
Screening of Complex Layered Chalcogenide Structures as High-Performance Thermoelectrics by High-Throughput Calculations

 , and
, and
Abstract
:1. Introduction
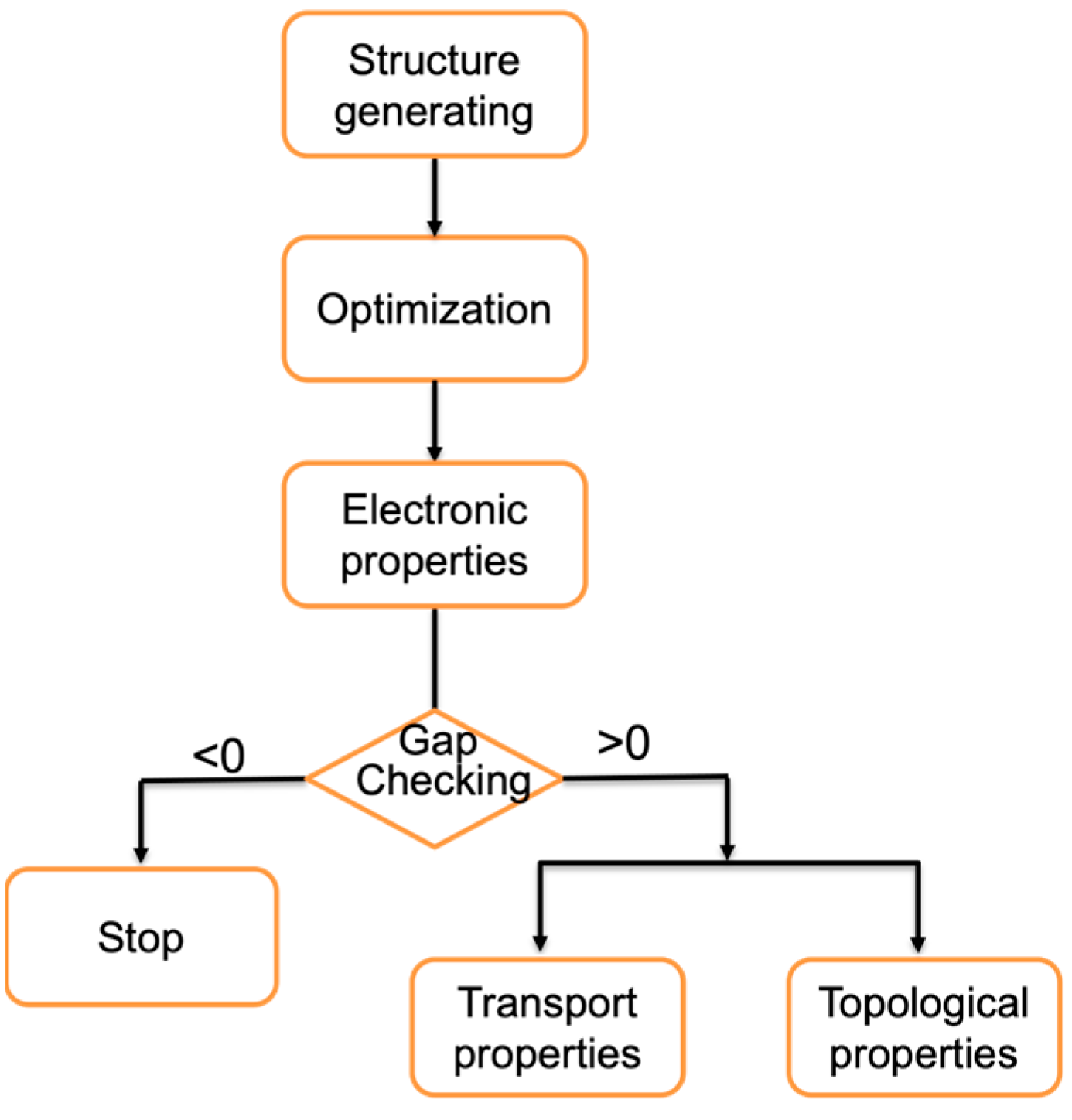
2. Computational Methods
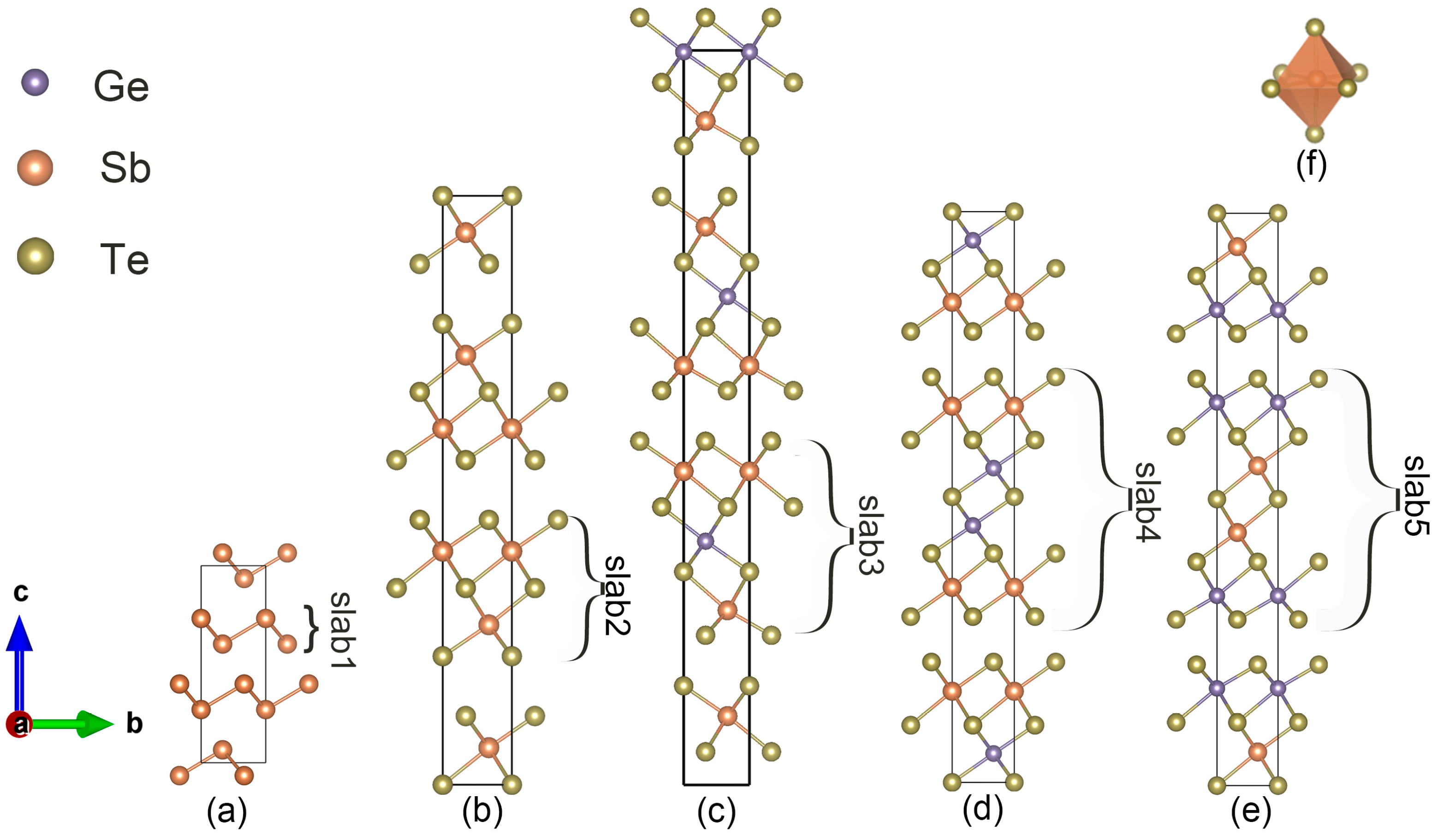
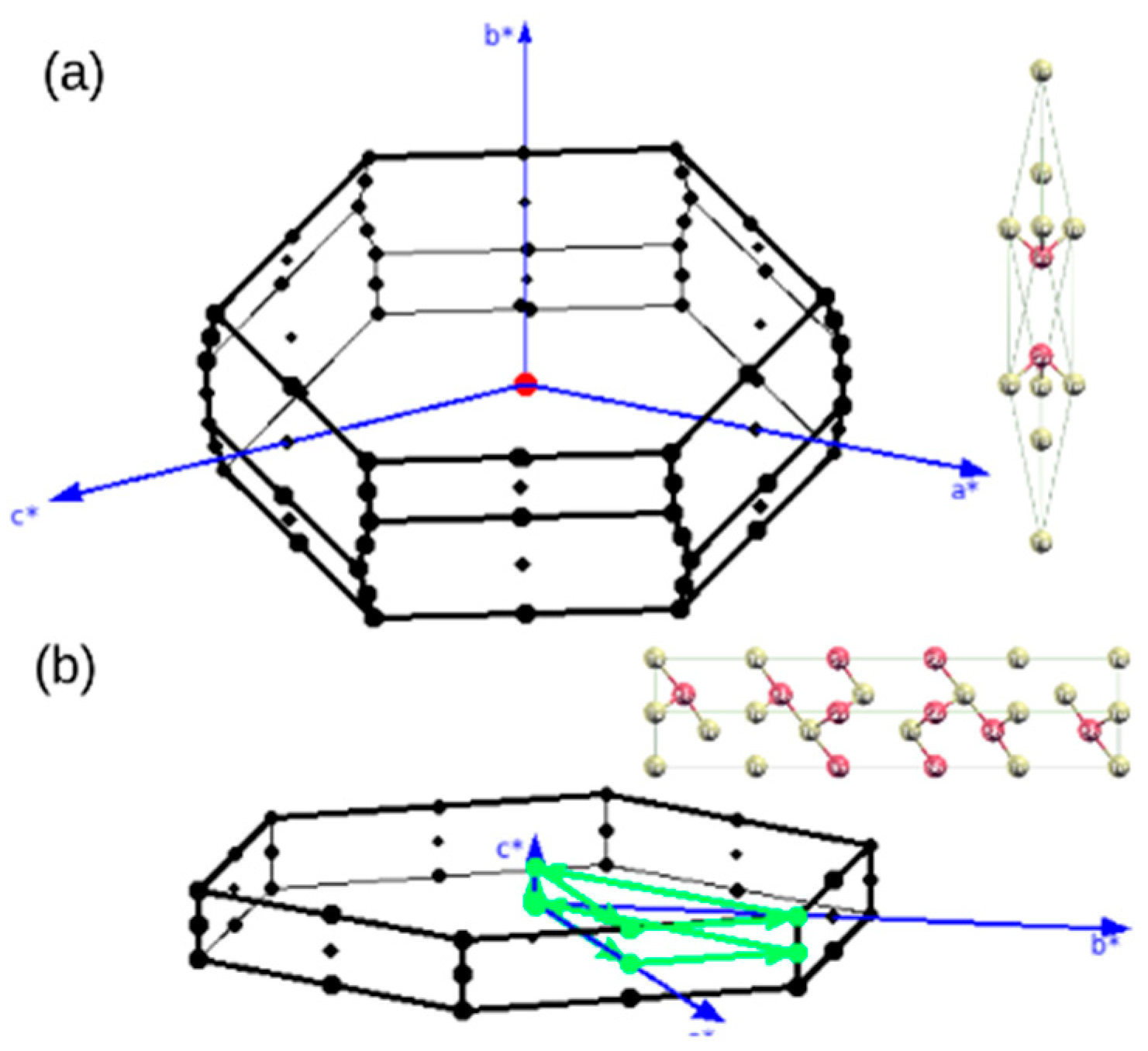
3. Structure Generation
4. Results
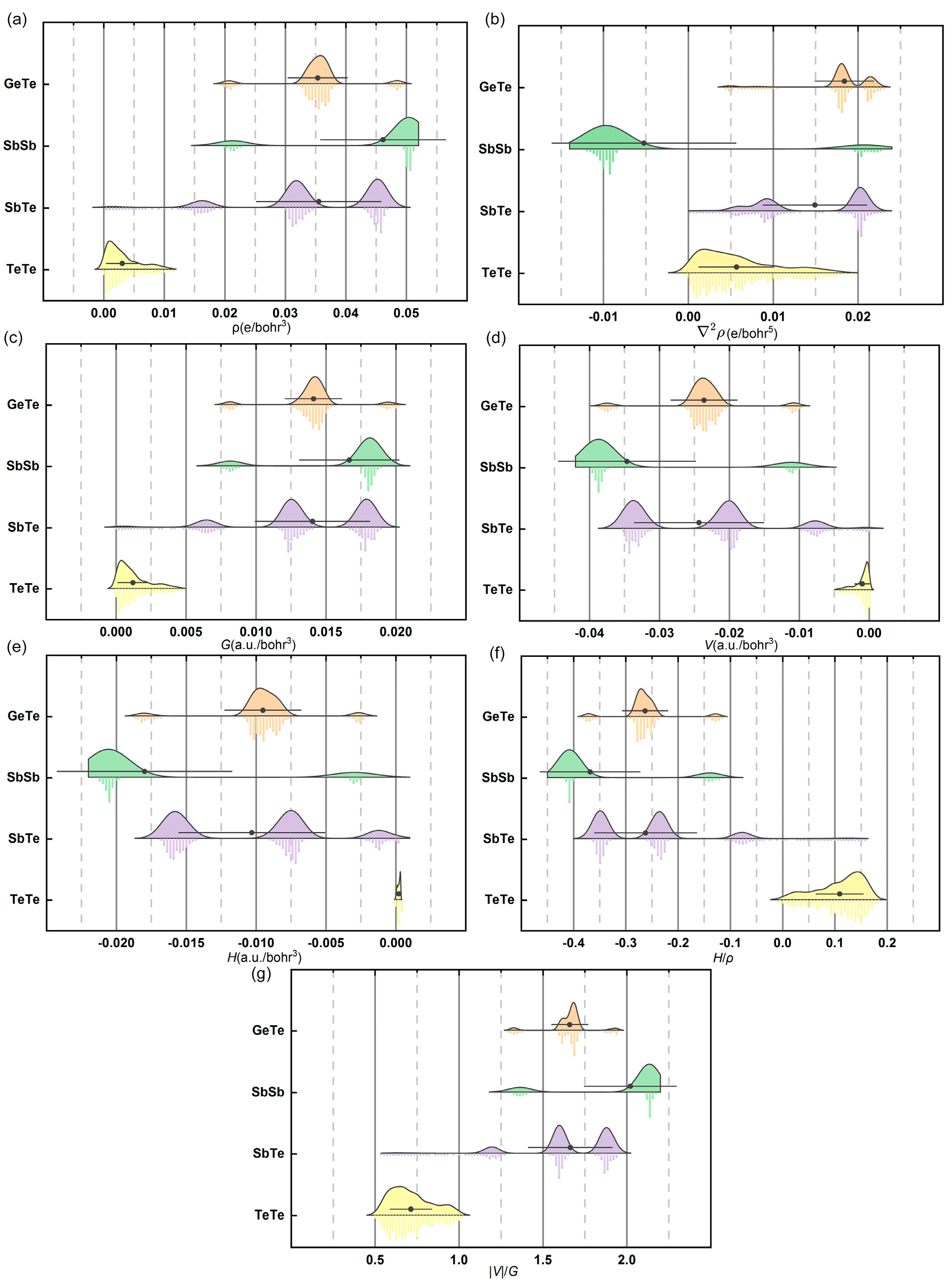
4.1. Electron Density Topology
4.2. Structure Stability
4.3. Electronic Properties
4.4. Transport Properties
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luo, Y.; Yang, J.; Jiang, Q.; Li, W.; Zhang, D.; Zhou, Z.; Cheng, Y.; Ren, Y.; He, X. Progressive Regulation of Electrical and Thermal Transport Properties to High-Performance CuInTe2 Thermoelectric Materials. Adv. Energy Mater. 2016, 6, 1600007. [Google Scholar] [CrossRef]
- Kumar, A.; Chaturvedi, K.M.; Bano, S.; Govind, B.; Misra, D.K. Enhanced thermoelectric performance of p-type ZrCoSb0.9Sn0.1 via Tellurium doping. Mater. Chem. Phys. 2021, 258, 123915. [Google Scholar] [CrossRef]
- Rausch, E.; Balke, B.; Deschauer, T.; Ouardi, S.; Felser, C. Charge carrier concentration optimization of thermoelectric p-type half-Heusler compounds. APL Mater. 2015, 3, 41516. [Google Scholar] [CrossRef]
- Wei, Z.; Li, Z.; Luo, P.; Zhang, J.; Luo, J. Simultaneously increased carrier concentration and mobility in p-type Bi0.5Sb1.5Te3 throng Cd doping. J. Alloy. Compd. 2020, 830, 154625. [Google Scholar] [CrossRef]
