
Pressure-Induced Monoclinic to Tetragonal Phase Transition in RTaO4 (R = Nd, Sm): DFT-Based First Principles Studies

Abstract
:1. Introduction
2. Computational Details
3. Results and Discussions
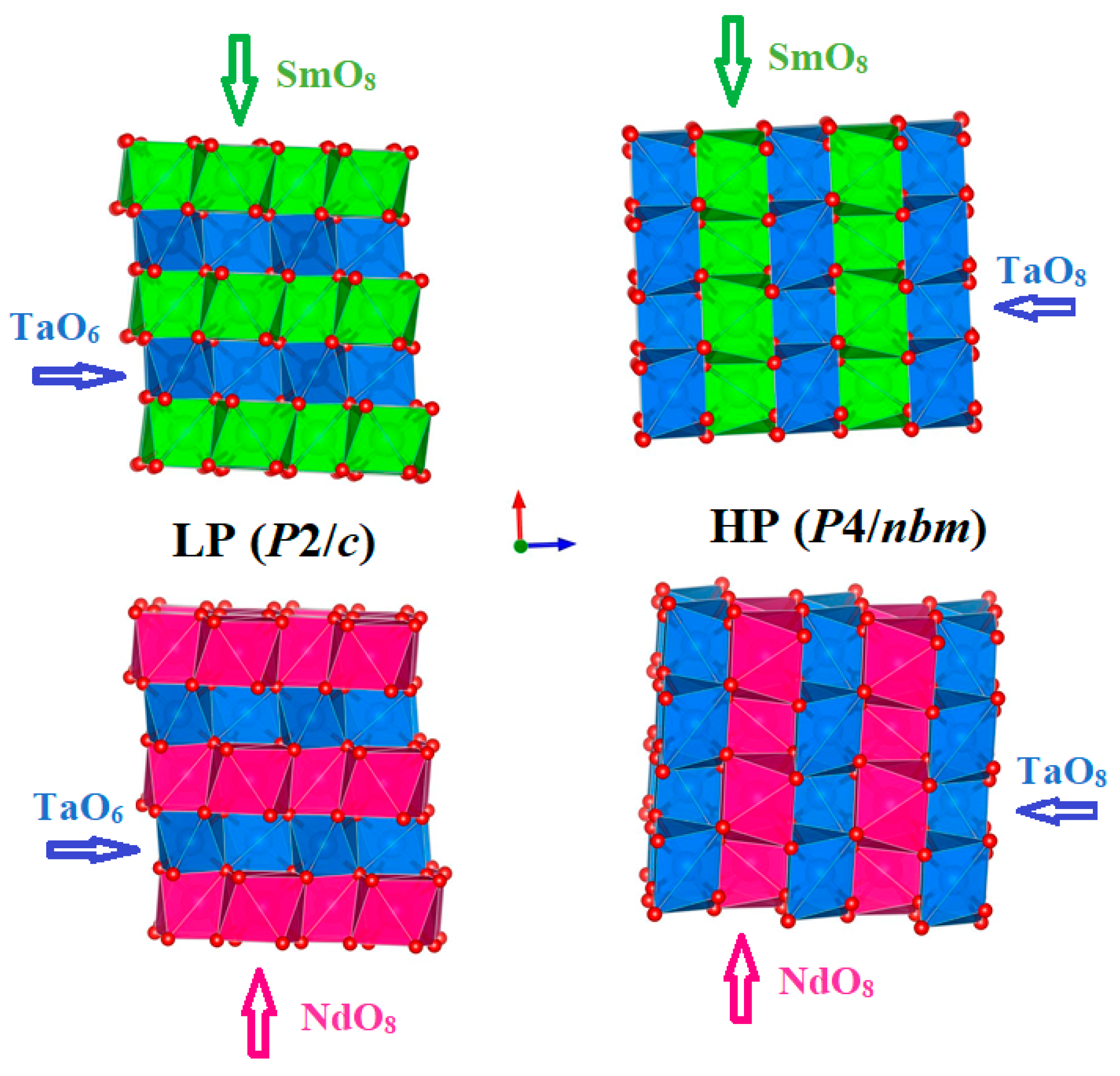
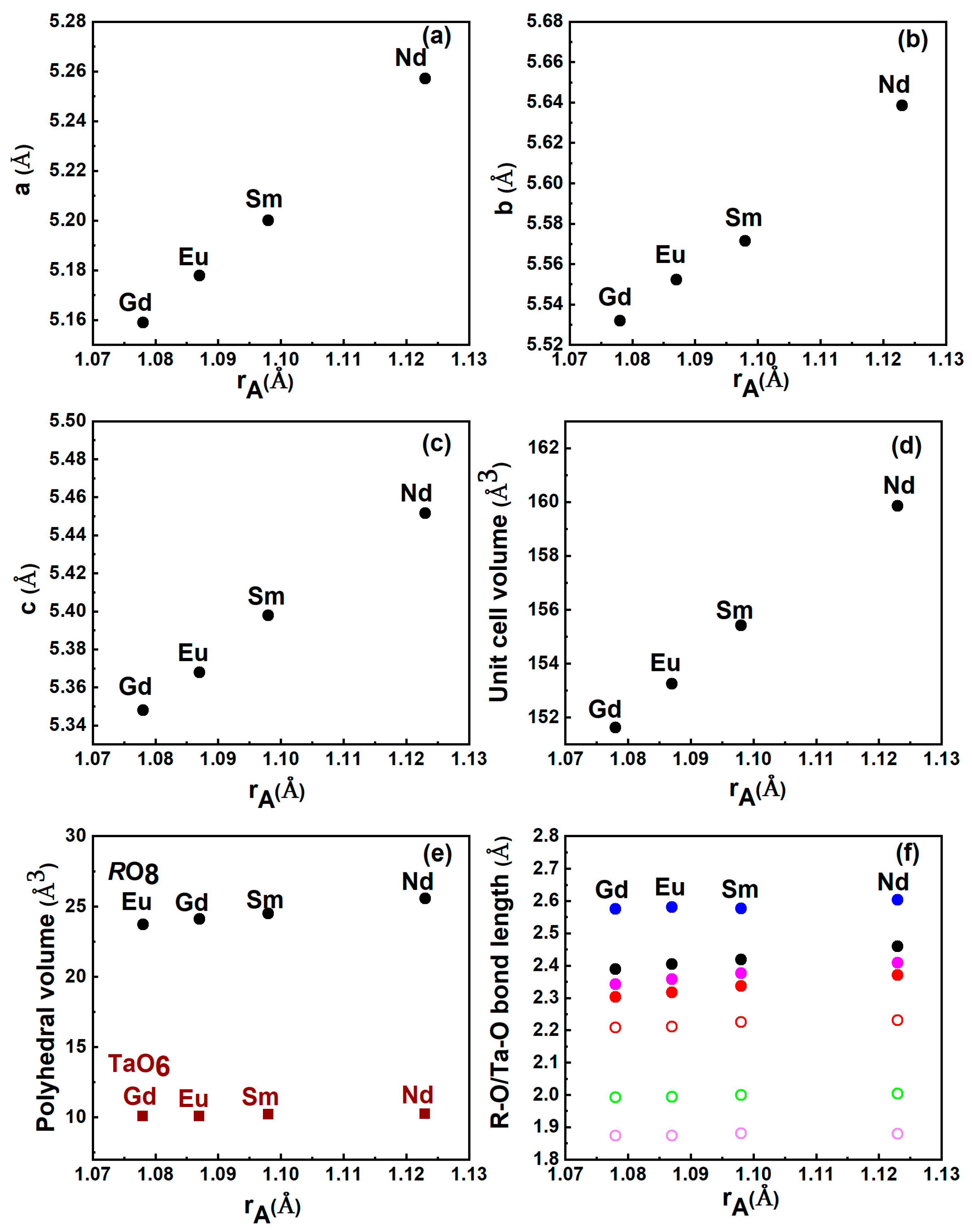
3.1. Ambient Structure
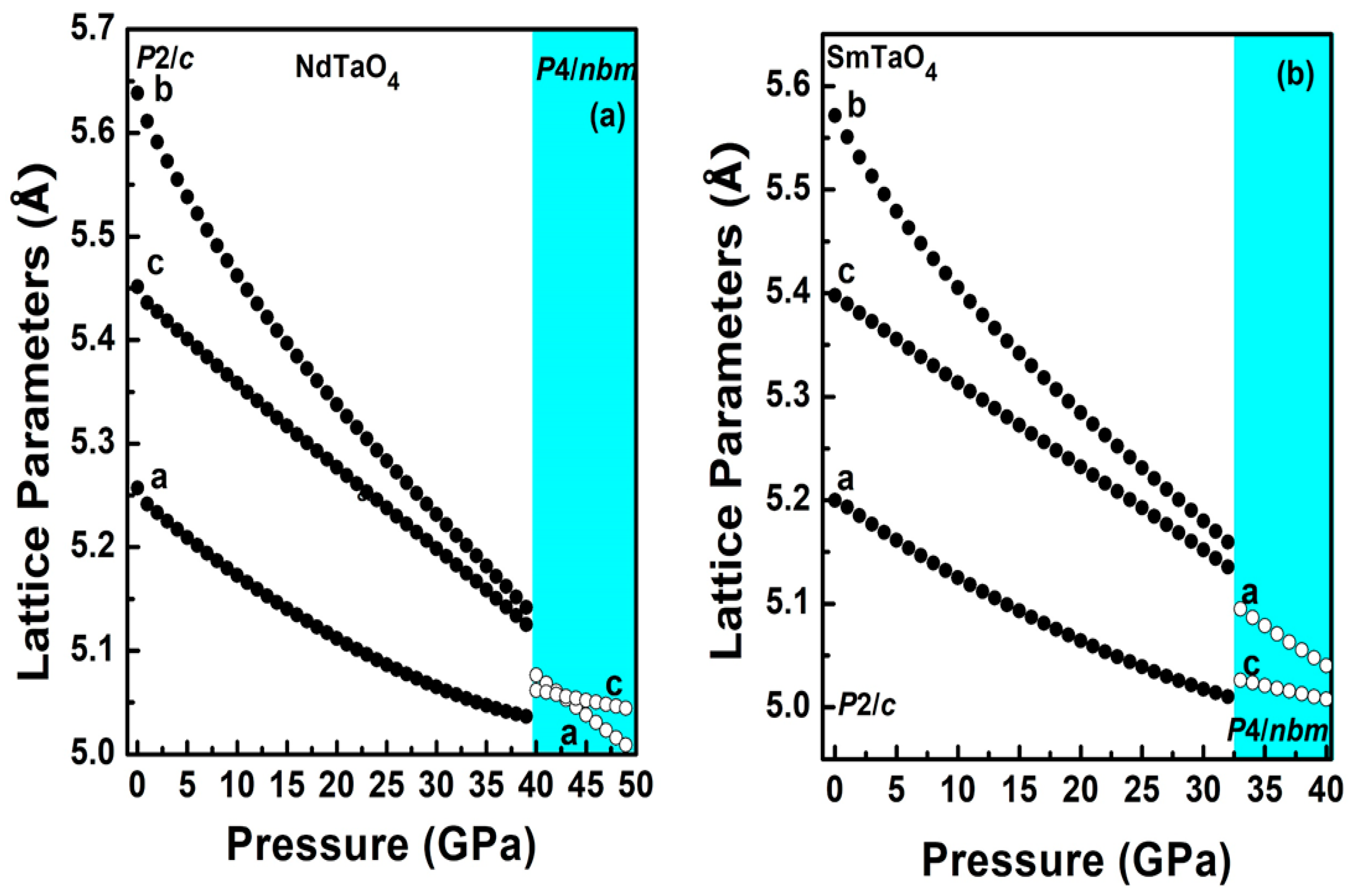
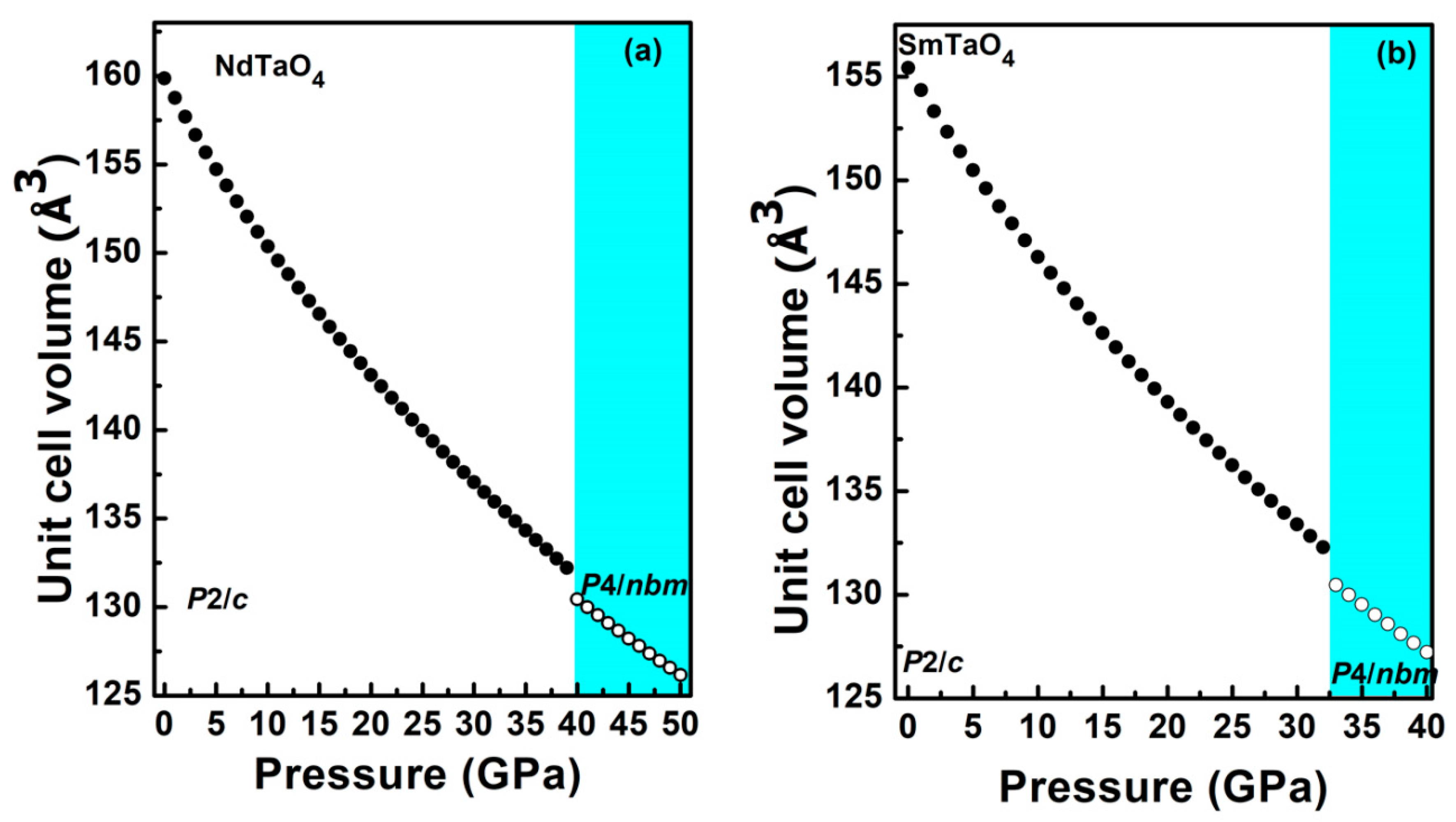
3.2. Structural Behavior under Compression
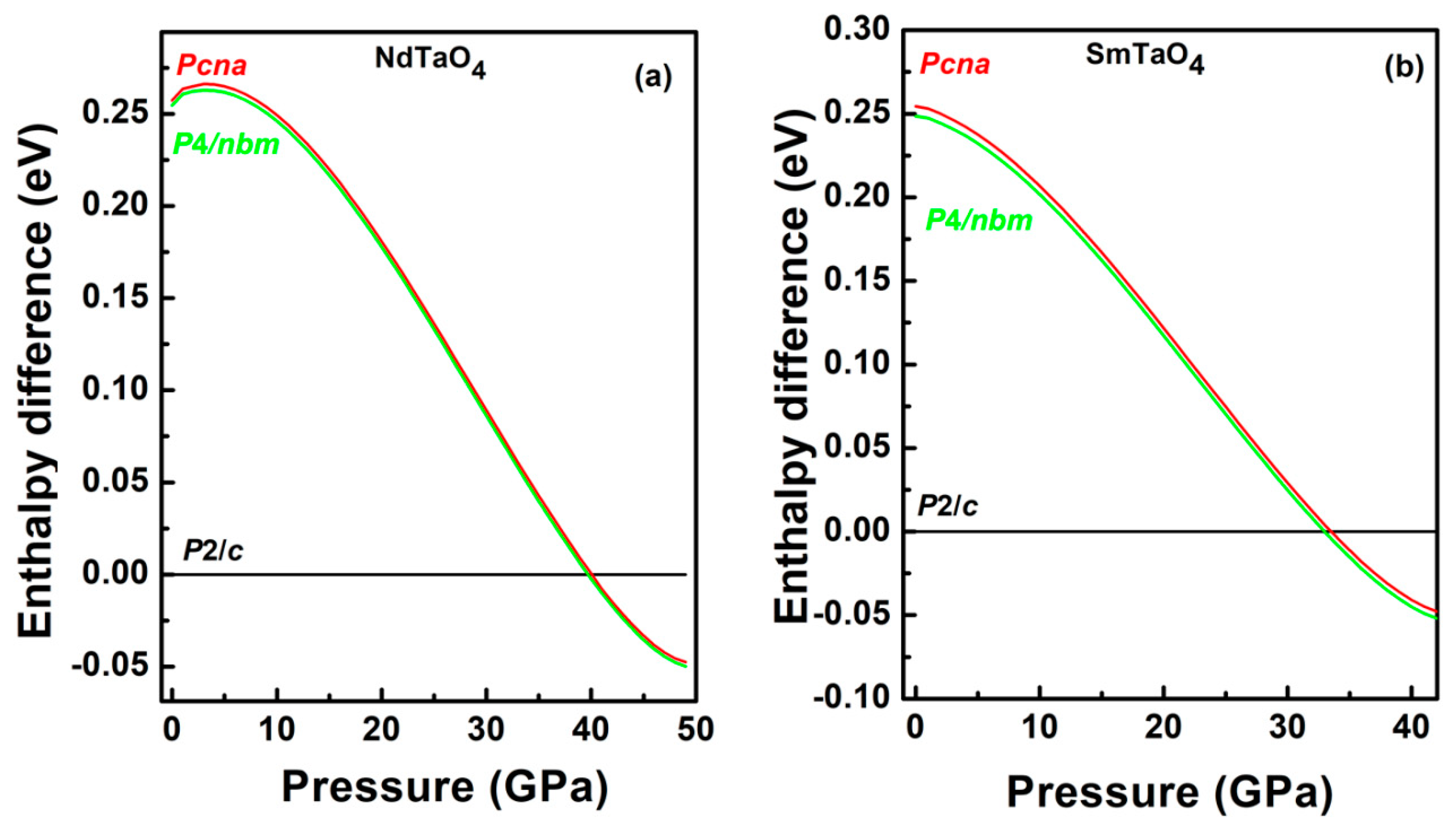
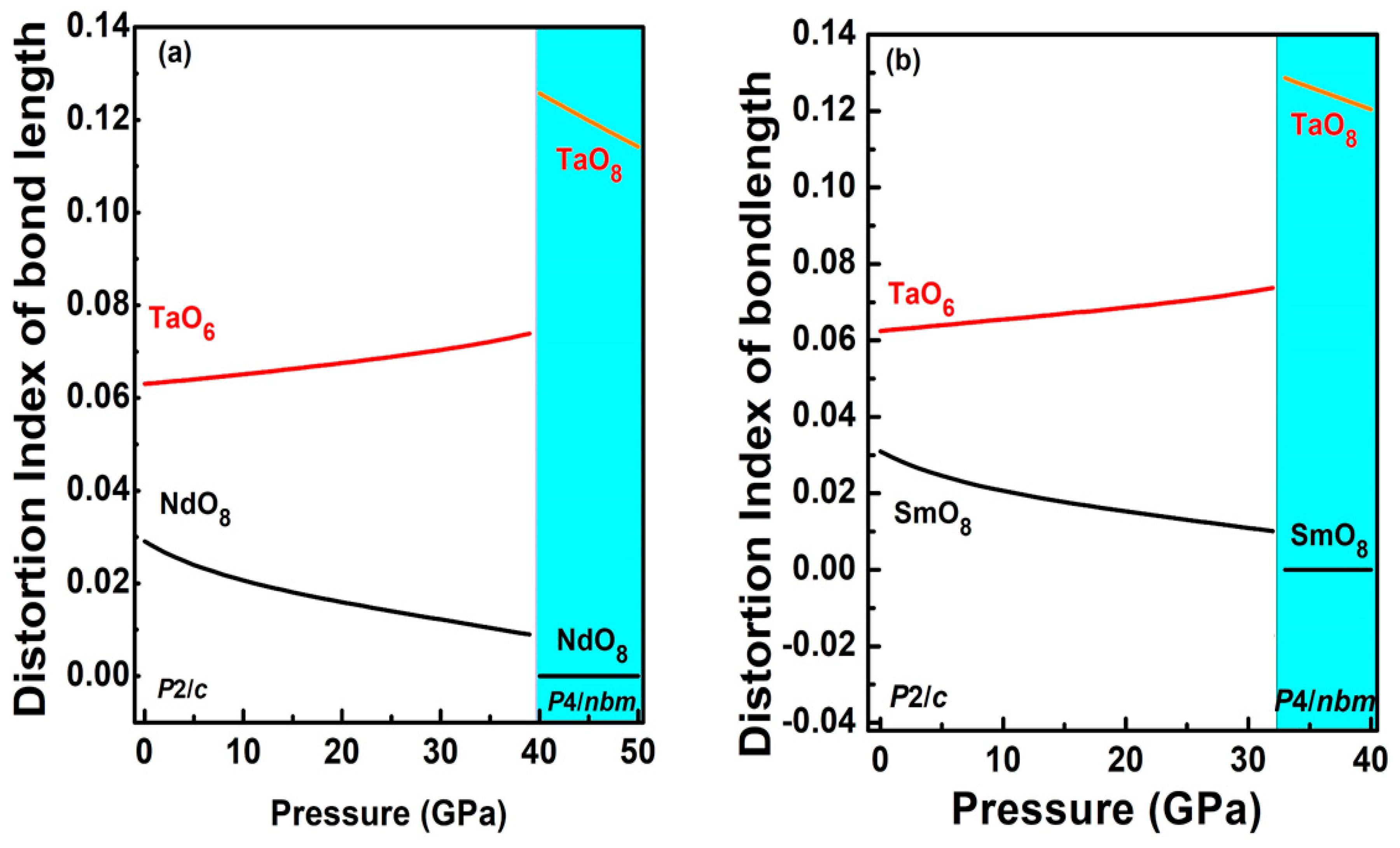
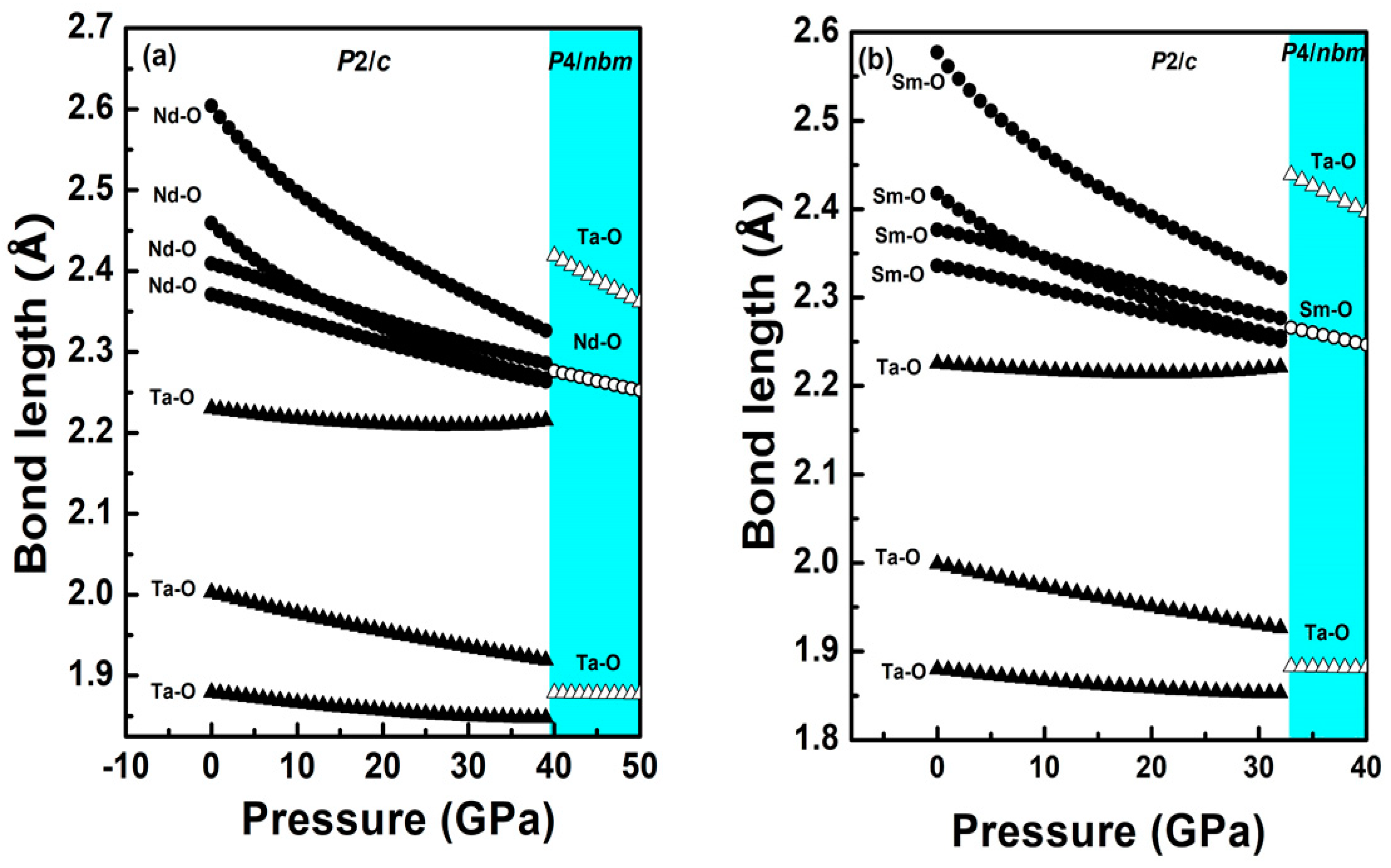
3.2.1. The Low-Pressure Phase
3.2.2. The High-Pressure Phase
3.3. Vibrational Properties under Compression
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haugsrud, R.; Norby, T. Proton conduction in rare-earth ortho-niobates and ortho-tantalates. Nat. Mater. 2006, 5, 193–196. [Google Scholar] [CrossRef]
- Yang, H.; Teng, Y.; Ren, X.; Wu, L.; Liu, H.; Wang, S.; Xu, L. Synthesis and crystalline phase of monazite-type Ce1−xGdxPO4 solid solutions for immobilization of minor actinide curium. J. Nucl. Mater. 2014, 444, 39–42. [Google Scholar] [CrossRef]
- Heuser, J.; Bukaemskiy, A.; Neumeier, S.; Neumann, A.; Bosbach, D. Raman and infrared spectroscopy of monazite-type ceramics used for nuclear waste conditioning. Prog. Nucl. Energy 2014, 72, 149–155. [Google Scholar] [CrossRef]
