
4-Methyl/Phenyl-1,2,5,6-tetraazafluoranthen-3(2H)-ones Synthesis: Mechanistic Pathway Study and Single-Crystal X-ray Analysis of the Intermediates


 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
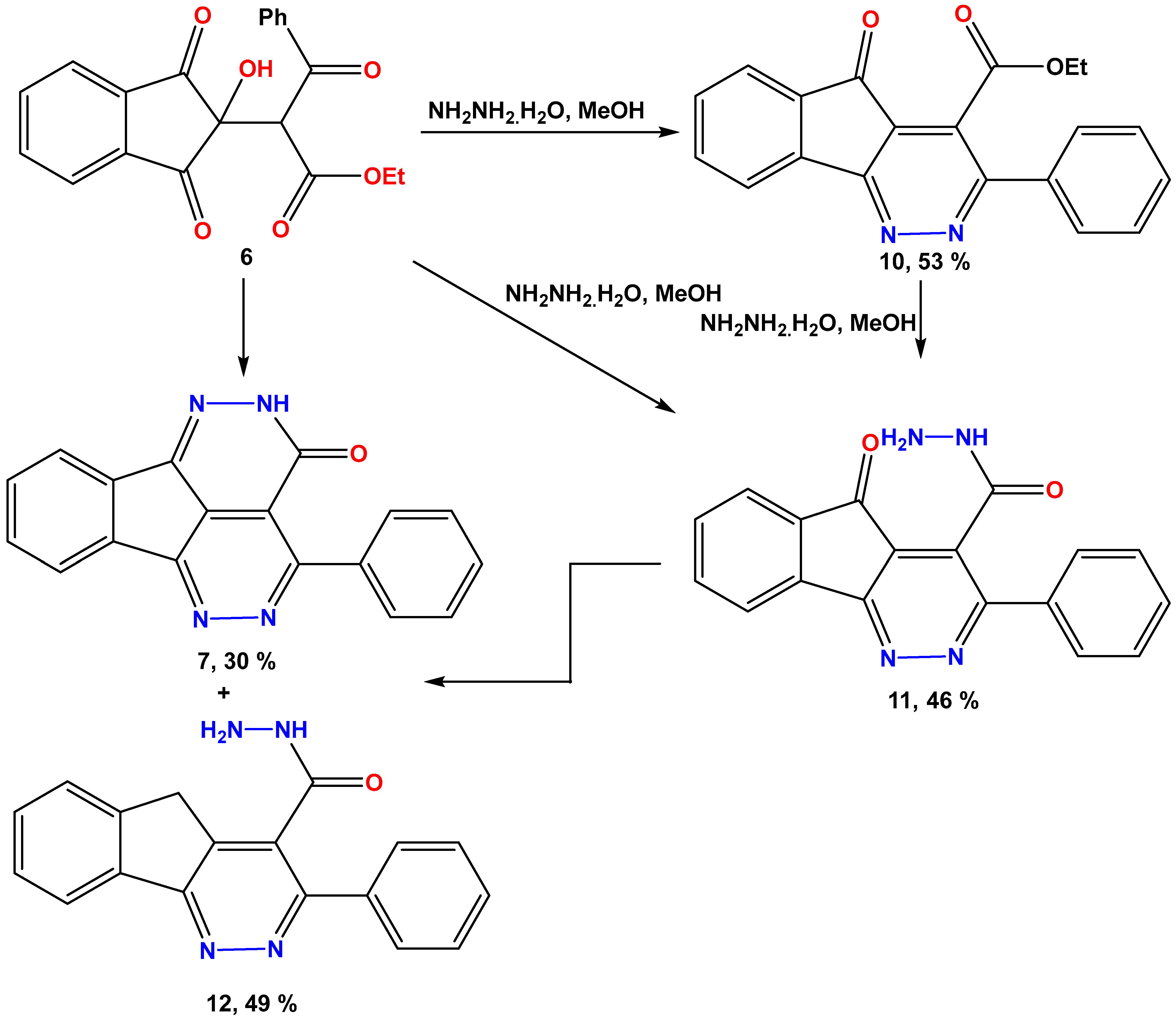
2.1. General
2.2. Procedures
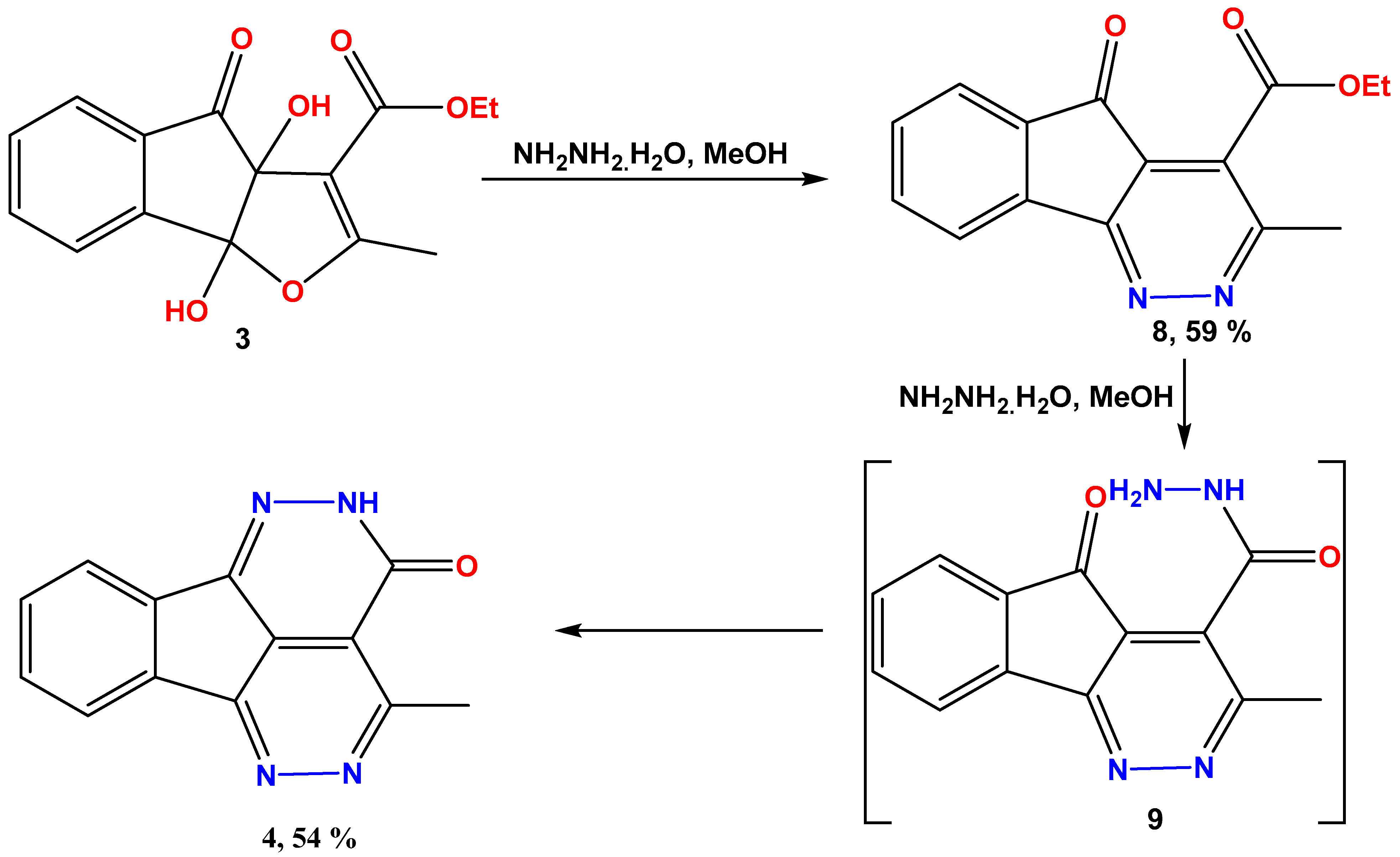
2.3. Synthesis of the Intermediates Indeno[1,2-c]Pyridazine-4-Carboxylate 8 and 9
2.4. Ethyl 3-Methyl-5-Oxo-5H-Indeno[1,2-c]Pyridazine-4-Carboxylate 8
2.5. Ethyl 5-Oxo-3-Phenyl-5H-Indeno[1,2-c]Pyridazine-4-Carboxylate 10
2.6. Synthesis of 5-Oxo-3-Phenyl-5H-Indeno[1,2-c]Pyridazine-4-Carbohydrazide 11
2.7. Synthesis of 3-Phenyl-5H-Indeno[1,2-c]Pyridazine-4-Carbohydrazide 12
2.8. X-ray Structure Determination
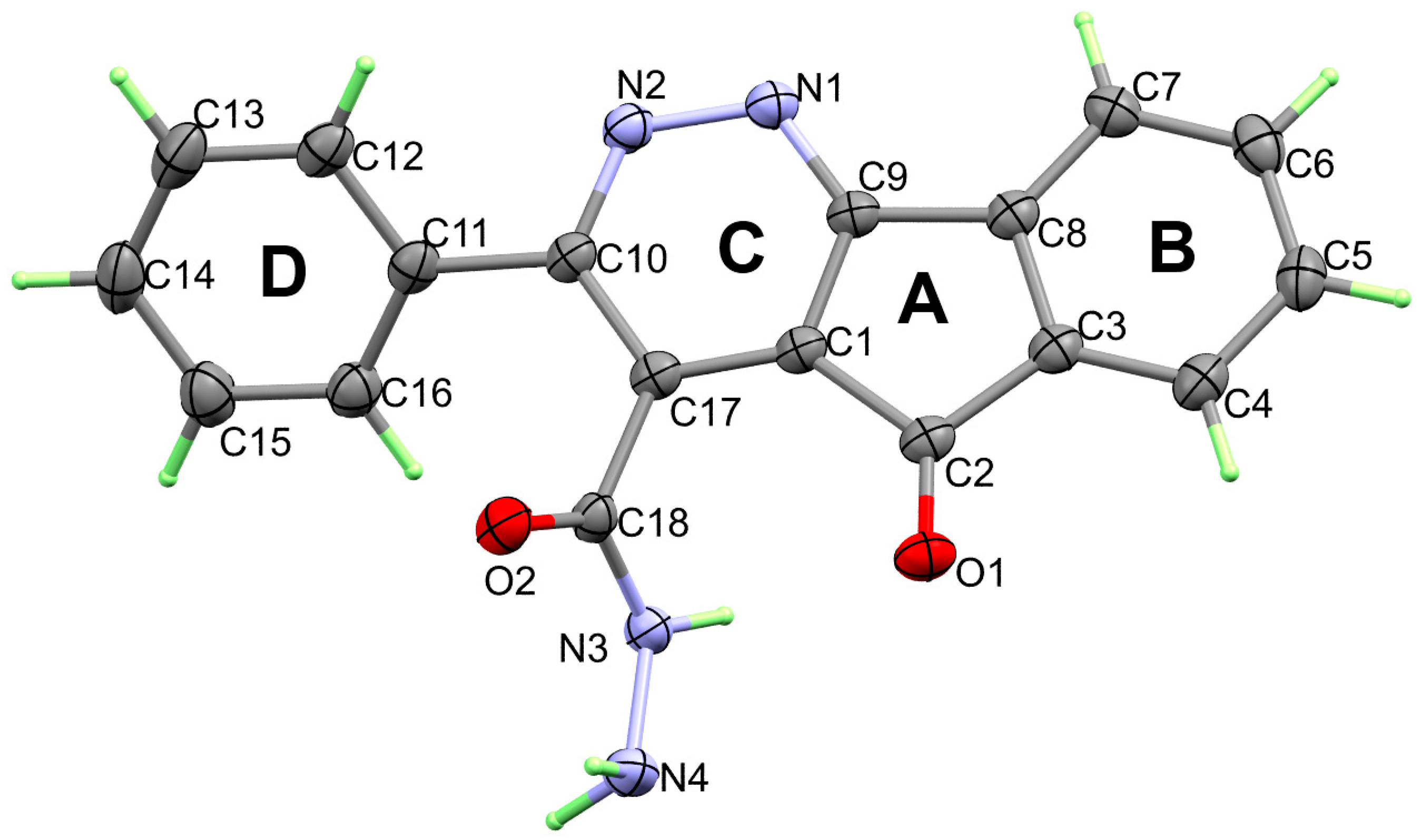
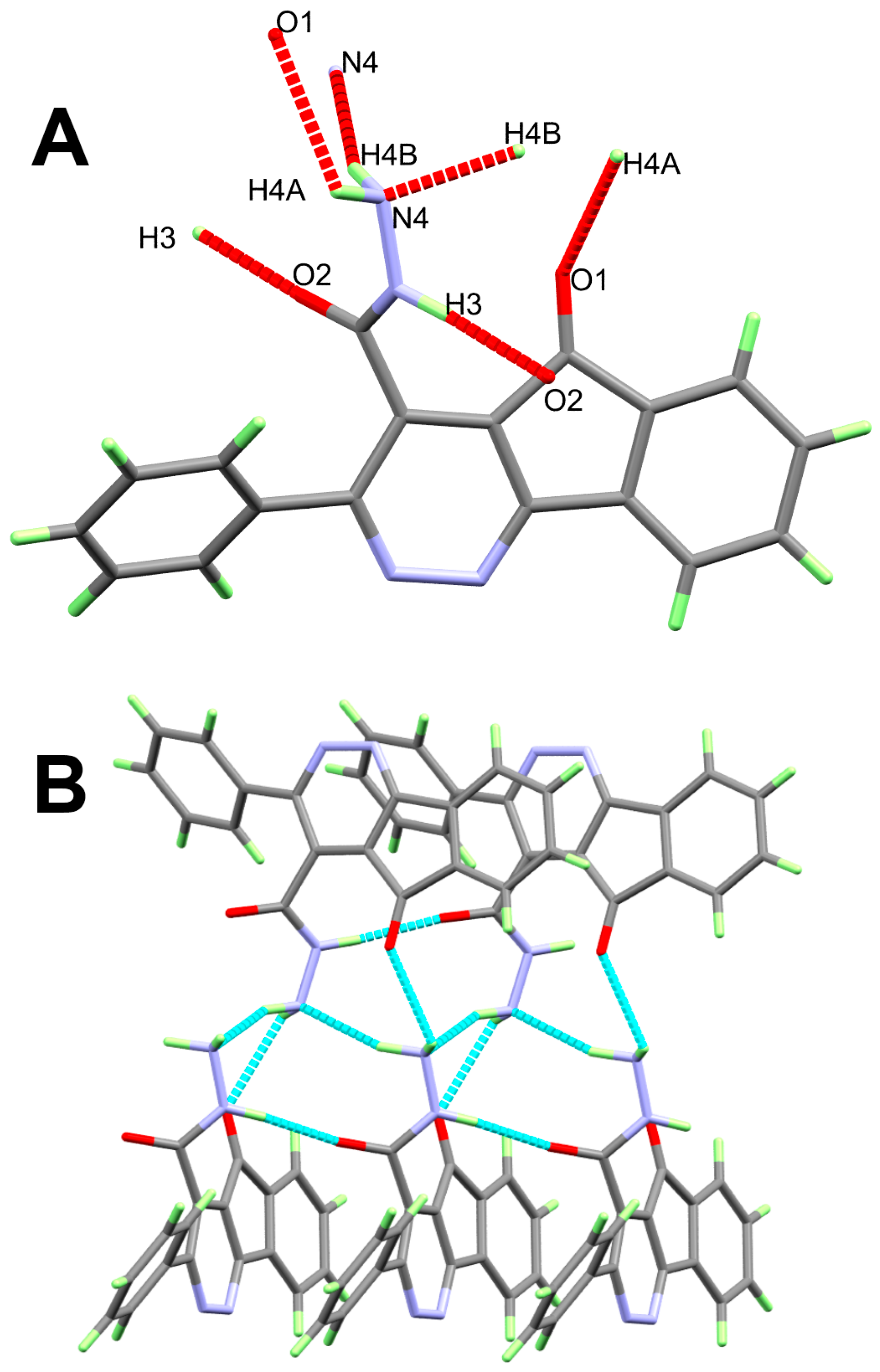
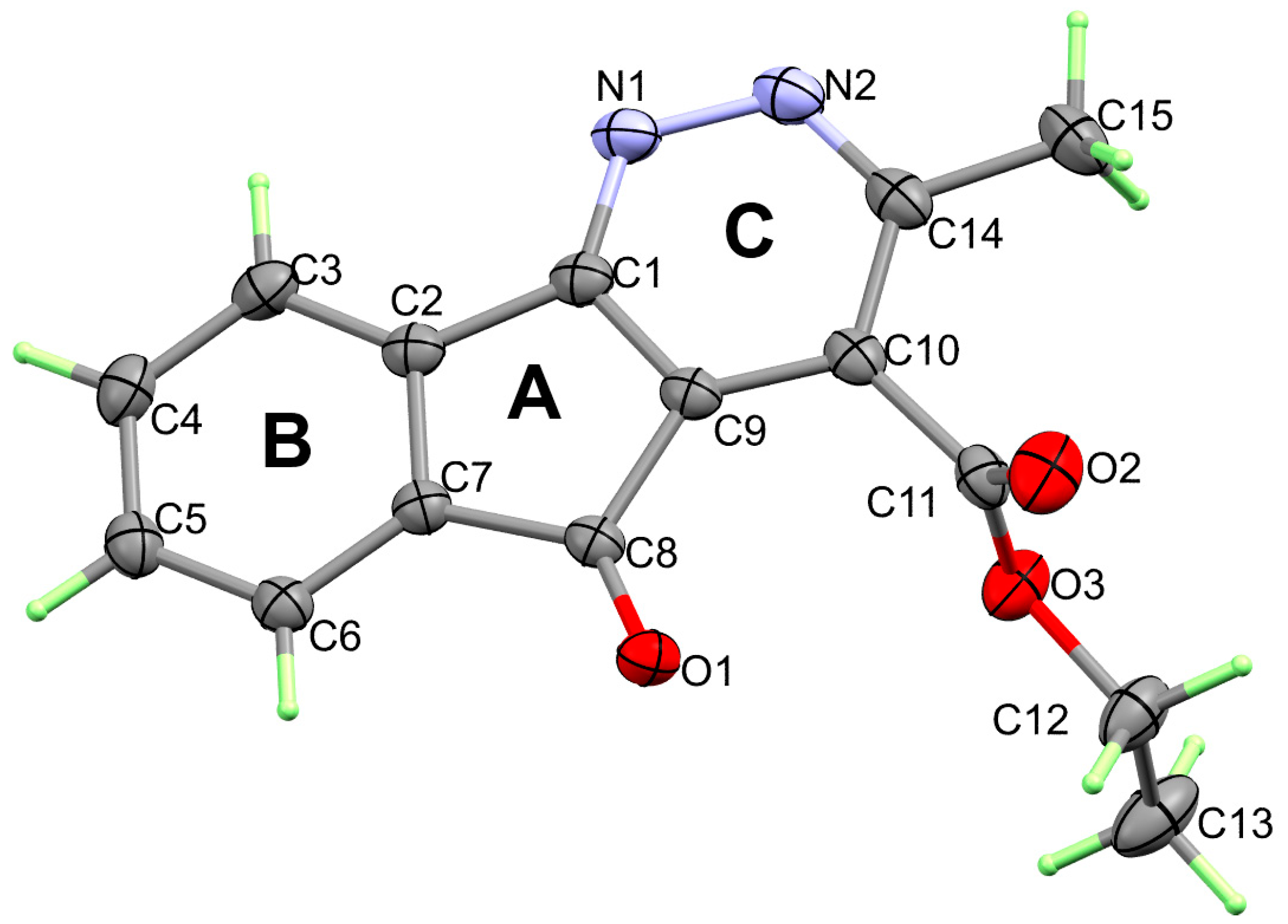
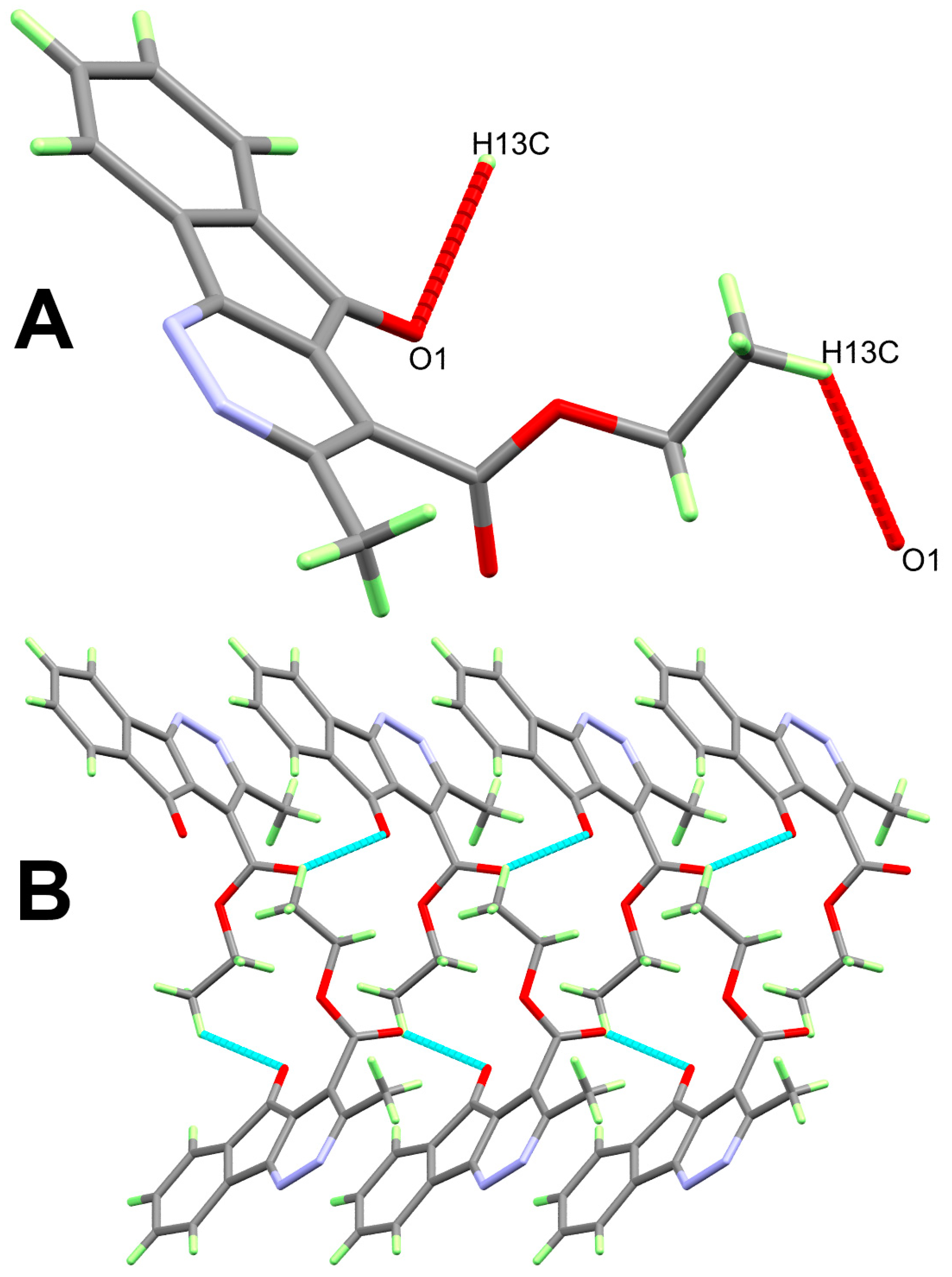
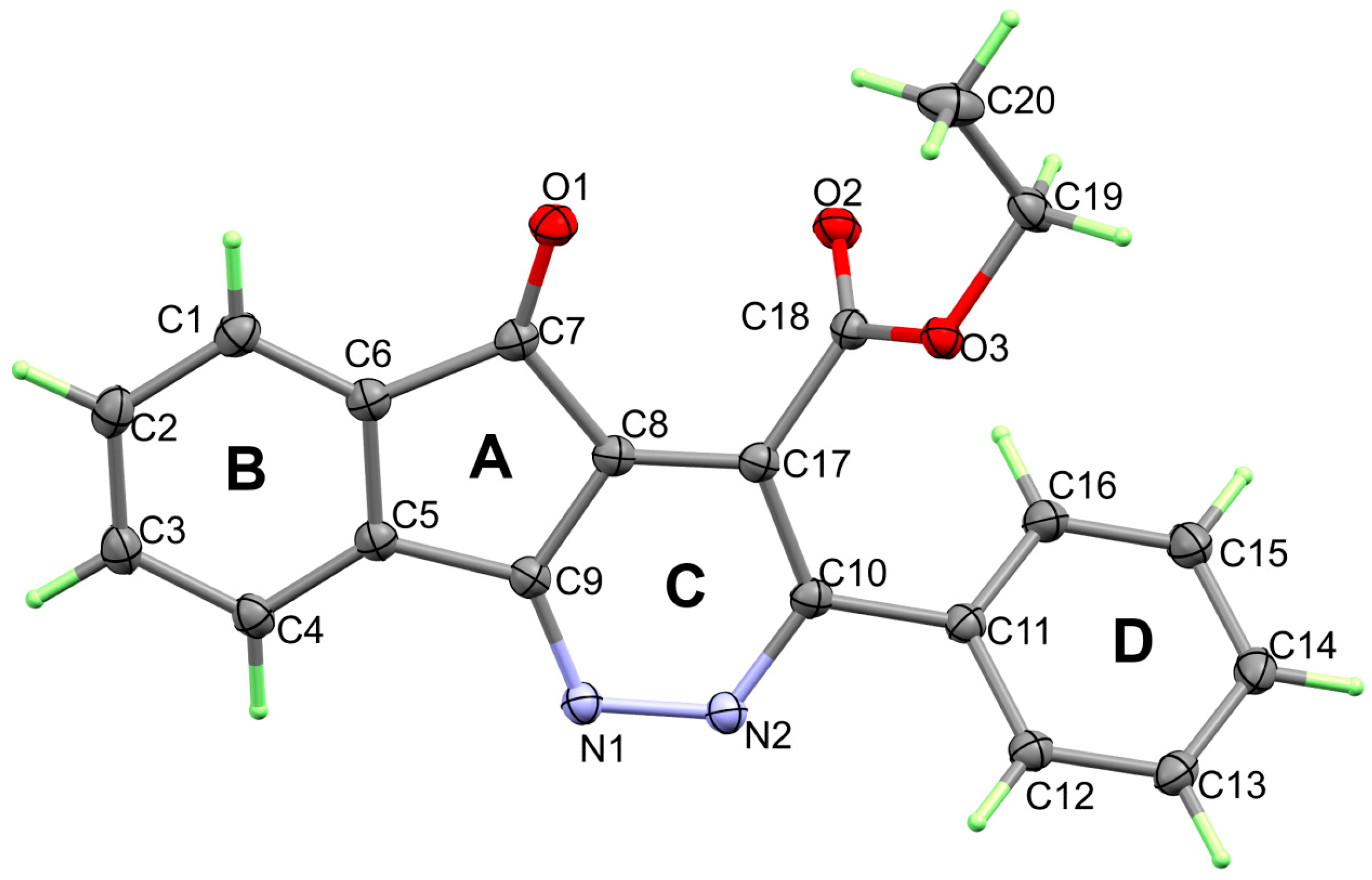
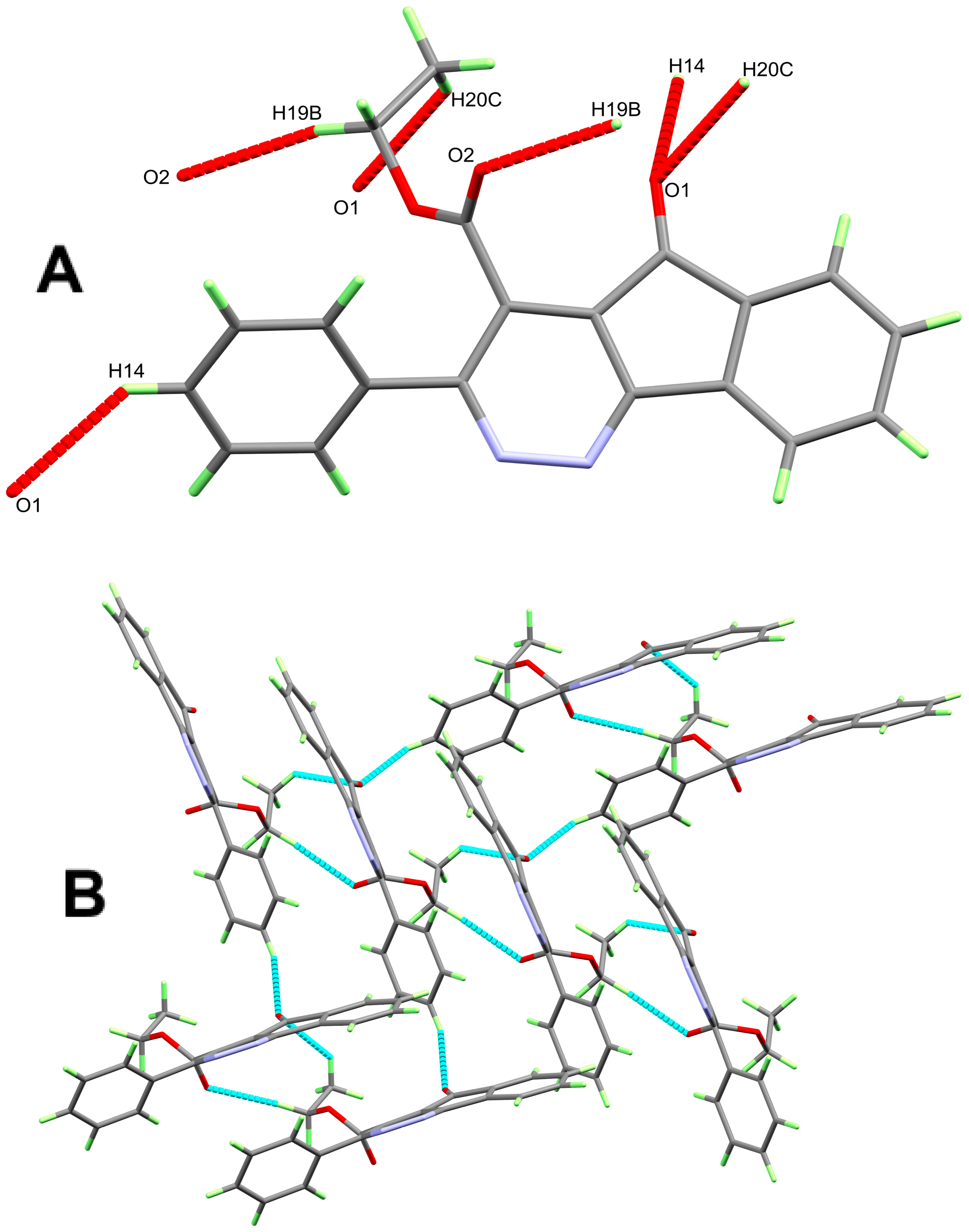
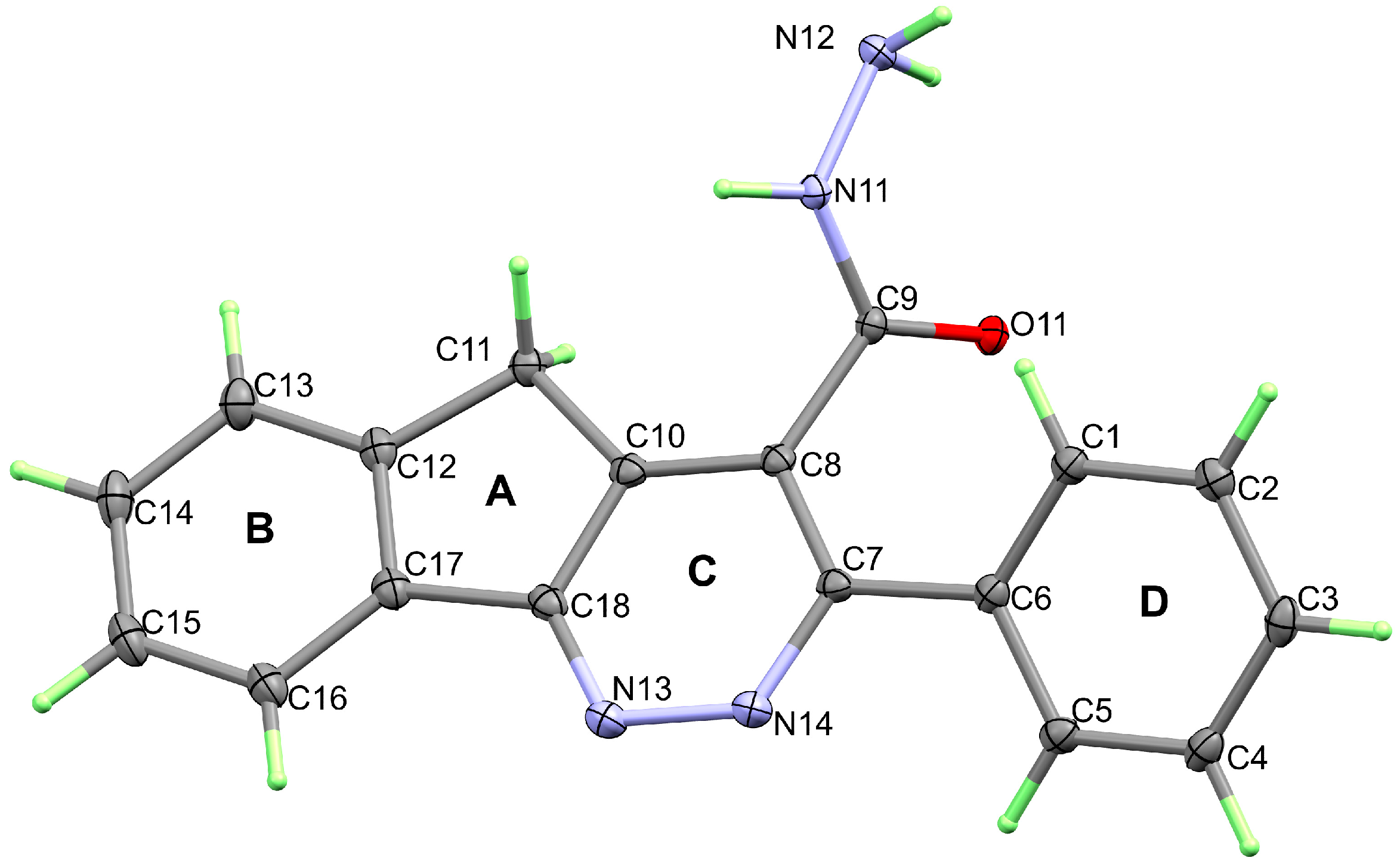
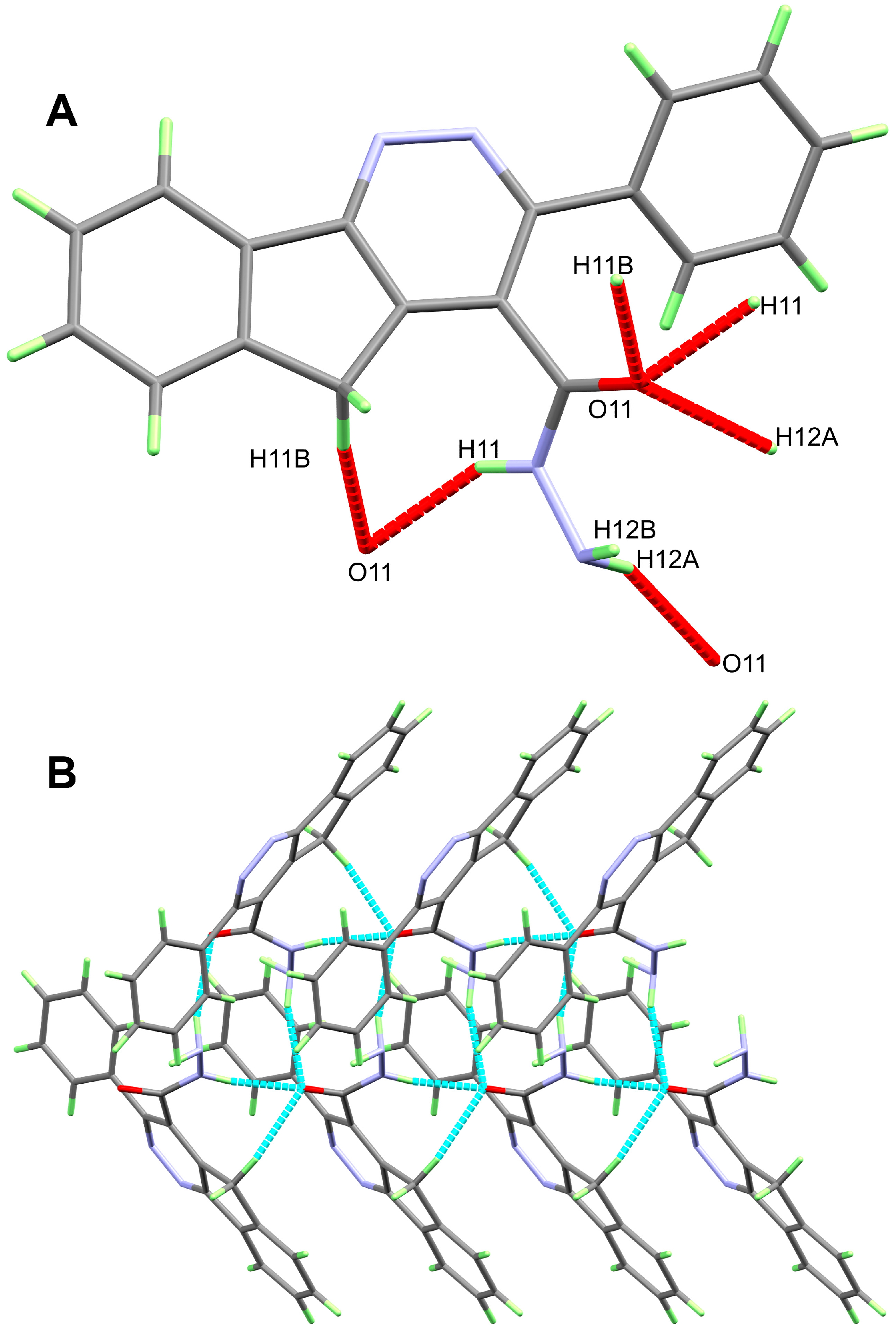
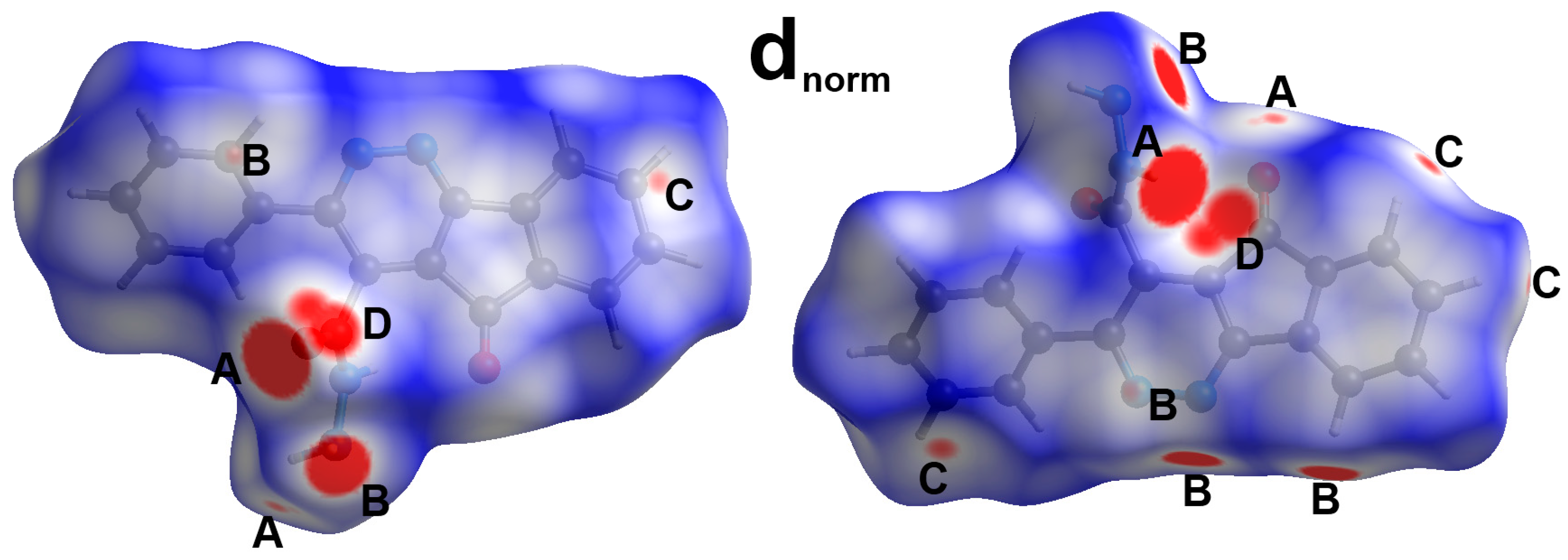
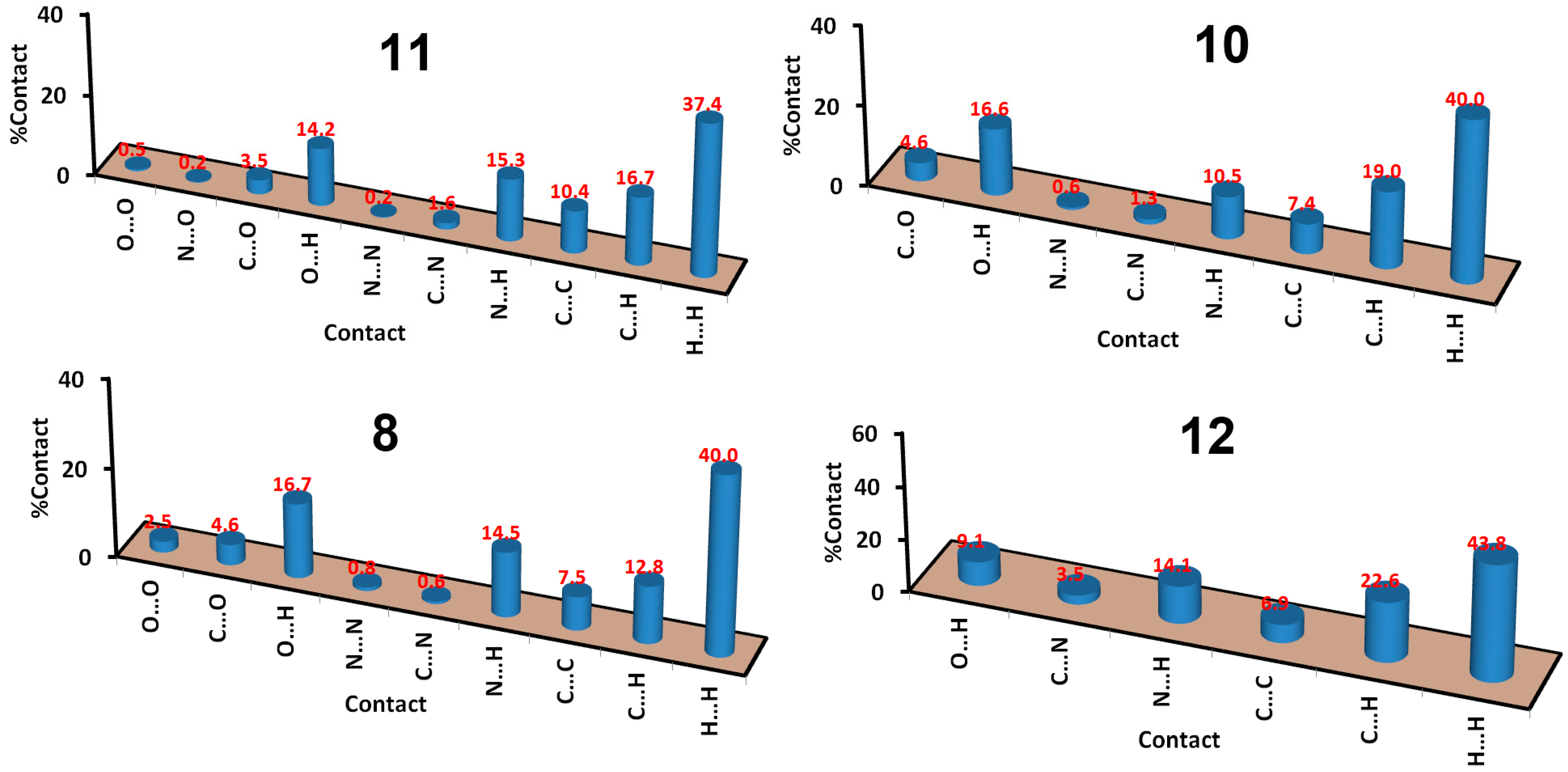
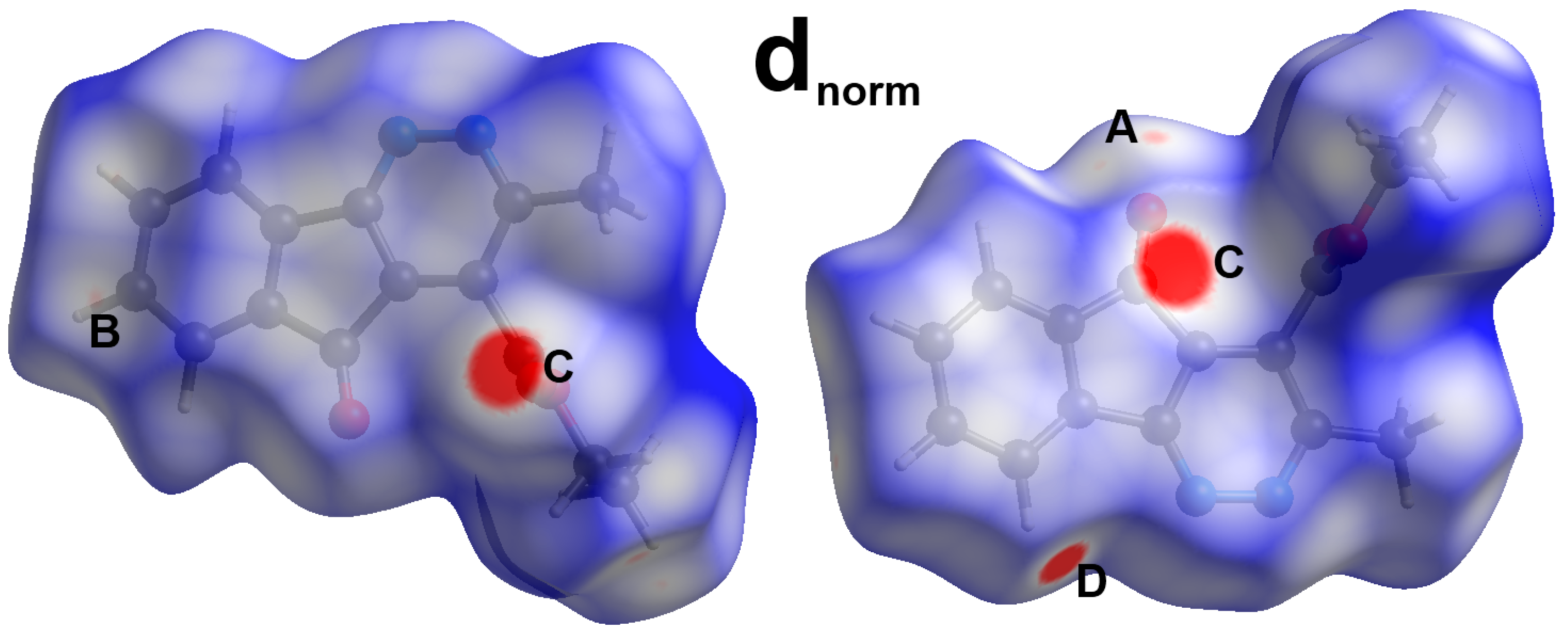
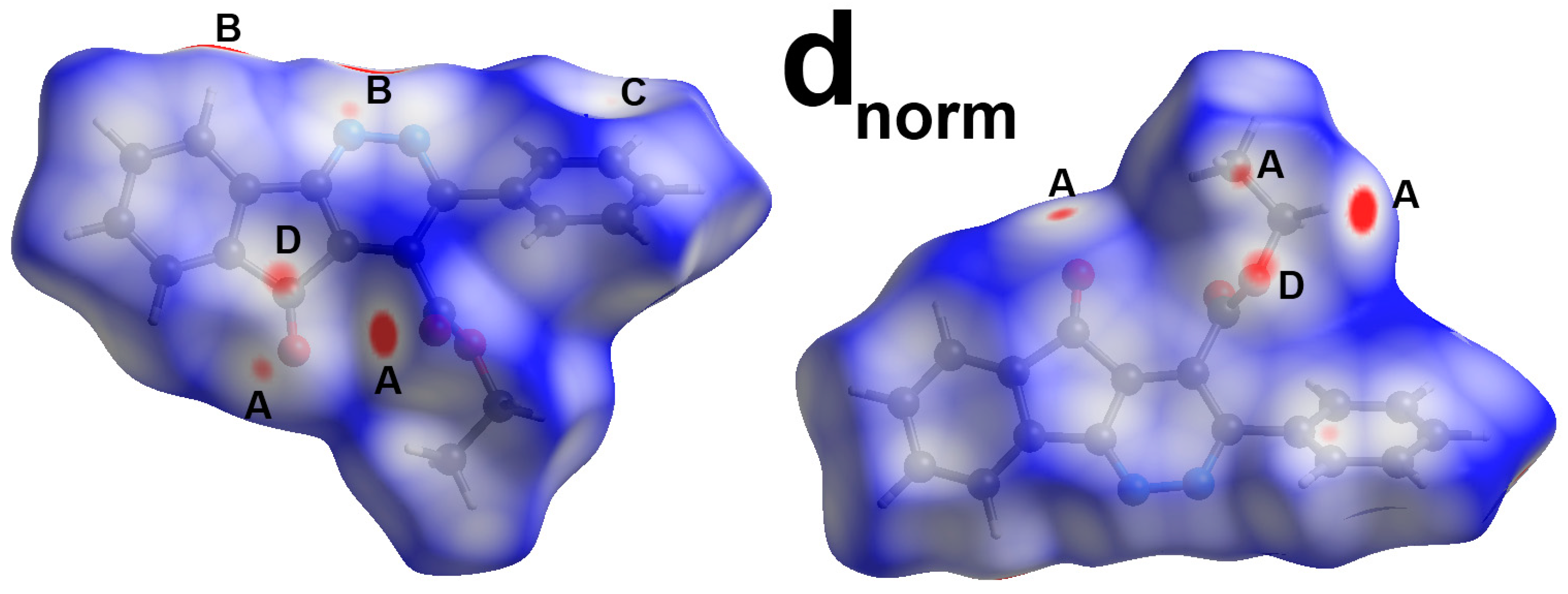
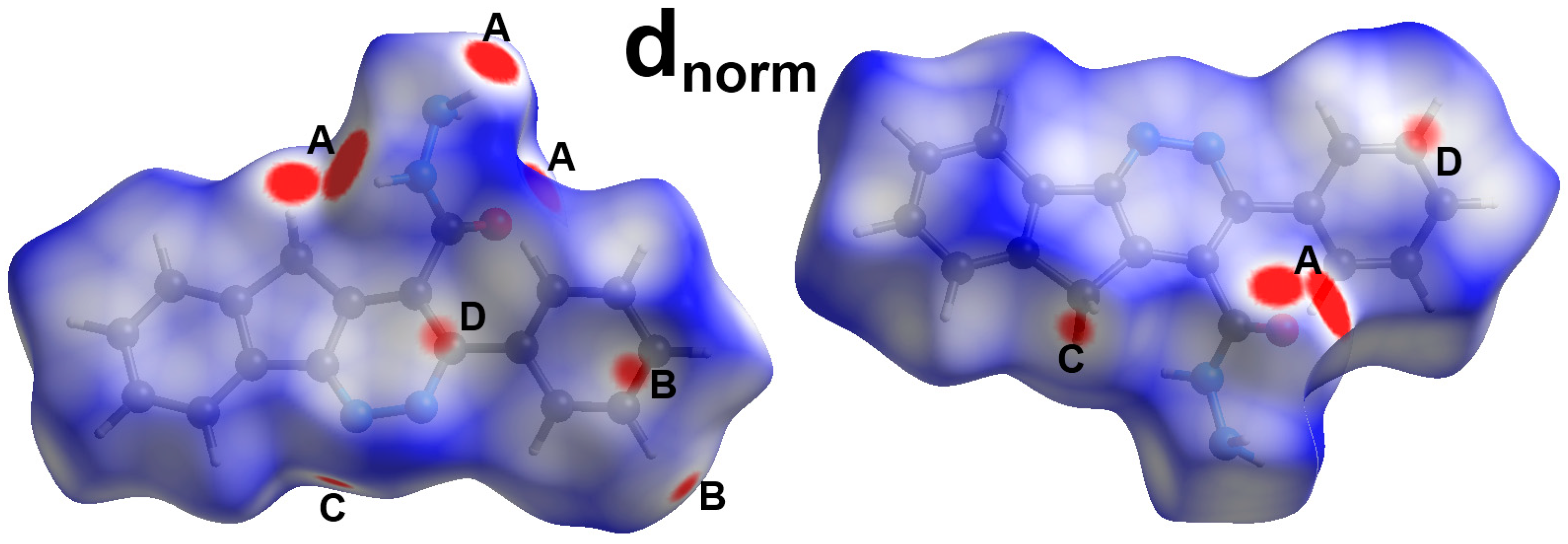
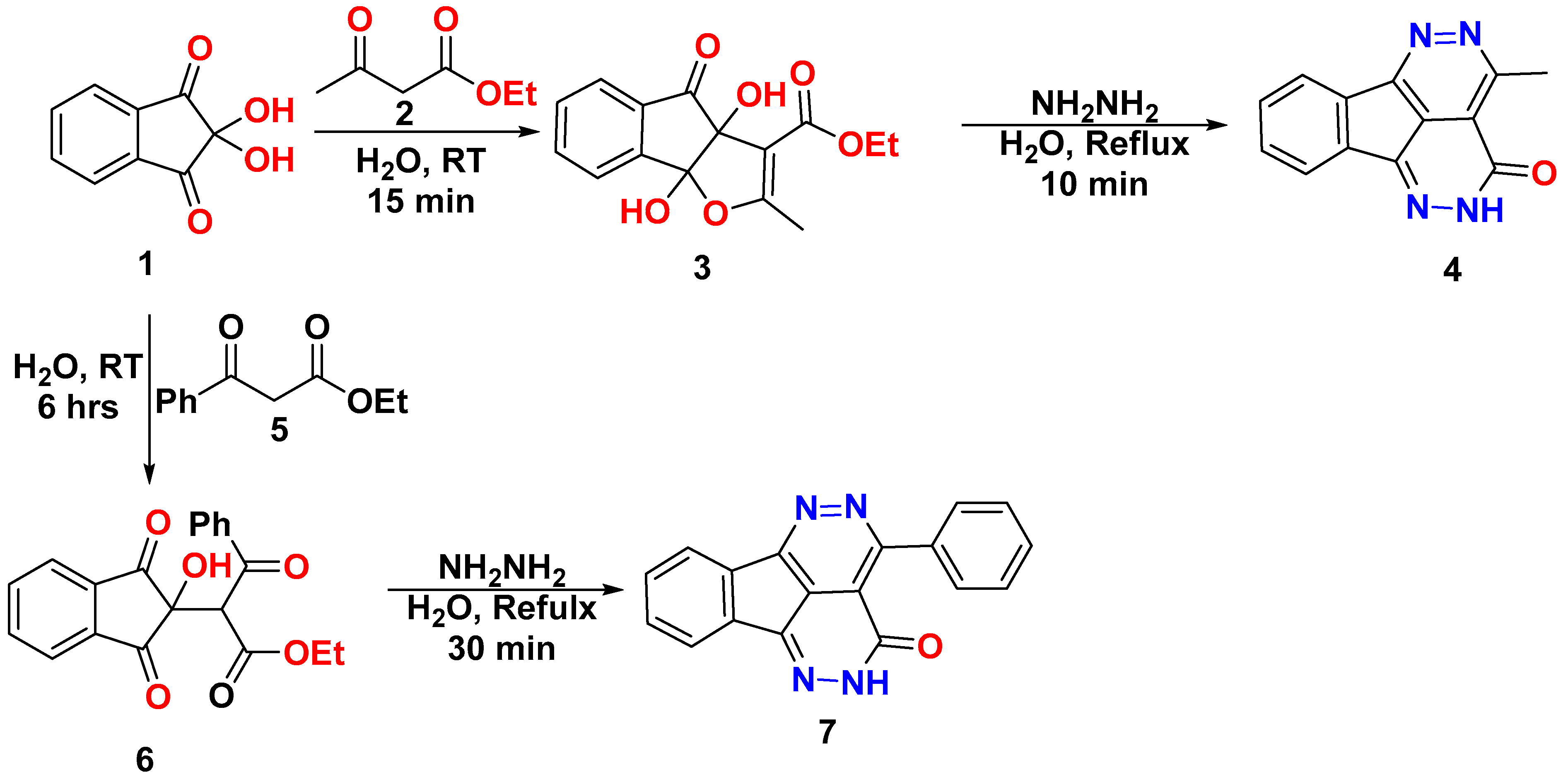
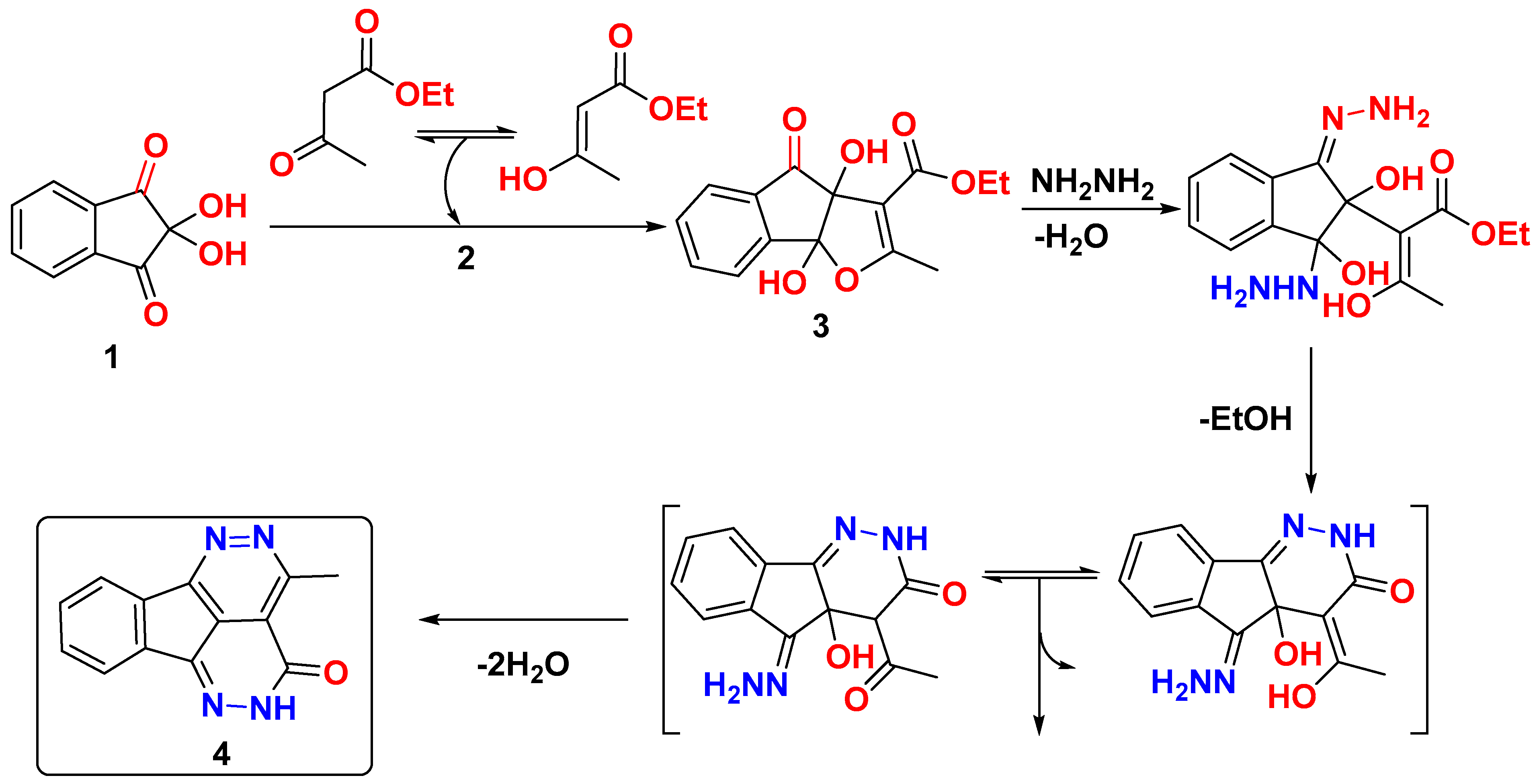
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Feng, X.; Pisula, W.; Müllen, K. Large polycyclic aromatic hydrocarbons: Synthesis and discotic organization. Pure Appl. Chem. 2009, 81, 2203–2224. [Google Scholar] [CrossRef]
- Stępień, M.; Gońka, E.; Żyła, M.; Sprutta, N. Heterocyclic nanographenes and other polycyclic heteroaromatic compounds: Synthetic routes, properties, and applications. Chem. Rev. 2017, 117, 3479–3716. [Google Scholar] [CrossRef]