- Kumar, A.; Bano, S.; Govind, B.; Bhardwaj, A.; Singh, V.N. Enhanced thermoelectric performance of n-type Zr0.66Hf0.34Ni1+xSn Heusler nanocomposites. J. Alloy. Compd. 2022, 900, 163454. [Google Scholar] [CrossRef]
- Yu, B.; Zebarjadi, M.; Wang, H.; Lukas, K.; Wang, H.; Wang, D.; Opeil, C.; Dresselhaus, M.; Chen, G.; Ren, Z. Enhancement of Thermoelectric Properties by Modulation-Doping in Silicon Germanium Alloy Nanocomposites. Nano Lett. 2012, 12, 2077–2082. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhang, B.; Zhang, X.; Wang, S.; Lu, X.; Han, G.; Wang, G.; Zhou, X. Enhancing the Thermoelectric Performance of p-Type Mg3Sb2 via Codoping of Li and Cd. ACS Appl. Mater. Interfaces 2020, 12, 8359–8365. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Shi, X.; LaLonde, A.; Wang, H.; Chen, L.; Snyder, G.J. Convergence of electronic bands for high performance bulk thermoelectrics. Nature 2011, 473, 66–69. [Google Scholar] [CrossRef]
- Balout, H.; Boulet, P.; Record, M.-C. Strain-induced electronic band convergence: Effect on the Seebeck coefficient of Mg2Si for thermoelectric applications. J. Mol. Model. 2017, 23, 130. [Google Scholar] [CrossRef]
- Tian, J.; Ma, W.; Record, M.-C.; Boulet, P. High Thermoelectric Performance of Ge-Sb-Te Nanosheets: A Density Functional Study. J. Electron. Mater. 2024. Submitted. [Google Scholar]
- Yu, J.; Fu, C.; Liu, Y.; Xia, K.; Aydemir, U.; Chasapis, T.C.; Snyder, G.J.; Zhao, X.; Zhu, T. Unique role of refractory Ta alloying in enhancing the figure of merit of NbFeSb thermoelectric materials. Adv. Energy Mater. 2018, 8, 1701313. [Google Scholar] [CrossRef]