- Errandonea, D.; Manjon, F.J. Pressure effects on the structural and electronic properties of ABX4 scintillating crystals. Prog. Mater. Sci. 2008, 53, 711–773. [Google Scholar] [CrossRef]
- Errandonea, D.; Garg, A.B. Recent progress on the characterization of the high-pressure behaviour of AVO4 orthovanadates. Prog. Mater. Sci. 2018, 97, 123–169. [Google Scholar] [CrossRef]
- López-Moreno, S.; Rodríguez-Hernández, P.; Muñoz, A.; Romero, A.H.; Errandonea, D. First-principles calculations of elec-tronic, vibrational, and structural properties of scheelite EuWO4 under pressure. Phys. Rev. B 2011, 84, 064108. [Google Scholar] [CrossRef]
- Errandonea, D.; Santamaria-Perez, D.; Achary, S.N.; Tyagi, A.K.; Gall, P.; Gougeon, P. High-pressure x-ray diffraction study of CdMoO4 and EuMoO4. J. Appl. Phys. 2011, 109, 043510. [Google Scholar] [CrossRef]
- Siqueira, K.P.F.; Moreira, R.L.; Dias, A. Synthesis and Crystal Structure of Lanthanide Orthoniobates Studied by Vibrational Spectroscopy. Chem. Mater. 2010, 22, 2668–2674. [Google Scholar] [CrossRef]