- Albarano, L.; De Rosa, I.; Santaniello, I.; Montuori, M.; Serafini, S.; Toscanesi, M.; Trifuoggi, M.; Lofrano, G.; Guida, M.; Libralato, G. Synergistic, antagonistic, and additive effects of naphthalene, phenanthrene, fluoranthene and benzo (k) fluoranthene on Artemia franciscana nauplii and adult. Environ. Pollut. 2023, 335, 122286. [Google Scholar] [CrossRef]
- Plunkett, K. What about the five-membered ring? Cyclopentafused polycyclic aromatic hydrocarbons as a building block for functional materials. Synlett 2013, 24, 898–902. [Google Scholar] [CrossRef]
- Brauers, G.; Ebel, R.; Edrada, R.; Wray, V.; Berg, A.; Gräfe, U.; Proksch, P. Hortein, a new natural product from the fungus Hortaea werneckii associated with the Sponge Aplysina aerophoba. J. Nat. Prod. 2001, 64, 651–652. [Google Scholar] [CrossRef]
- Xie, S.; Chen, W.; Liu, S.; Zong, H.; Ming, B.; Zhou, G. Facile synthesis and functionalization of fluoranthenes via intramolecular [4+2] annulations between thiophenes and alkynes. Chin. Chem. Lett. 2023, 34, 107642. [Google Scholar] [CrossRef]
- Goel, A.; Sharma, A.; Kathuria, M.; Bhattacharjee, A.; Verma, A.; Mishra, P.R.; Nazir, A.; Mitra, K. New fluoranthene FLUN-550 as a fluorescent probe for selective staining and quantification of intracellular lipid droplets. Org. Lett. 2014, 16, 756–759. [Google Scholar] [CrossRef]
- Du, L.; King, J.B.; Cichewicz, R.H. Chlorinated polyketide obtained from a Daldinia sp. Treated with the epigenetic modifier suberoylanilide hydroxamic Acid. J. Nat. Prod. 2014, 77, 2454–2458. [Google Scholar] [CrossRef]