- Fu, T.; Xin, J.; Zhu, T.; Shen, J.; Fang, T.; Zhao, X. Approaching the minimum lattice thermal conductivity of p-type SnTe thermoelectric materials by Sb and Mg alloying. Sci. Bull. 2019, 14, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, Y.; Yang, J.; Duan, B.; Wu, L.; Zhang, W.; Yang, J. High Thermoelectric Performance in Te-free (Bi,Sb)2Se3 via Structural Transition Induced Band Convergence and Chemical Bond Softening. Energy Environ. Sci. 2016, 9, 3436–3447. [Google Scholar] [CrossRef]
- Ying, P.; Li, X.; Wang, Y.; Yang, J.; Fu, C.; Zhang, W.; Zhao, X.; Zhu, T. Hierarchical Chemical Bonds Contributing to the Intrinsically Low Thermal Conductivity in α-MgAgSb Thermoelectric Materials. Adv. Funct. Mater. 2017, 27, 1604145. [Google Scholar] [CrossRef]
- Li, W.; Lin, S.; Ge, B.; Yang, J.; Zhang, W.; Pei, Y. Low Sound Velocity Contributing to the High Thermoelectric Performance of Ag8SnSe6. Adv. Sci. 2016, 3, 1600196. [Google Scholar] [CrossRef] [PubMed]
- Charoenphakdee, A.; Kurosaki, K.; Muta, H.; Uno, M.; Yamanaka, S. Ag8SiTe6: A New Thermoelectric Material with Low Thermal Conductivity. Jpn. J. Appl. Phys. 2009, 48, 011603. [Google Scholar] [CrossRef]
- Zhu, T.J.; Zhang, S.N.; Yang, S.H.; Zhao, X.B. Improved Thermoelectric Figure of Merit of Self-Doped Ag8−xGeTe6 Compounds with Glass-like Thermal Conductivity. Phys. Status Solidi RRL 2010, 4, 317–319. [Google Scholar] [CrossRef]
- Zhao, L.-D.; Lo, S.-H.; Zhang, Y.; Sun, H.; Tan, G.; Uher, C.; Wolverton, C.; Dravid, V.P.; Kanatzidis, M.G. Ultralow Thermal Conductivity and High Thermoelectric Figure of Merit in SnSe Crystals. Nature 2014, 508, 373–377. [Google Scholar] [CrossRef]
