1. Introduction
Zeolites are crystalline, microporous aluminosilicates from interlinked tetrahedra of alumina (AlO
4) and silica (SiO
4) with a relatively open, three-dimensional crystal structure, cavities and channels of molecular dimensions in the different directions. The tetrahedra are linked together to form cages connected by pore openings of defined size. Depending on the structural type, the pore sizes range from approximately 0.3 to 1.0 nm. The presence of aluminium atoms in the framework induces an electrical imbalance leading to a negatively charged framework that is compensated by additional non-framework cations, such as calcium, sodium or potassium. Due to their exceptional properties, zeolites have been widely-used in numerous technical applications as catalysts, adsorbents and ion exchangers [
1,
2]. Zeolite A with LTA structure and lowest Si/Al ratio of 1.0 is one of the best known, most important and widely-used industrial zeolites. It has a three-dimensional pore system with larger cavity of minimum free diameter 1.14 nm and much smaller 8-ring windows of approximately 0.4 nm for NaA and 0.5 nm for CaA. The huge amounts of zeolite A are produced every year for applications as diverse as water softening in detergents; as an additive in polyvinyl chloride (PVC) thermoplastics; industrial gas drying; separation of linear and branched hydrocarbons, etc. Besides, the catalytic properties of zeolite A were also paid attention. Selective dehydration of n-butanol in the presence of
i-butanol and selective cracking of n-alkanes in the presence of branched alkanes over the CaA were early examples for the applications of zeolite LTA in catalytic reaction [
3,
4]. The high selectivity over CaA is attributed to the fact that only
n-butanol and n-alkanes can penetrate into the pore system of the zeolite, while branched reactants cannot. Because of this reason, branched products are essentially absent in products. Moreover, the mechanism and product distribution for catalytic cracking of n-octane on small-pore CaA zeolite were investigated; high selectivity to C
1 and C
2 products were shown because of the influence of the pore dimensions [
5], and catalytic combustion of hexane over zeolite CaA was studied [
6].
Like other zeolites, most of the active centers in zeolites A with small windows are located in the inner cavities. The presence of active sites in the zeolite micropores gives rise to shape-selective catalysis, but imposes steric limitations to the diffusion of the reactant molecular to the interior of the pores and reduces the diffusion rate of the reaction products outside the inner cavities, rendering a large portion of the actual zeolite crystals ineffective and the occurrence of undesirable second reaction. It is well-known that the incorporation of additional meso(macro)pores in zeolite can enhance the accessibility to the active sites, overcoming the steric limitations and shortening diffusion pathways. Hence, as the optimal solution, the synthesis of hierarchically structured zeolitic materials with two or three levels of porosities in zeolite has obtained intensive attention [
7,
8,
9,
10,
11,
12,
13,
14,
15]. Compared with other zeolites, there are fewer reports on the synthesis of hierarchically structured zeolites LTA, either by using organosilane surfactants such as 3-(trimethoxysilyl)propyl hexadecyl dimethyl ammonium chloride (TPHAC) as a soft-template [
16], by amino acid as a mesopore generating agent [
17], or by using organofunctionalized silica as an Si-source, which was reported by our research group [
18]. The enhancement effect of mesopores in zeolite LTA on mass transport has been proved. For example, Xenon uptake revealed that the xenon diffusion into the highly mesoporous LTA zeolite could occur 200 times more rapidly compared with that of a solely microporous LTA, and therefore two zeolites show a dramatic difference in product selectivity, catalytic activity, and lifetime. Moreover, it is also known that the diffusion rate of hydrated Mg
2+ and n-alkanes [
19] increased with mesoporosity in zeolite 5A. In addition, LTA zeolites exhibited a remarkably higher adsorption capacity towards large biomolecules, and retained greater enzyme activity [
17].