- Ding, L.; Ying, H.-Z.; Zhou, Y.; Lei, T.; Pei, J. Polycyclic imide derivatives: Synthesis and effective tuning of lowest unoccupied molecular orbital levels through molecular engineering. Org. Lett. 2010, 12, 5522–5525. [Google Scholar] [CrossRef]
- Chiechi, R.C.; Tseng, R.J.; Marchioni, F.; Yang, Y.; Wudl, F. Efficient Blue-Light-Emitting Electroluminescent Devices with a Robust Fluorophore: 7,8,10-Triphenylfluoranthene. Adv. Mater. 2006, 18, 325–328. [Google Scholar] [CrossRef]
- Yan, Q.; Zhou, Y.; Ni, B.-B.; Ma, Y.; Wang, J.; Pei, J.; Cao, Y. Organic semiconducting materials from sulfur-hetero benzo[k]-fluoranthene derivatives: Synthesis, photophysical properties, and thin film fransistor fabrication. J. Org. Chem. 2008, 73, 5328–5339. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Kumar, V.; Chaurasia, S.; Rawat, M.; Prasad, R.; Anand, R.S. Synthesis, electrochemical and optical properties of stable yellow fluorescent fluoranthenes. J. Org. Chem. 2010, 75, 3656–3662. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Fang, R.; Tao, J.; Wang, D.; Qiao, X.; Yang, X.; Hartl, F.; Li, H. Diacenaphthylene-fused benzo [1, 2-b: 4, 5-b’] dithiophenes: Polycyclic heteroacenes containing full-carbon five-membered aromatic rings. Chem. Commun. 2017, 53, 751–754. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.E.; Cai, Z.W. An intramolecular arene-triflate coupling reaction for the regiospecific synthesis of substituted benzofluoranthenes. J. Org. Chem. 1993, 58, 1415–1424. [Google Scholar] [CrossRef]
- Gu, X.; Luhman, W.A.; Yagodkin, E.; Holmes, R.J.; Douglas, C.J. Diarylindenotetracenes via a selective cross-coupling/C–H functionalization: Electron donors for organic photovoltaic cells. Org. Lett. 2012, 14, 1390–1393. [Google Scholar] [CrossRef]