- Lacomba-Perales, R.; Errandonea, D.; Meng, Y.; Bettinelli, M. High-pressure stability and compressibility of APO4 (A = La, Nd, Eu, Gd, Er, and Y) orthophosphates: An x-ray diffraction study using synchrotron radiation. Phys. Rev. B 2010, 81, 064113. [Google Scholar] [CrossRef]
- Errandonea, D.; Pellicer-Porres, J.; Manjon, F.J.; Segura, A.; Ferrer Roca, C.; Kumar, R.; Tschauner, O.; Lopez-Solano, J.; Ro-driguez, P.; Radescu, S.; et al. Determination of the High-Pressure Crystal Structure of BaWO4 and PbWO4. Phys. Rev. B 2006, 73, 224103. [Google Scholar] [CrossRef] [Green Version]
- Siqueira, K.P.F.; Dias, A. Effect of the processing parameters on the crystalline structure of lanthanide orthotantalates. Mater. Res. 2013, 17, 167–173. [Google Scholar] [CrossRef]
- Siqueira, K.P.F.; Carvalho, G.B.; Dias, A. Influence of the processing conditions and chemical environment on the crystal structures and phonon modes of lanthanide orthotantalates. Dalton Trans. 2011, 40, 9454–9460. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Lee, W.; Melis, A.; Elmughrabi, A.; Lee, K.; Park, C.; Yeom, J.-Y. A Review of Inorganic Scintillation Crystals for Extreme Environments. Crystals 2021, 11, 669. [Google Scholar] [CrossRef]