Dimethyl ether (DME), as a new synthetic fuel commodity, as well as a momentous chemical intermediate, is a potential alternative for the energy and fuels of the future. Therefore, there is a growing demand to produce a large amount of DME in the near future to meet the global requirements. In general, DME can be produced by methanol dehydration over solid porous acid catalysts, such as most typical catalysts H-ZSM-5 zeolite and γ-Al
2O
3. Although the DME selectivity is high for methanol dehydration on γ-Al
2O
3 [
20,
21], the γ-Al
2O
3 exhibits much lower activity due to its weak Lewis acidity [
21]. On the contrary, H-ZSM-5 has high activity, but it also has some disadvantages, such as low selectivity and short catalytic lifetime. One of the reasons for this is that the existence of strong acidic sites on the zeolite surface, such as high acidity, can result in the significant production of undesirable hydrocarbons byproducts, coke formation and consequently fast deactivation [
22,
23]. The second reason arises from the small microporous channel of zeolite, in which dehydration catalytic reaction occurs. Because the size of the reacting molecules and the micropore diameter are similar, methanol and DME cannot diffuse quickly enough in the narrow and slender micropores. This consequence severely limits the activity, and causes secondary reactions to produce hydrocarbons and carbon deposits in the catalytic process (i.e., coke deposition) [
24]. Therefore, it would be desirable to develop a solid acid zeolite catalyst with the proper acidity and favorable pore structure for mass transfer.
In the present work, we report the preparation of hierarchical zeolite CaA single crystals with micro-, meso- and macroporous structure by using organo-functionalized mesoporous silica (MS) as silica sources in a one-pot hydrothermal system, which is based on bond-blocking principles of organic moiety linked on the surface of the silica through Si–C covalent bonds established by our research group [
25,
26]. The resultant CaA zeolites are characterized by XRD, SEM/TEM, N
2 adsorption/desorption and NH
3-TPD techniques to investigate the influence of silanization degree over the MS surface on the crystalline and pore structure as well as acidity of synthesized samples. Moreover, the actual performances of these materials are assessed by methanol dehydration to DME, to further prove the existence of hierarchical pore structure and to expand their potential application as a solid acid catalyst.
2. Results and Discussion
The X-ray powder diffraction (XRD) patterns of microporous NaA (MP-NaA) and hierarchical NaA zeolite samples (assigned as HP-NaA-1, HP-NaA-2 and HP-NaA-3), which are precursors of CaA zeolites, are shown in
Figure 1. All synthesized samples exhibit the characteristic diffraction reflections located at 2θ value of 7.1°, 10.1° and 12.4°, 21.6°, 24.0°, 29.9°, and 34.1°, which are exclusively ascribed to the NaA zeolite structure of a LTA topology. A view of the 2θ diffraction range of 5°–35° of these samples shows that there is no evidence of the formation of other zeolites or amorphous silica in the synthesized NaA zeolites. Compared with the zeolite MP-NaA, obtained from unsilanized Mesoporous Silica (MS), the intensity of the diffraction peaks of HP-NaA-1, HP-NaA-2 and HP-NaA-3 show the trend of consecutive decrease, which indicates that the introduction of an organo-functional group on the surface of silica source MS slightly decreases the crystallinity of CaA zeolites with the increase of silanization degree used in zeolite synthesis. However, HP-NaA-1,2,3 samples exhibit no distinct broadening of the peaks, suggesting that there is no decrease in the crystal sizes and formation of NaA aggregates of nanocrystals for hierarchical HP-NaA-1,2,3 samples, unlike surfactant mediated mesoporous LTA zeolites [
16]. After Ca
2+ exchange, the obtained CaA samples with different ion exchange degrees of 65.8%, 72.3%, 76.5% and 78.6% (the corresponding molar ratios of Ca/Al and Na/Al are 0.33, 0.36, 0.38, 0.39 and 0.34, 0.28, 0.24, 0.21) for MP-CaA and HP-CaA-1, -2, -3, respectively, persist their crystalline structure of LTA, except from the change of relative strength of peaks, for example I
12.4/I
10.2.