- Wegner, H.A.; Scott, L.T.; de Meijere, A. A new Suzuki– Heck-Type coupling cascade: Indeno [1, 2, 3]-annelation of polycyclic aromatic hydrocarbons. J. Org. Chem. 2003, 68, 883–887. [Google Scholar] [CrossRef]
- Pascual, S.; Bour, C.; de Mendoza, P.; Echavarren, A.M. Synthesis of fluoranthenes by hydroarylation of alkynes catalyzed by gold (I) or gallium trichloride. Beilstein J. Org. Chem. 2011, 7, 1520–1525. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, W.; Wang, B.; Ren, H. Friedel–Crafts arylation for the formation of Csp2-Csp2 bonds: A route to unsymmetrical and functionalized polycyclic aromatic hydrocarbons from aryl triazenes. Angew. Chem., Int. Ed. 2012, 51, 12293–12297. [Google Scholar] [CrossRef]
- Reisch, H.A.; Bratcher, M.S.; Scott, L.T. Imposing curvature on a polyarene by intramolecular palladium-catalyzed arylation reactions: A simple synthesis of dibenzo[a,g]corannulene. Org. Lett. 2000, 2, 1427–1430. [Google Scholar] [CrossRef]
- Kawasumi, K.; Mochida, K.; Kajino, T.; Segawa, Y.; Itami, K. Pd(OAc)2/o-Chloranil/M(OTf)n: A catalyst for the direct C–H arylation of polycyclic aromatic hydrocarbons with boryl-, silyl-, and unfunctionalized arenes. Org. Lett. 2012, 14, 418–421. [Google Scholar] [CrossRef]
- Ogawa, N.; Yamaoka, Y.; Yamada, K.-i.; Takasu, K. Synthesis of π-extended fluoranthenes via a KHMDS-promoted anionic-radical reaction cascade. Org. Lett. 2017, 19, 3327–3330. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Nagashima, Y.; Tanaka, J.; Tanaka, K. Room Temperature Fluoranthene Synthesis through Cationic Rh(I)/H8-BINAP-Catalyzed [2+2+2] Cycloaddition: Unexpected Acceleration due to Noncovalent Interactions. ACS Catal. 2023, 13, 1604–1613. [Google Scholar] [CrossRef]