- Yang, H.; Peng, F.; Zhang, Q.; Guo, C.; Shi, C.; Liu, W.; Sun, G.; Zhao, Y.; Zhang, D.; Sun, D.; et al. A promising high-density scintillator of GdTaO4 single crystal. Crystengcomm 2014, 16, 2480–2485. [Google Scholar] [CrossRef]
- Wang, J.; Chong, X.; Zhou, R.; Feng, J. Microstructure and thermal properties of RETaO4 (RE = Nd, Eu, Gd, Dy, Er, Yb, Lu) as promising thermal barrier coating materials. Scr. Mater. 2017, 126, 24–28. [Google Scholar] [CrossRef]
- Banerjee, S.; Garg, A.B.; Poswal, H.K. Pressure driven structural phase transition in EuTaO4: Experimental and first principles investigations. J. Phys. Condens. Matter 2022, 34, 135401. [Google Scholar] [CrossRef]
- Banerjee, S.; Garg, A.B.; Poswal, H.K. Structural and vibrational properties of GdTaO 4 under compression: An insight from experiment and first principles simulations. J. Appl. Phys. 2023, 133, 025902. [Google Scholar] [CrossRef]
- Xiao, W.; Yang, Y.; Pi, Z.; Zhang, F. Phase Stability and Mechanical Properties of the Monoclinic, Monoclinic-Prime and Tetragonal REMO4 (M = Ta, Nb) from First-Principles Calculations. Coatings 2022, 12, 73. [Google Scholar] [CrossRef]
- Garg, A.B.; Errandonea, D.; Rodríguez-Hernández, P.; López-Moreno, S.; Muñoz, A.; Popescu, C. High-pressure structural behaviour of HoVO4: Combined XRD experiments and ab initio calculations. J. Phys. Condens. Matter 2014, 26, 265402. [Google Scholar] [CrossRef]