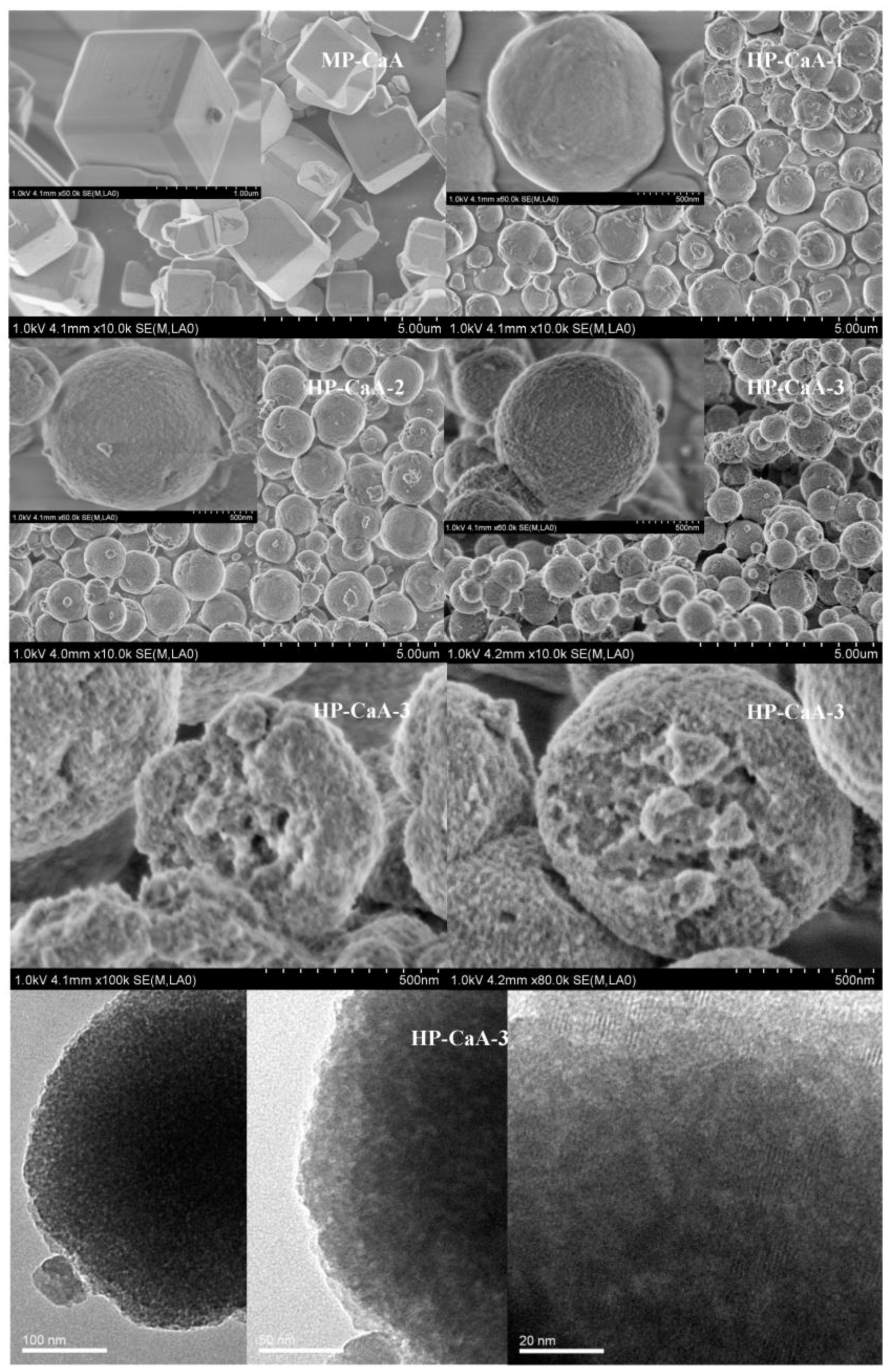
The evolution of the morphology of LTA zeolites synthesized by using mesoporous silica with different silanization degrees as silica sources is clearly demonstrated in the SEM images (
Figure 2). The conventional MP-CaA zeolite exhibits a typical crystalline LTA zeolite shape of well-defined cubic morphology with truncated edges and a smooth surface. The size of particles is around 1.5–2 μm. The application of organo-functionalized mesoporous silica into the synthesis indeed has an obvious effect on the morphology of LTA zeolite, which suggests that the presence of organic moiety affects the crystal growth process. All obtained HP-CaA-1,2,3 samples exhibit uniform spherical particle morphology with an average size of 1–2 μm. Different from the smooth surfaces of microporous MP-CaA, HP-CaA-1, 2, 3 samples exhibit obviously rugged surfaces, and an increasing roughness of particle surface is observed with an increasing silanization degree on the MS surface, in which the mesopores and/or macropores within the particles can be obviously found. The most typical sample is HP-CaA-3; there exists a large number of mesopores and macropores in the interior of the entire zeolite particles. The presence of the mesopores in the synthesized HP-CaA zeolite is also directly identified from the TEM investigation (
Figure 2). From the TEM of the HP-CaA-3 sample, both the abundant worm-like mesopores of 3 to 5 nm and the ordered micropores of around 0.5 nm related to zeolite CaA are visible in zeolite crystals (at the bottom of
Figure 2), indicating the presence of hierarchical intracrystalline mesoporous and the interconnectivity of these micro-, meso- and macropores.