- Wu, Y.-T.; Linden, A.; Siegel, J.S. Formal [(2+ 2)+ 2] and [(2+2)+(2+ 2)] nonconjugated dienediyne cascade cycloadditions. Org.Lett. 2005, 7, 4353–4355. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lu, P.; Wang, Y. Four iodine-mediated electrophilic cyclizations of rigid parallel triple bonds Mapped from 1, 8-Dialkynylnaphthalenes. Chem.–Eur. J. 2011, 17, 8105–8114. [Google Scholar] [CrossRef] [PubMed]
- Quimby, J.M.; Scott, L.T. Expanding the Suzuki–Heck-type coupling cascade: A new indeno [1, 2, 3]-annelation of polycyclic aromatic hydrocarbons. Adv. Synth. Catal. 2009, 351, 1009–1013. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Higuchi, M.; Tazawa, K.; Manabe, K. Three-step synthesis of fluoranthenes through Pd-catalyzed inter-and intramolecular C–H arylation. J. Org. Chem. 2016, 81, 3967–3974. [Google Scholar] [CrossRef]
- Pal, S.; Metin, Ö.; Türkmen, Y.E. Synthesis of fluoranthene derivatives via tandem Suzuki–Miyaura and intramolecular C–H Arylation reactions under both homogeneous and heterogeneous catalytic conditions. ACS Omega 2017, 2, 8689–8696. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Loizou, G.; Lo Re, D. Synthesis of triazafluoranthenones via silver (I)-mediated nonoxidative and oxidative intramolecular palladium-catalyzed cyclizations. J. Org. Chem. 2011, 76, 5793–5802. [Google Scholar] [CrossRef]
- Pathak, S.; Debnath, K.; Pramanik, A. Silica sulfuric acid: A reusable solid catalyst for one pot synthesis of densely substituted pyrrole-fused isocoumarins under solvent-free conditions. Beilstein J. Org. Chem. 2013, 9, 2344–2353. [Google Scholar] [CrossRef]
- Dong, P.; Majeed, K.; Wang, L.; Guo, Z.; Zhou, F.; Zhang, Q. Transition metal-free approach to azafluoranthene scaffolds by aldol condensation/[1+2+3] annulation tandem reaction of isocyanoacetates with 8-(alkynyl)-1-naphthaldehydes. Chem. Commun. 2021, 39, 4855–4858. [Google Scholar] [CrossRef]