- Curtarolo, S.; Hart, G.L.W.; Nardelli, M.B.; Mingo, N.; Sanvito, S.; Levy, O. The High-Throughput Highway to Computational Materials Design. Nat. Mater. 2013, 12, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Oganov, A.R. Discovery of High Performance Thermoelectric Chalcogenides through First-Principles High-Throughput Screening. J. Mater. Chem. C 2021, 9, 13226–13235. [Google Scholar] [CrossRef]
- Xu, G.; Xin, J.; Deng, H.; Shi, R.; Zhang, G.; Zou, P. High-Throughput Screening of High- Performance Thermoelectric Materials with Gibbs Free Energy and Electronegativity. Materials 2023, 16, 5399. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, Z.; Setyawan, W.; Mingo, N.; Curtarolo, S. Assessing the Thermoelectric Properties of Sintered Compounds via High-Throughput Ab-Initio Calculations. Phys. Rev. X 2011, 1, 021012. [Google Scholar]
- Yang, J.; Li, H.; Wu, T.; Zhang, W.; Chen, L.; Yang, J. Evaluation of Half-Heusler Compounds as Thermoelectric Materials Based on the Calculated Electrical Transport Properties. Adv. Funct. Mater. 2008, 18, 2880–2888. [Google Scholar] [CrossRef]
- Carrete, J.; Mingo, N.; Wang, S.; Curtarolo, S. Nanograined Half-Heusler Semiconductors as Advanced Thermoelectrics: An Ab Initio High-Throughput Statistical Study. Adv. Funct. Mater. 2014, 24, 7427–7432. [Google Scholar] [CrossRef]