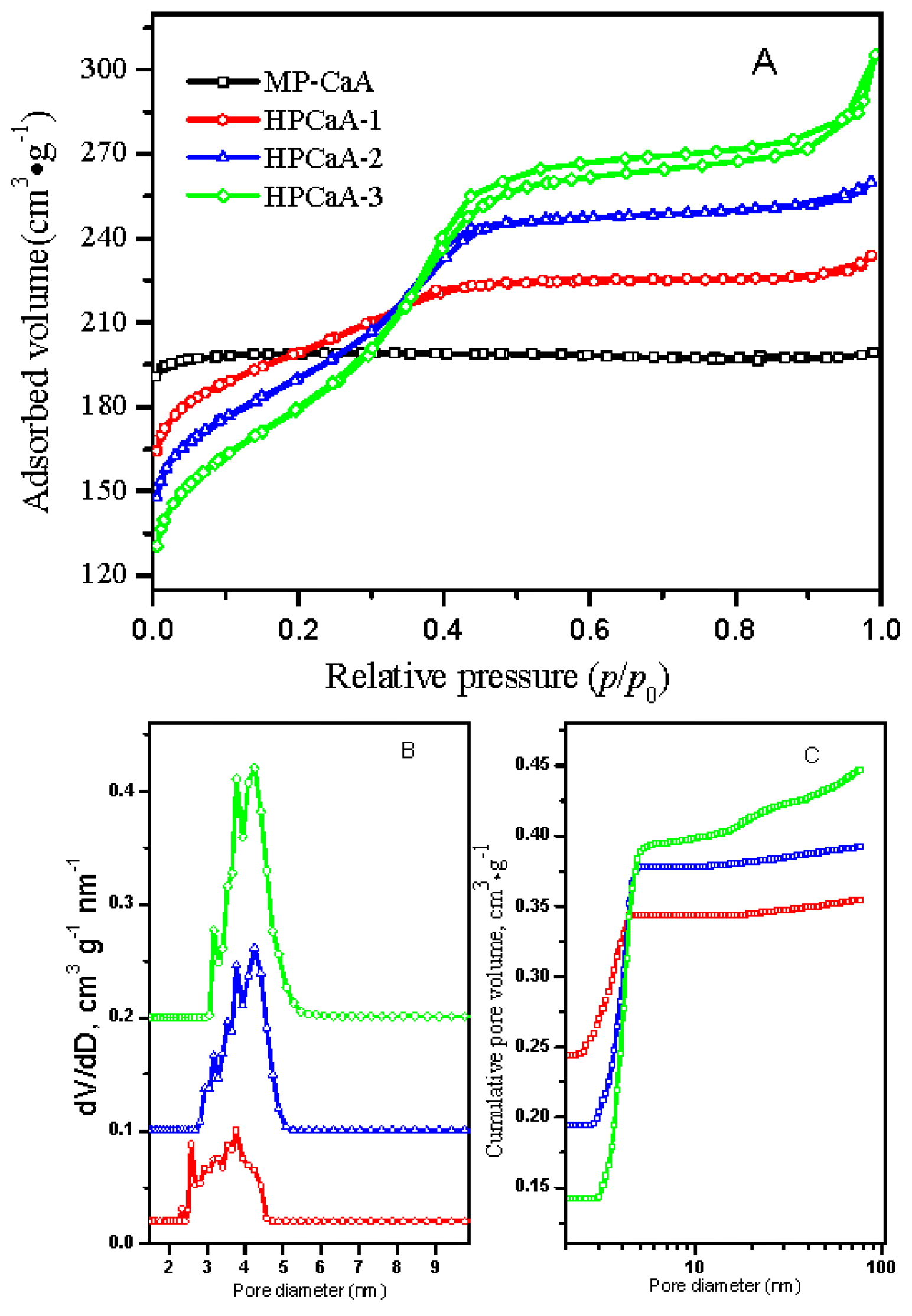
The specific surface area and porosity of series hierarchical CaA zeolite samples were evaluated from the nitrogen adsorption/desorption characterization. The nitrogen adsorption isotherms and corresponding pore distributions of all samples are shown in
Figure 3, and the textural parameters calculated are summarized in
Table 1. Obviously, all samples present a sharp increase in nitrogen uptake at a relatively low pressure, which is ascribed to the nitrogen filling within the micropore. In addition, the nitrogen adsorption/desorption behavior of MP-CaA and HP-CaA-1,-2,-3 is totally different. The zeolite MP-CaA exhibits a classic type I isotherm, but the adsorption isotherms of HP-CaA-1,-2,-3 samples exhibit characteristics of type I and type IV isotherms, indicative of micropore filling and pore condensation. For the HP-CaA-1,-2,-3 samples, besides micropore adsorption at low pressure, a significant continual uptake in the
p/p0 range of lower than 0.4 is also obvious. This uptake strongly suggests that there are mesopores in these samples. In addition, the reversible pore condensation in this pressure range is observed, reflecting the existence of smaller mesopores with pore diameters smaller than 4 nm [
27].
The pore size distribution of these mesopores, calculated with a NLDFT model from the adsorption branch, clearly evidence the presence of mesoporosity centered at around 4 nm in these samples. The mesopore volume can be systematically controlled by the variation of the silanization degree over the surface of mesoporous silica used in zeolite synthesis. Besides these small mesopores, it is seen, from