- Boraei, A.T.A.; Soliman, S.M.; Haukka, M.; Salama, E.E.; Sopaih, M.; Barakat, A.; Sarhan, A.A.M. Straightforward green synthesis of indeno-furan carboxylates from ninhydrin and β-ketoesters: X-Ray crystal structure, Hirshfeld and DFT investigations. J. Mol. Struct. 2022, 1255, 132433. [Google Scholar] [CrossRef]
- Boraei, A.T.; Ghabbour, H.A.; Sarhan, A.A.; Barakat, A. Expeditious green synthesis of novel 4-methyl-1, 2, 5, 6-tetraazafluoranthen-3 (2H)-one analogue from ninhydrin: N/S-alkylation and aza-Michael addition. ACS Omega 2020, 5, 5436–5442. [Google Scholar] [CrossRef] [PubMed]
- Boraei, A.T.A.; Haukka, M.; Sopaih, M.; Al-Majid, A.M.; Soliman, S.M.; Barakat, A.; Sarhan, A.M. Straightforward One-Pot Synthesis of New 4-Phenyl-1,2,5,6-tetraazafluoranthen-3(2H)-one Derivatives: X-ray Single Crystal Structure and Hirshfeld Analyses. Crystals 2022, 12, 262. [Google Scholar] [CrossRef]
- Rikagu Oxford Diffraction. CrysAlisPro; Rikagu Oxford Diffraction Inc.: Yarnton, Oxfordshire, UK, 2020. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Cryst. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer17, University of Western Australia. 2017. Available online: https://crystalexplorer.net/ (accessed on 30 July 2020).


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 8 | 10 | 11 | 12 | |
|---|---|---|---|---|
| CCDC | 2297471 | 2297472 | 2297470 | 2299618 |
| Empirical formula | C15H12N2O3 | C20H14N2O3 | C18H12N4O2 | C18H14N4O |
| fw | 268.27 | 330.33 | 316.32 | 302.33 |
| Temp (K) | 120(2) | 120(2) | 120(2) | 120(2) |
| λ(Å) | 1.54184 | 1.54184 | 1.54184 | 1.54184 |
| Cryst syst | Monoclinic | Monoclinic | Monoclinic | Orthorhombic |