- Gan, Y.; Wang, G.; Zhou, J.; Sun, Z.; Qiu, D.; Singh, D.J.; Xi, J.; Yang, J.; Xi, L. Prediction of Thermoelectric Performance for Layered IV-V-VI Semiconductors by High-Throughput Ab Initio Calculations and Machine Learning. npj Comput. Mater. 2021, 7, 176. [Google Scholar] [CrossRef]
- Jin, Y.; Wang, X.; Yao, M. High-Throughput Deformation Potential and Electrical Transport Calculations. npj Comput. Mater. 2023, 9, 190. [Google Scholar] [CrossRef]
- Kanatzidis, M.G. The Role of Solid-State Chemistry in the Discovery of New Thermoelectric Materials. Semicond. Semimet. 2001, 69, 51–100. [Google Scholar]
- Harker, D. The Crystal Structure of the Mineral Tetradymite, Bi2Te2S. Z. Für Krist.-Cryst. Mater. 1934, 89, 175–181. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W.; Beddall, P.M.; Cade, P.E. Partitioning and Characterization of Molecular Charge Distributions. J. Am. Chem. Soc. 1971, 93, 3095–3107. [Google Scholar] [CrossRef]
- Otero-de-la-Roza, A.; Blanco, M.A.; Pendaás, A.M.; Luaña, V. Critic: A New Program for the Topological Analysis of Solid-State Electron Densities. Comput. Phys. Commun. 2009, 180, 157–166. [Google Scholar] [CrossRef]
- Madsen, G.K.H.; Carrete, J.; Verstraete, M.J. BoltzTraP2, a Program for Interpolating Band Structures and Calculating Semi-Classical Transport Coefficients. Comput. Phys. Commun. 2018, 231, 140–145. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From Weak to Strong Interactions: A Comprehensive Analysis of the Topological and Energetic Properties of the Electron Density Distribution Involving X–H⋯F–Y Systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Harrison, W.A. Coulomb Interactions in Semiconductors and Insulators. Phys. Rev. B 1985, 31, 2121–2132. [Google Scholar] [CrossRef]