Figure 3, that the cumulative pore volume of hierarchical zeolite CaA samples presents a slow and continuous increase after a step increase in a pore size range between 3.0 and 5.0 nm, which corresponds to nitrogen adsorption in the
p/p0 range of around 0.4. This phenomenon reflects that there is a certain amount of large meso and macro-pores in these samples. Thereinto, the particularly evident sample is HP-CaA-3 synthesized from MS with the highest silanization degree, which is also shown visually in SEM photographs. With respect to the Brunauer-Emmett-Teller (BET) surface areas, they slightly decrease with the introduction of secondary porosities into zeolite, though the surface areas of micropores present a larger reduction, due to the complementary of more mesopore surface. The smaller microporosity is due to the lower crystallinity in these samples, which is also confirmed by the XRD characterization.
Ammonia temperature programmed desorption (NH
3-TPD) in the range of 120–600 °C was performed to evaluate the acidity of various CaA zeolitic samples with different mesoporosity. As presented in
Figure 4 and
Table 1, all samples showed mainly two peaks at around of 225 and 295 °C related to weak and medium acid sites respectively, meaning nearly no effect of introduction of second meso(macro)porosity on the strength of acid sites over the surface of CaA zeolites. Compared between microporous MP-CaA and hierarchical HP-CaA-1,-2,-3 samples, the difference is that the total amounts of acidic sites for HP-CaA zeolites are relatively lower than that microporous CaA zeolite, in which the main contribution comes from weak acid sites. For HP-CaA-1,-2,-3 samples, the amounts of weak and medium acid sites (acidity) remain slightly changed with increasing mesoporosity, reflecting the similarity of acidity over these samples.