- Garg, A.B.; Errandonea, D. High-pressure powder x-ray diffraction study of EuVO4. J. Solid State Chem. 2015, 226, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, B.; Tan, D.; Xiao, W.; Song, M. Phase transformations of zircon-type DyVO4 at high pressures up to 36.4 GPa: X-ray diffraction measurements. J. Alloy. Compd. 2021, 875, 159926. [Google Scholar] [CrossRef]
- Garg, A.B.; Rao, M.R.; Errandonea, D.; Pellicer-Porres, J.; Martinez-Garcia, D.; Popescu, C. Pressure-induced instability of the fergusonite phase of EuNbO4 studied by in situ Raman spectroscopy, x-ray diffraction, and photoluminescence spectroscopy. J. Appl. Phys. 2020, 127, 175905. [Google Scholar] [CrossRef]
- Pellicer-Porresa, J.; Garg, A.B.; Vázquez-Socorroa, D.; Martínez-García, D.; Popescu, C.; Errandonea, D. Stability of the fer-gusonite phase in GdNbO4 by high pressure XRD and Raman experiments. J. Solid State Chem. 2017, 251, 14–18. [Google Scholar] [CrossRef]
- Garg, A.B.; Liang, A.; Errandonea, D.; Rodríguez-Hernández, P.; Muñoz, A. Monoclinic–triclinic phase transition induced by pressure in fergusonite-type YbNbO4. J. Phys. Condens. Matter 2022, 34, 174007. [Google Scholar] [CrossRef]