- Stevanović, V.; Lany, S.; Zhang, X.; Zunger, A. Correcting Density Functional Theory for Accurate Predictions of Compound Enthalpies of Formation: Fitted Elemental-Phase Reference Energies. Phys. Rev. B 2012, 85, 115104. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | 31 | 222 | 333 | 4455 | 54432 | 313452 |
| a | 4.34 | 4.32 | 4.30 | 4.28 | 4.29 | 4.30 |
| c | 17.40 | 31.94 | 42.84 | 75.27 | 81.47 | 81.94 |
| Formation energy | −0.32 | −0.27 | −0.28 | −0.31 | −0.30 | −0.30 |
| Cohesive energy | −2.80 | −2.70 | −2.84 | −2.91 | −2.87 | −2.84 |
| Compounds | CBM-VBM (eV) | CBM-VBM Type | Compounds | CBM-VBM (eV) | CBM-VBM Type |
|---|---|---|---|---|---|
| 4 | 0.6553 | Indirect | 32 | 0.4727 | Direct |
| 34334 | 0.6457 | Indirect | 34232 | 0.4548 | Direct |
| 43334 | 0.6424 | Indirect | 432234 | 0.4441 | Direct |
| 3433 | 0.6242 | Indirect | 23342 | 0.4372 | Direct |
| 2434 | 0.5899 | Indirect | 42242 | 0.4364 | Indirect |
| 44423 | 0.5899 | Indirect | 23432 | 0.4246 | Indirect |
| 3443443 | 0.5833 | Indirect | 232233 | 0.4243 | Direct |
| 342 | 0.5755 | Indirect | 422424 | 0.4235 | Indirect |
| 432 | 0.5749 | Indirect | 3223 | 0.4166 | Direct |
| 333432 | 0.5748 | Direct | 4222 | 0.3872 | Direct |
| 44243 | 0.5747 | Indirect | 233223 | 0.3855 | Direct |
| 424233 | 0.5707 | Indirect | 242232 | 0.3752 | Direct |
| 4423 | 0.5574 | Indirect | 224232 | 0.3732 | Direct |