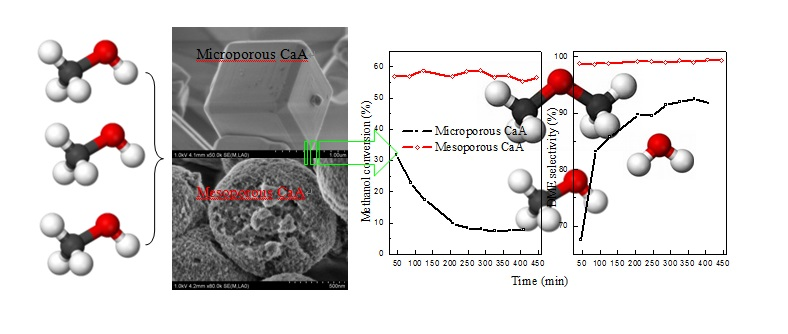
Figure 5 shows the activity, stability, and selectivity in methanol dehydration to dimethyl ether reaction (MTD) over various CaA samples. In general, all catalysts are active and selective for DME formation. The conversion of methanol and the selectivity of dimethyl ether in reaction products as a function of reaction time display clearly the effect of hierarchical meso(macro)pores in CaA zeolites on their catalytic performance. Firstly, as indicated in
Figure 5A, the order of conversion of methanol, both in the initial and steady period, is HP-CaA-3 > HP-CaA-2 > HP-CaA-1 > MP-CaA, which is the same order as the external surface area and secondary meso(macro)porosity, but the reverse order as acidity. Secondly, for the MP-CaA sample, the conversion of methanol decreases more quickly from 31.7% to 17.3% at the early stage of 40–120 min, followed by a slower decrease. However, the conversion of methanol decreases only from 37.2% to 27.6% and from 48.3% to 41.4% for the hierarchical mesoporous HP-CaA-1 and HP-CaA-2 samples, respectively, and then, for the HP-CaA-3 sample, the conversion of methanol shows a very slight change in the conversion of methanol and remains at nearly 58%. Third, the comparison of DME selectivity obtained over all the CaA catalysts in
Figure 5B exhibits distinctly that DME selectivities over hierarchical CaA catalysts are much higher than that over a microporous CaA catalyst. For the HP-CaA-3 sample, nearly no hydrocarbons are formed, thus giving a total selectivity for DME of 100%. In one word, CaA catalysts with hierarchical meso(macro)pores exhibit excellent catalytic performance in the MTD process.
It is generally accepted that the acidity and pore structure of zeolite catalyst are two main influence factors for the catalytic activity, selectivity and stability of the methanol dehydration to dimethyl ether. With respect to acidity, DME formation is mainly related to sites with weak and medium acidity, and strong acid sites are recognized as a main cause for undesirable byproducts in DME synthesis, such as hydrocarbons and even coke, which in turn affect the catalytic activity. It is known, from NH
3-TPD results, that there are considerably weak and medium acid sites, but no strong acid sites over both microporous and hierarchical CaA catalysts, and thus they are just preferable in the Methanol to Dimethyl ether (MTD) reaction. However, the MP-CaA catalyst with more acid sites displays lower catalytic activity, selectivity and stability than the hierarchical HPCaA-1,-2,-3 catalysts. This indicates that the acidity of CaA zeolite is not the main reason that influences catalytic activity, selectivity and stability, whereas the catalytic behavior of CaA zeolitic catalysts for MTD strongly depends on the hierarchical pore structure of the catalysts. For the microporous MP-CaA catalyst, small microporous channels, in which the size of the reacting molecules and the micropore diameter are similar, give a limitation for the accessibility of acid sites due to slower diffusion, resulting in lower catalytic activity. At the same time, the produced DME cannot diffuse quickly enough in the narrow and slender micropore and this consequently causes secondary reactions to produce hydrocarbons and carbon deposits [
28]. Nevertheless, for HP-CaA catalysts with a higher external surface area and intra-meso(macro)porosity, more pore entrances and shorter diffusion lengths within catalysts make methanol conversion easier to occur inside pore channels, due to enhancing diffusions of reactants and products to/from the active site located in the micropore. Therefore, it is concluded that the introduction of meso(macro)porosity into zeolite CaA crystals increases accessibility to acid sites, enhances the diffusion rate of reactants and products and avoids the formation of undesirable byproducts. Consequently, catalytic activity, stability and selectivity can be improved accordingly.
