.; Sheldon, R.A.; Švedas, V.K. Application of aminoacylase I to the enantioselective resolution of α-amino acid esters and amides. Tetrahedron Asymmetry 2004, 15, 1933–1936. [Google Scholar] [CrossRef]
- Stelkes-Ritter, U.; Beckers, G.; Bommarius, A.; Drauz, K.; Günther, K.; Kottenhahn, M.; Schwarm, M.; Kula, M.R. Kinetics of Peptide Amidase and its Application for the Resolution of Racemates. Biocatal. Biotransform. 2009, 15, 205–219. [Google Scholar] [CrossRef]
- Mahon, C.S.; O’Donoghue, A.J.; Goetz, D.H.; Murray, P.G.; Craik, C.S.; Tuohy, M.G. Characterization of a multimeric, eukaryotic prolyl aminopeptidase: An inducible and highly specific intracellular peptidase from the non-pathogenic fungus Talaromyces emersonii. Microbiology 2009, 155, 3673–3682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, T.; Ito, K.; Tozaka, T.; Hatakeyama, S.; Tanaka, N.; Nakamura, K.T.; Yoshimoto, T. Novel inhibitor for prolyl aminopeptidase from Serratia marcescens and studies on the mechanism of substrate recognition of the enzyme using the inhibitor. Arch. Biochem. Biophys. 2003, 416, 147–154. [Google Scholar] [CrossRef]
- Tamura, T.; Tamura, N.; Lottspeich, F.; Baumeister, W. Tricorn protease (TRI) interacting factor 1 from Thermoplasma acidophilum is a proline iminopeptidase. FEBS Lett. 1996, 398, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, Y.; Miyanaga, A.; Kudo, F.; Eguchi, T. The crystal structure of the amidohydrolase VinJ shows a unique hydrophobic tunnel for its interaction with polyketide substrates. FEBS Lett. 2014, 588, 995–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezawa, T.; Jung, S.; Kawashima, Y.; Noguchi, T.; Imai, N. Convenient green preparation of dipeptides using unprotected α-amino acids. Tetrahedron Asymmetry 2017, 28, 75–83. [Google Scholar] [CrossRef]
- Riley, J.P. The spectrophotometric determination of ammonia in natural waters with particular reference to sea-water. Anal. Chim. Acta 1953, 9, 575–589. [Google Scholar] [CrossRef]
- Erlanger, B.F.; Kokowsky, N.; Cohen, W. The preparation and properties of two new chromogenic substrates of trypsin. Arch. Biochem. Biophys. 1961, 95, 271–278. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Cryst. D Biol. Cryst. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Cryst. D Biol. Cryst. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunkóczi, G.; Echols, N.; McCoy, A.J.; Oeffner, R.D.; Adams, P.D.; Read, R.J. Phaser.MRage: Automated molecular replacement. Acta Cryst. D Biol. Cryst. 2013, 69, 2276–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Cryst. D Biol. Cryst. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst. D Biol. Cryst. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. D Biol. Cryst. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Painter, J.; Merritt, E.A. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Cryst. D Biol. Cryst. 2006, 62, 439–450. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Cryst. D Biol. Cryst. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, L. DALI and the persistence of protein shape. Protein Sci. 2020, 29, 128–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Pymol Molecular Graphics System, Schrödinger, LLC. Available online: https://www.scirp.org/(S(vtj3fa45qm1ean45vvffcz55))/reference/ReferencesPapers.aspx?ReferenceID=1958992 (accessed on 25 November 2021).