| 333423 | 0.5566 | Indirect | 422232 | 0.3717 | Direct |
| 333 | 0.5526 | Indirect | 222432 | 0.3411 | Direct |
| 434232 | 0.5429 | Indirect | 22244 | 0.3406 | Direct |
| 32324 | 0.5359 | Direct | 22232 | 0.3199 | Direct |
| 24233 | 0.5294 | Indirect | 333222 | 0.3156 | Direct |
| 33233 | 0.5251 | Direct | 2323222 | 0.2964 | Direct |
| 433323 | 0.5083 | Direct | 222 | 0.2922 | Direct |
| 2442442 | 0.4940 | Direct | 342222 | 0.2882 | Indirect |
| 4332334 | 0.4841 | Direct |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, J.; Ma, W.; Carenzi, M.; Boulet, P.; Record, M.-C. Screening of Complex Layered Chalcogenide Structures as High-Performance Thermoelectrics by High-Throughput Calculations. Crystals 2024, 14, 403. https://doi.org/10.3390/cryst14050403
Tian J, Ma W, Carenzi M, Boulet P, Record M-C. Screening of Complex Layered Chalcogenide Structures as High-Performance Thermoelectrics by High-Throughput Calculations. Crystals. 2024; 14(5):403. https://doi.org/10.3390/cryst14050403
Chicago/Turabian StyleTian, Jing, Weiliang Ma, Manuela Carenzi, Pascal Boulet, and Marie-Christine Record. 2024. "Screening of Complex Layered Chalcogenide Structures as High-Performance Thermoelectrics by High-Throughput Calculations" Crystals 14, no. 5: 403. https://doi.org/10.3390/cryst14050403
APA StyleTian, J., Ma, W., Carenzi, M., Boulet, P., & Record, M. -C. (2024). Screening of Complex Layered Chalcogenide Structures as High-Performance Thermoelectrics by High-Throughput Calculations. Crystals, 14(5), 403. https://doi.org/10.3390/cryst14050403