3. Experimental Section
3.1. Synthesis
The synthesis procedure was adopted according to similar procedures proposed by our research group. Mesoporous silica (MS, surface area 800 m2/g) was firstly functionalized by organosilane phenylaminopropyl-trimethoxysilane (PHAPTMS, Sigma-Aldrich, Saint Louis, MI, USA) at the molar ratio: SiO2:60H2O:x (x = 0, 0.065, 0.12, or 0.20) PHAPTMS:6CH3OH to obtain organo-functionalized mesoporous silica with a different silanization degree. In a typical organic-functionalization process, 10 g fumed silica was added into 350 mL of distilled water. The mixture was stirred (100 rpm) under reflux at 373 K. Then 10 mL PHAPTMS in methanol solution was added into the former mixture. After refluxing for 8 h at 373 K, the silica was washed with ethanol several times, and then dried at 373 K. The weight losses of the silanized (organo-functionalized) MS samples in thermogravimetric analysis (TGA) were 0%, 14%, 21% and 28%, respectively. Next, hierarchical LTA zeolites were synthesized by employing organo-functionalized mesoporous silica (org-MS) as a silica source at the molar ratio of 5Na2O:Al2O3:2SiO2:180H2O in the hydrothermal system. In a typical synthesis procedure, org-MS was mixed with sodium hydroxide (AR), sodium aluminate (AR) and deionized water under stirring at room temperature. The resultant gel was hydrothermally crystallized in Teflon-lined stainless steel autoclaves at 100 °C for 24 h and then the product samples were filtered and dried in air, and calcined in oxygen at 550 °C for 6 h to remove the organic moiety. The weight losses of the organics in the as-synthesized MS samples (before calcinations) in TG analysis were 0%, 7.4%, 11.8% and 15.3%, respectively. The resultant zeolites A in the Na+ form were labelled as MP-NaA, HP-NaA-1, HP-NaA-2 and HP-NaA-3 respectively, which corresponds to the products prepared from different silica source (x = 0, 0.065, 0.12, or 0.20). The corresponding zeolites A in the Ca2+ form were obtained through Ca2+ exchange of zeolite NaA with 0.5 mol/L CaCl2 solution. Zeolite CaA samples were washed with distilled water until there was no remaining Cl−. The samples are denoted as MP-CaA, HP-CaA-1, HP-CaA-2 and HP-CaA-3 respectively.
3.2. Characterization
Powder XRD analysis was conducted on a Shimadzu XRD-6000 (Shimadzu, Nakagyo-ku, Kyoto, Japan) instrument with CuKα radiation. Nitrogen adsorption/desorption isotherms were obtained in a Quantachrome NOVA 1200e (Quantachrome Instruments, Boynton Beach, FL, USA) at 77 K. Before analysis, the samples were outgassed under vacuum at 300 °C for 5 h. The BET surface areas were calculated by adsorption data in the relative pressure (p/p0) range of 0.02–0.10, while the external surface area and micropore volume were obtained by the t-plot method at the relative pressure range of 0.10–0.20. Pore size distributions were obtained by the Density Functional Theory (DFT) method on the adsorption branch using the cylindrical pore NLDFT model. Field emission scanning electron microscopy (SEM) images were taken on a HITACHI S-4800 (Hitachi HighTechnolo-gies, Tokyo, Japan) instrument. Transmission electron microscopy (TEM) images were collected by JEOL JEM-2010 instruments (Jeol, Tokyo, Japan). NH3-TPD was performed on a TP-5076 TPD/TPR chemisorption analyzer with a thermal conductivity detector (TCD) (Xianquan, Tianjin, China). Typically, 0.1 g of the sample (40–60 mesh) was pretreated at 550 °C in a He flow for 120 min and then saturated with NH3 at 110 °C for 60 min. After saturation, the sample was purged by He flow to remove physically adsorbed NH3 for 60 min. Finally, desorption of NH3 was carried out from 110 to 600 °C with a heating rate of 10 °C/min under the He flow.
3.3. Catalytic Tests
Catalytic conversion of methanol to DME was conducted in a fixed-bed micro-reactor apparatus with a quartz tube (i.d. 6 mm) at 400 °C over 0.2 g Ca2+ ion-exchanged LTA zeolite. Prior to each experiment, the catalyst CaA was activated at 550 °C for 2 h in air flow (50 mL·min−1) and then kept at 400 °C. Methanol (99.5%, AR) was introduced as a saturated vapor in N2 flow (50 mL·min−1) at 25 °C. The weight hourly space velocity (WHSV) of methanol was 4.2g·g−1·h−1. Products were analyzed by an online gas chromatograph equipped with a flame ionization detector and HP-Plot-Q column (Agilent, Santa Clara, CA, USA).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}