- Stetefeld, J.; McKenna, S.A.; Patel, T.R. Dynamic light scattering: A practical guide and applications in biomedical sciences. Biophys. Rev. 2016, 8, 409–427. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, C.; Zhang, Z.; Zheng, R.; Zheng, Y. Amidase as a versatile tool in amide-bond cleavage: From molecular features to biotechnological applications. Biotechnol. Adv. 2020, 43, 107574. [Google Scholar] [CrossRef]
- Wagner, F.W.; Wilkes, S.H.; Prescott, J.M. Specificity of Aeromonas aminopeptidase toward amino acid amides and dipeptides. J. Biol. Chem. 1972, 247, 1208–1210. [Google Scholar] [CrossRef]
- Nandan, A.; Nampoothiri, K.M. Molecular advances in microbial aminopeptidases. Bioresour. Technol. 2017, 245, 1757–1765. [Google Scholar] [CrossRef]
- Nandan, A.; Nampoothiri, K.M. Therapeutic and biotechnological applications of substrate specific microbial aminopeptidases. Appl. Microbiol. Biotechnol. 2020, 104, 5243–5257. [Google Scholar] [CrossRef]
- Inoue, A.; Komeda, H.; Asano, Y. Asymmetric Synthesis of L-α-Methylcysteine with the Amidase from Xanthobacter flavus NR303. Adv. Synth. Catal. 2005, 347, 1132–1138. [Google Scholar] [CrossRef]
- Matsushima, M.; Takahashi, T.; Ichinose, M.; Miki, K.; Kurokawa, K.; Takahashi, K. Structural and immunological evidence for the identity of prolyl aminopeptidase with leucyl aminopeptidase. Biochem. Biophys. Res. Commun. 1991, 178, 1459–1464. [Google Scholar] [CrossRef]
- Nakamura, T.; Yu, F. Amidase Gene. U.S. Patent 6617139, 9 September 2003. [Google Scholar]
- Katoh, O.; Akiyama, T.; Nakamura, T. Novel Amide Hydrolase Gene. EP Patent Application EP1428876A1, 16 June 2004. [Google Scholar]
- Asano, Y. L-Amino Acid Amide Asymmetric Hydrolase and Dna Encoding the Same. EP1770166A4, 19 December 2007. [Google Scholar]
- Matsushima, M.; Takahashi, T.; Ichinose, M.; Miki, K.; Kurokawa, K.; Takahashi, K. Prolyl aminopeptidases from pig intestinal mucosa and human liver: Purification, characterization and possible identity with leucyl aminopeptidase. Biomed. Res. 1991, 12, 323–333. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Holmquist, M. Alpha/Beta-hydrolase fold enzymes: Structures, functions and mechanisms. Curr. Protein Pept. Sci. 2000, 1, 209–235. [Google Scholar] [CrossRef] [PubMed]
- Marchot, P.; Chatonnet, A. Enzymatic activity and protein interactions in alpha/beta hydrolase fold proteins: Moonlighting versus promiscuity. Protein Pept. Lett. 2012, 19, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Goettig, P.; Groll, M.; Kim, J.S.; Huber, R.; Brandstetter, H. Structures of the tricorn-interacting aminopeptidase F1 with different ligands explain its catalytic mechanism. EMBO J. 2002, 21, 5343–5352. [Google Scholar] [CrossRef] [Green Version]
- Lenfant, N.; Hotelier, T.; Velluet, E.; Bourne, Y.; Marchot, P.; Chatonnet, A. ESTHER, the database of the α/β-hydrolase fold superfamily of proteins: Tools to explore diversity of functions. Nucleic Acids Res. 2013, 41, D423–D429. [Google Scholar] [CrossRef] [Green Version]