| Space group | P21/c | P21/n | P21/c | P212121 |
| a (Å) | 15.3400(4) | 16.89752(13) | 14.9862(2) | 5.04710(10) |
| b (Å) | 5.02920(10) | 5.76627(4) | 4.86410(10) | 12.5114(3) |
| c (Å) | 16.7042(4) | 17.97633(13) | 20.3991(3) | 22.2915(5) |
| β (deg) | 100.545(2) | 113.9469(9) | 101.697(2) | 90 |
| V (Å3) | 1266.93(5) | 1600.77(2) | 1456.10(4) | 1407.63(5) |
| Z | 4 | 4 | 4 | 4 |
| ρcalc (Mg/m3) | 1.406 | 1.371 | 1.443 | 1.427 |
| μ(Mo Kα) (mm−1) | 0.825 | 0.766 | 0.804 | 0.745 |
| No. of reflns. | 14014 | 35632 | 17874 | 10944 |
| Unique reflns. | 2666 | 3465 | 3050 | 2929 |
| Completeness to θ = 67.684° | 100% | 100% | 100% | 100% |
| GOOF (F2) | 1.054 | 1.053 | 1.047 | 1.046 |
| Rint | 0.0361 | 0.0237 | 0.0295 | 0.0462 |
| R1 a (I ≥ 2σ) | 0.0346 | 0.0358 | 0.0397 | 0.0332 |
| wR2 b (I ≥ 2σ) | 0.0863 | 0.0927 | 0.1041 | 0.0768 |
| D-H...A | d(D-H) | d(H...A) | d(D...A) | <(DHA) | Symm. Code |
|---|---|---|---|---|---|
| 11 | |||||
| N3-H3...O2#1 | 0.94(2) | 1.93(2) | 2.8614(16) | 175.0(19) | x, y + 1, z |
| N4-H4A...O1#2 | 0.98(2) | 2.52(2) | 3.0425(16) | 113.3(15) | −x + 1, y − 1/2, −z + 3/2 |
| N4-H4B...N4#2 | 1.01(2) | 2.16(2) | 3.1152(16) | 157.7(19) | −x + 1, y − 1/2, −z + 3/2 |
| 8 | |||||
| C13…H13C…O1 | 0.98 | 2.6368 | 3.331(2) | 128 | 1 − x, −1/2 + y,1.5 − z |
| 10 | |||||
| C4-H4...N1 | 0.95 | 2.44 | 3.3764(14) | 167 | 1 − x, −y, 1 − z |
| C4-H4...N2 | 0.95 | 2.6 | 3.4325(14) | 146 | 1 − x, −y, 1 − z |
| C14-H14...O1 | 0.95 | 2.58 | 3.3447(15) | 137 | −1/2 + x, 3/2 − y, −1/2 + z |
| C19-H19B...O2 | 0.99 | 2.44 | 3.4061(14) | 165 | x, 1 + y, z |
| C20-H20C...O1 | 0.98 | 2.58 | 3.3182(19) | 132 | x, 1 + y, z |
| 12 | |||||
| N11-H11...O11 | 0.89(3) | 2.06(3) | 2.852(3) | 147(2) | −1 + x, y, z |