- Garg, A.B.; Errandonea, D.; Rodríguez-Hernández, P.; Muñoz, A. High-pressure monoclinic–monoclinic transition in fergu-sonite-type HoNbO4. J. Phys. Condens. Matter 2021, 33, 195401. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Baseggio, O.; Bonfà, P.; Brunato, D.; Car, R.; Carnimeo, I.; Cavazzoni, C.; de Gironcoli, S.; Delugas, P.; Ruffino, F.F.; et al. Quantum ESPRESSO toward the exascale. J. Chem. Phys. 2020, 152, 154105. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Corso, A.D. Pseudopotentials periodic table: From H to Pu. Comput. Mater. Sci. 2014, 95, 337–350. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Head, J.D.; Zerner, M.C. A Broyden—Fletcher—Goldfarb—Shanno optimization procedure for molecular geometries. Chem. Phys. Lett. 1985, 122, 264–270. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; Corso, A.D.; Giannozzi, P. Phonons and related crystal properties from density-functional per-turbation theory. Rev. Mod. Phys. 2001, 73, 515. [Google Scholar] [CrossRef]
- Hartenbach, I.; Lissner, F.; Nikelski, T.; Meier, S.F.; Müller-Bunz, H.; Schleid, T. About Lanthanide Oxotantalates with the Formula MTaO4 (M = La–Nd, Sm–Lu). Z. Anorg. Und Allg. Chem. 2005 631, 2377–2382.