- Medrano, F.J.; Alonso, J.; García, J.L.; Romero, A.; Bode, W.; Gomis-Rüth, F.X. Structure of proline iminopeptidase from Xanthomonas campestris pv. citri: A prototype for the prolyl oligopeptidase family. EMBO J. 1998, 17, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, T.; Kabashima, T.; Uchikawa, K.; Inoue, T.; Tanaka, N.; Nakamura, K.T.; Tsuru, M.; Ito, K. Crystal structure of prolyl aminopeptidase from Serratia marcescens. J. Biochem. 1999, 126, 559–565. [Google Scholar] [CrossRef]
- Okai, M.; Miyauchi, Y.; Ebihara, A.; Lee, W.C.; Nagata, K.; Tanokura, M. Crystal structure of the proline iminopeptidase-related protein TTHA1809 from Thermus thermophilus HB8. Proteins 2008, 70, 1646–1649. [Google Scholar] [CrossRef] [PubMed]
- Goettig, P.; Brandstetter, H.; Groll, M.; Göhring, W.; Konarev, P.V.; Svergun, D.I.; Huber, R.; Kim, J.S. X-ray snapshots of peptide processing in mutants of tricorn-interacting factor F1 from Thermoplasma acidophilum. J. Biol. Chem. 2005, 280, 33387–33396. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Inoue, T.; Kabashima, T.; Kanada, N.; Huang, H.S.; Ma, X.; Azmi, N.; Azab, E.; Yoshimoto, T. Substrate recognition mechanism of prolyl aminopeptidase from Serratia marcescens. J. Biochem. 2000, 128, 673–678. [Google Scholar] [CrossRef] [PubMed]







| Data Collection | |
|---|---|
| Source | ESRF ID30B |
| Space group | P 21 21 21 |
| Cell dimensions | |
| a, b, c (Å) | 49.428, 65.033, 85.016 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Resolution (Å) | 42.73–1.95 (2.02–1.95) |
| Unique reflections | 20,565 (2016) |
| I/σI | 7.2 (1.8) |
| Completeness (%) | 99.70 (99.75) |
| Redundancy | 5.1 (5.1) |
| Rmerge | 15.7 (91.9) |
| CC1/2 | 0.991 (0.636) |
| Refinement | |
| Rwork/Rfree | 15.68/22.25 |
| No. atoms | 2824 |
| Protein | 2571 |
| Ligand/ion | 5 |
| Water | 248 |
| B-factors | 21.97 |
| Protein | 20.95 |
| Ligand/ion | 30.16 |
| Water | 32.35 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 0.85 |
| PDB ID | 7A6G |
| Substrate | Km (mM) | kcat (s−1) | kcat/Km (s−1·mM−1) |
|---|---|---|---|
| pN-Pro | ND * | 445.38 ± 19.17 | ND * |
| pN-Leu | 1.14 ± 0.21 | 71.25 ± 3.34 | 62.5 ± 3.37 |
| L-Pro amide | 9.48 ± 1.6 | 14.03 ± 1.26 | 1.48 ± 0.13 |
| L-Leu amide | ND ** | ND ** | 0.03 ± 0.00 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Rodríguez, S.; Contreras-Montoya, R.; Torres, J.M.; de Cienfuegos, L.Á.; Gavira, J.A. A New L-Proline Amide Hydrolase with Potential Application within the Amidase Process. Crystals 2022, 12, 18. https://doi.org/10.3390/cryst12010018
Martinez-Rodríguez S, Contreras-Montoya R, Torres JM, de Cienfuegos LÁ, Gavira JA. A New L-Proline Amide Hydrolase with Potential Application within the Amidase Process. Crystals. 2022; 12(1):18. https://doi.org/10.3390/cryst12010018
Chicago/Turabian StyleMartinez-Rodríguez, Sergio, Rafael Contreras-Montoya, Jesús M. Torres, Luis Álvarez de Cienfuegos, and Jose Antonio Gavira. 2022. "A New L-Proline Amide Hydrolase with Potential Application within the Amidase Process" Crystals 12, no. 1: 18. https://doi.org/10.3390/cryst12010018
APA StyleMartinez-Rodríguez, S., Contreras-Montoya, R., Torres, J. M., de Cienfuegos, L. Á., & Gavira, J. A. (2022). A New L-Proline Amide Hydrolase with Potential Application within the Amidase Process. Crystals, 12(1), 18. https://doi.org/10.3390/cryst12010018