| N12-H12A...O11 | 0.89(3) | 2.34(3) | 3.174(3) | 156(2) | −1/2 + x, 3/2 − y, 1 − z |
| C11-H11B...O11 | 0.99 | 2.36 | 3.317(3) | 163 | −1 + x, y, z |
| Contact | Distance | Contact | Distance |
|---|---|---|---|
| C1…O2 | 3.050 | O1…H4A | 2.509 |
| C2…O2 | 2.929 | O2…H3 | 1.855 |
| N4…H4B | 2.158 | C13…H4 | 2.655 |
| N1…H7 | 2.244 | C6…H5 | 2.717 |
| N2…H7 | 2.610 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarhan, A.A.M.; Haukka, M.; Barakat, A.; Soliman, S.M.; Boraei, A.T.A.; Sopaih, M.; Salama, E.E. 4-Methyl/Phenyl-1,2,5,6-tetraazafluoranthen-3(2H)-ones Synthesis: Mechanistic Pathway Study and Single-Crystal X-ray Analysis of the Intermediates. Crystals 2023, 13, 1537. https://doi.org/10.3390/cryst13111537
Sarhan AAM, Haukka M, Barakat A, Soliman SM, Boraei ATA, Sopaih M, Salama EE. 4-Methyl/Phenyl-1,2,5,6-tetraazafluoranthen-3(2H)-ones Synthesis: Mechanistic Pathway Study and Single-Crystal X-ray Analysis of the Intermediates. Crystals. 2023; 13(11):1537. https://doi.org/10.3390/cryst13111537
Chicago/Turabian StyleSarhan, Ahmed A. M., Matti Haukka, Assem Barakat, Saied M. Soliman, Ahmed T. A. Boraei, Manar Sopaih, and Eid E. Salama. 2023. "4-Methyl/Phenyl-1,2,5,6-tetraazafluoranthen-3(2H)-ones Synthesis: Mechanistic Pathway Study and Single-Crystal X-ray Analysis of the Intermediates" Crystals 13, no. 11: 1537. https://doi.org/10.3390/cryst13111537
APA StyleSarhan, A. A. M., Haukka, M., Barakat, A., Soliman, S. M., Boraei, A. T. A., Sopaih, M., & Salama, E. E. (2023). 4-Methyl/Phenyl-1,2,5,6-tetraazafluoranthen-3(2H)-ones Synthesis: Mechanistic Pathway Study and Single-Crystal X-ray Analysis of the Intermediates. Crystals, 13(11), 1537. https://doi.org/10.3390/cryst13111537