- Birch, F. Finite Elastic Strain of Cubic Crystals. Phys. Rev. 1947, 71, 809–824. [Google Scholar] [CrossRef]
- Panchal, V.; Errandonea, D.; Manjón, F.; Muñoz, A.; Rodríguez-Hernández, P.; Achary, S.; Tyagi, A. High-pressure lattice-dynamics of NdVO. J. Phys. Chem. Solids 2017, 100, 126–133. [Google Scholar] [CrossRef]
- Garg, A.B.; Errandonea, D.; Rodríguez-Hernández, P.; Muñoz, A. ScVO4 under non-hydrostatic compression: A new meta-stable polymorph. J. Phys. Condens. Matter 2017, 29, 055401. [Google Scholar] [CrossRef] [PubMed]
- Hazen, R.M.; Finger, L.W. Bulk modulus-volume relationship for cation-anion polyhedra. J. Geophys. Res. Solid Earth 1979, 84, 6723–6728. [Google Scholar] [CrossRef]
- Baur, W.H. The geometry of polyhedral distortions. Predictive relationships for the phosphate group. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1974, 30, 1195–1215. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Errandonea, D.; Popescu, C.; Garg, A.B.; Botella, P.; Martinez-García, D.; Pellicer-Porres, J.; Rodríguez-Hernández, P.; Muñoz, A.; Cuenca-Gotor, V.; Sans, J.A. Pressure-induced phase transition and band-gap collapse in the wide-band-gap semiconductor InTaO4. Phys. Rev. B 2016, 93, 035204. [Google Scholar] [CrossRef]
- Errandonea, D.; Ruiz-Fuertes, J. A Brief Review of the Effects of Pressure on Wolframite-Type Oxides. Crystals 2018, 8, 71. [Google Scholar] [CrossRef]
- Marqueño, T.; Errandonea, D.; Pellicer-Porres, J.; Martinez-Garcia, D.; Santamaria-Pérez, D.; Muñoz, A.; Rodríguez-Hernández, P.; Mujica, A.; Radescu, S.; Achary, S.N.; et al. High-pressure polymorphs of gadolinium orthovanadate: X-ray diffraction, Raman spectroscopy, and ab initio calculations. Phys. Rev. B 2019, 100, 064106. [Google Scholar] [CrossRef]
- Ruiz-Fuertes, J.; Errandonea, D.; López-Moreno, S.; González, J.; Gomis, O.; Vilaplana, R.; Manjón, F.J.; Muñoz, A.; Rodríguez-Hernández, P.; Friedrich, A.; et al. High-pressure Raman spectroscopy and lat-tice-dynamics calculations on scintillating MgWO4: A comparison with isomorphic compounds. Phys. Rev. B 2011, 83, 214112. [Google Scholar] [CrossRef]
- Errandonea, D.; Achary, S.N.; Pellicer-Porres, J.; Tyagi, A.K. Pressure-Induced Transformations in PrVO4 and SmVO4 and Isolation of High-Pressure Metastable Phases. Inorg. Chem. 2013, 52, 5464–5469. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lattice Parameters | NdTaO4 | SmTaO4 | ||
|---|---|---|---|---|
| Experiment [32] | Calculated | Experiment [32] | Calculated | |
| a (Å) | 5.2437(4) | 5.257 | 5.2065(4) | 5.206 |
| b (Å) | 5.5969(4) | 5.638 | 5.5542(4) | 5.571 |
| c (Å) | 5.4275(4) | 5.451 | 5.3947(4) | 5.397 |
| β (degree) | 96.767(9) | 96.79 | 97.721(9) | 96.74 |
| Bond length | 2× | 2× | 2× | 2× |
| R-O2 (Å) | 2.454 | 2.459 | 2.414 | 2.4179 |
| R-O1(Å) | 2.371 | 2.371 | 2.346 | 2.33622 |
| R-O1(Å) | 2.608 | 2.603 | 2.586 | 2.57709 |
| R-O2(Å) | 2.408 | 2.409 | 2.377 | 2.37666 |
| Ta-O1(Å) | 1.864 | 1.879 | 1.874 | 1.88026 |
| Ta-O2(Å) | 2.201 | 2.230 | 2.211 | 2.22584 |
| Ta-O2(Å) | 1.996 | 2.003 | 1.990 | 1.99934 |
| NdTaO4: LP Monoclinic Phase (P2/c) @ Ambient Pressure a = 5.2504 Å; b = 5.6312 Å; c = 5.4446 Å; β = 96.79° | |||
| Nd (2e) | 0.0000 | 0.23488 | 0.2500 |
| Ta (2f) | 0.5000 | 0.31247 | 0.7500 |
| O1 (4g) | 0.74508 | 0.90645 | 0.39125 |
| O2 (4g) | 0.27392 | 0.56319 | 0.49246 |
| NdTaO4: HP Tetragonal Phase (P4/nbm) @ 40 GPa a = 5.0765 Å; c = 5.0616 Å | |||
| Nd (2b) | 0.75000 | 0.75000 | 0.500 |
| Ta (2c) | 0.75000 | 0.25 | 0.0 |
| O (8m) | 0.45494 | 0.54506 | 0.23076 |
| SmTaO4: LP Monoclinic Phase (P2/c) @ Ambient Pressure a = 5.2025 Å; b = 5.5722 Å; c = 5.3985 Å; β = 96.74° | |||
| Sm (2e) | 0.0000 | 0.23438 | 0.2500 |
| Ta (2f) | 0.5000 | 0.30982 | 0.7500 |
| O1 (4g) | 0.74726 | 0.91026 | 0.39507 |
| O2 (4g) | 0.27135 | 0.56359 | 0.49281 |
| SmTaO4: HP Tetragonal Phase (P4/nbm) @ 33 GPa a = 5.0949 Å; c = 5.0262 Å | |||
| Sm (2b) | 0.75000 | 0.75000 | 0.500 |
| Ta (2c) | 0.75000 | 0.25 | 0.0 |
| O (8m) | 0.45372 | 0.54628 | 0.23465 |
| Raman Frequency | NdTaO4 | SmTaO4 | EuTaO4 [16] | GdTaO4 [17] | ||||
|---|---|---|---|---|---|---|---|---|
| ω | dω/dP | ω | dω/dP | ω | dω/dP | ω | dω/dP | |
| Bg | 99.3 | 1.42 | 101.6 | 1.83 | 102.3 | 1.25 | 100.6 | 1.64 |
| Ag | 108.7 | 1.40 | 107.5 | 2.45 | 107.1 | 1.14 | 103.5 | 1.84 |
| Bg | 119.6 | 1.28 | 119.7 | 1.53 | 119.8 | 1.10 | 119.1 | 1.27 |
| Bg | 138.7 | 2.22 | 138.4 | 2.95 | 138.5 | 1.67 | 138.5 | 2.49 |
| Bg | 164.3 | 0.88 | 168.7 | 1.43 | 172.0 | 0.94 | 171 | 0.92 |
| Ag | 176.7 | 0.76 | 177.2 | 1.05 | 177.6 | 0.54 | 177.9 | 0.41 |
| Ag | 220.6 | 4.07 | 219 | 4.7 | 216.9 | 4.17 | 211.1 | 4.74 |
| Bg | 257.1 | 2.41 | 261.9 | 2.99 | 263.6 | 2.65 | 261.6 | 2.66 |
| Ag | 257.6 | 2.85 | 262.5 | 3.04 | 264.3 | 2.70 | 263.4 | 2.18 |
| Bg | 314.1 | 4.22 | 321.9 | 5.19 | 325.2 | 4.62 | 322 | 4.78 |
| Bg | 383.5 | 4.20 | 392.5 | 5.3 | 396.0 | 3.96 | 395.3 | 4.80 |
| Ag* | 392.6 | 1.36 | 397.1 | 1.8 | 400.3 | 1.68 | 402.6 | 1.76 |
| Bg | 466.9 | 1.7 | 475.1 | 2.3 | 480.8 | 1.88 | 486.7 | 1.53 |
| Ag* | 472.9 | 1.42 | 478.7 | 1.97 | 482.2 | 1.84 | 487.6 | 1.61 |
| Bg* | 600.3 | 3.65 | 613 | 4.11 | 620.5 | 3.93 | 626.3 | 4.29 |
| Ag* | 609.3 | 3.62 | 620.8 | 4.05 | 627.6 | 3.77 | 633.4 | 4.15 |
| Bg* | 627.4 | 4.78 | 643.4 | 4.93 | 651.1 | 4.17 | 661.7 | 4.76 |
| Ag* | 762.2 | 3.55 | 771.5 | 3.58 | 777.1 | 3.11 | 785.9 | 3.69 |
| IR Frequency | NdTaO4 @ Ambient Pressure | SmTaO4 @ Ambient Pressure | ||
|---|---|---|---|---|
| ω | dω/dP | ω | dω/dP | |
| Au | 135.5 | 2.13 | 134.3 | 2.88 |
| Bu | 138.6 | 1.58 | 137 | 2.92 |
| Bu | 156.4 | 3.85 | 155.1 | 5.03 |
| Bu | 207.5 | −1.44 | 204.2 | −1.54 |
| Au | 249.1 | Nonlinear | 253.7 | Nonlinear |
| Bu | 257.2 | 2.24 | 257.2 | 2.97 |
| Bu | 275.8 | 2.24 | 281.9 | 2.39 |
| Au | 311.1 | 0.87 | 310.8 | 1.53 |
| Au | 354.7 | 3.33 | 359.4 | 3.81 |
| Bu | 382 | 3.26 | 387.3 | 3.59 |
| Bu | 480.7 | 4.12 | 491.5 | 4.65 |
| Au | 503.6 | 3.74 | 512.4 | 4.23 |
| Au | 561.3 | 4.33 | 572.8 | 4.68 |
| Bu | 607.5 | 4.23 | 616.7 | 4.66 |
| Au | 743.2 | 3.77 | 752.2 | 3.96 |
| Raman Frequency | NdTaO4(HP) @ 40 GPa | SmTaO4(HP) @ 33 GPa | ||
|---|---|---|---|---|
| ω | dω/dP | ω | dω/dP | |
| Eg | 103.1 | Nonlinear | 99.6 | Nonlinear |
| B2g | 163.6 | Nonlinear | 159.4 | Nonlinear |
| Eg | 208.7 | 1.1 | 199.9 | 1.2 |
| B1g | 357.5 | 0.92 | 343.9 | 1.26 |
| Eg | 412.3 | 1.93 | 397.2 | 2.16 |
| A1g | 412.9 | Nonlinear | 408.7 | Nonlinear |
| B2g | 513 | 2.77 | 501 | 2.73 |
| Eg | 546.6 | 2.9 | 533.6 | 3.09 |
| Eg | 741.8 | 1.55 | 738.4 | 1.85 |
| B2g | 750 | 1.85 | 743.8 | 2.09 |
| A1g | 823.7 | 1.12 | 815.7 | 1.25 |
| IR Frequency | NdTaO4(HP)40 GPa | SmTaO4(HP)33 GPa | ||
|---|---|---|---|---|
| ω | dω/dP | ω | dω/dP | |
| A2u | 109 | Nonlinear | 110.6 | Nonlinear |
| Eu | 136.2 | 1.92 | 126.2 | 2.24 |
| Eu | 183.2 | 1.36 | 183.8 | 1.33 |
| A2u | 281.2 | 1.80 | 264.1 | 1.89 |
| Eu | 509.3 | 2.26 | 494.6 | 2.52 |
| A2u | 669.1 | 0.62 | 659.6 | 0.85 |
| Eu | 699.9 | 1.53 | 687.7 | 1.72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, S.; Tyagi, A.; Garg, A.B. Pressure-Induced Monoclinic to Tetragonal Phase Transition in RTaO4 (R = Nd, Sm): DFT-Based First Principles Studies. Crystals 2023, 13, 254. https://doi.org/10.3390/cryst13020254
Banerjee S, Tyagi A, Garg AB. Pressure-Induced Monoclinic to Tetragonal Phase Transition in RTaO4 (R = Nd, Sm): DFT-Based First Principles Studies. Crystals. 2023; 13(2):254. https://doi.org/10.3390/cryst13020254
Chicago/Turabian StyleBanerjee, Saheli, Amit Tyagi, and Alka B. Garg. 2023. "Pressure-Induced Monoclinic to Tetragonal Phase Transition in RTaO4 (R = Nd, Sm): DFT-Based First Principles Studies" Crystals 13, no. 2: 254. https://doi.org/10.3390/cryst13020254
APA StyleBanerjee, S., Tyagi, A., & Garg, A. B. (2023). Pressure-Induced Monoclinic to Tetragonal Phase Transition in RTaO4 (R = Nd, Sm): DFT-Based First Principles Studies. Crystals, 13(2), 254. https://doi.org/10.3390/cryst13020254