1. Introduction
The field of plasmonics has grown in the last two decades, driven by the prospect of controlling the properties of light with nanometre-scale precision by coupling electromagnetic fields to the oscillation of free electrons in metals. This has led to gain fundamental understanding into the interaction between light and matter at the nanoscale, allowed the diffraction limit to be beaten, produced ways to modify the properties of light emitters and enabled materials with optical properties with no counterpart in nature to be developed [
1,
2].
Single metal nanoparticles are the smallest unit to support plasmon-based interactions. Isolated or randomly aggregated metal nanoparticles display peculiar opto-electronic properties useful for numerous applications. However, the real interest in the last decade has concentrated on development of methodologies for the controlled chemical assembly of plasmonic nanoparticles into well-defined superstructures with predictable spatial geometry. Slow and controlled solvent evaporation of surfactant stabilized metal nanoparticle solutions has been used by our group and others to fabricate ordered large two- and three-dimensional superstructures with controlled geometries [
3,
4,
5]. Another approach for fabrication of plasmonic structures of controlled dimension and geometry is the bottom-up assembly of chemically synthesized metal nanocrystals into higher-order structures [
6,
7]. The interest for this approach relies on the numerous applications of resulting structures ranging from surface enhanced spectroscopies [
8,
9,
10], optical circuitry [
11], biological sensors [
12,
13] and metamaterials [
14,
15]. In addition, new and complex self-assembled materials may exhibit novel properties arising from the combination of the individual component properties. In fact, these structures can be treated as plasmonic molecules, where the combination of localized surface plasmon resonances (LSPR) of individual nanostructures interact to form new coupled excitations, similar to the way electrons interact in molecules to form bonds [
16]. The most common approach for self-assembly of metal nanoparticles exploits the known affinity between gold and sulfur groups thus relying on the use of thiol-functionalized linking molecules. Numerous molecules containing thiol and dithiol groups were used as linkers including aliphatic [
17] and aromatic thiols [
18], biological compounds [
19,
20] and single strand oligonucleotides [
21,
22]. The addition of controlled aliquots of such linkers to nanocrystal solutions gave rise to controlled growth and formation of geometrically well-defined nanocrystal clusters. Isothiocyanide (–N=C=S) groups are also known to possess high affinity versus gold. So far this affinity has mostly been exploited to attach organic chromophores to colloidal gold nanocrystals or to nanoscale electrodes for surface-enhanced Raman scattering (SERS) applications [
23], formation of metal junctions for molecular electronic applications [
24], bioimmunoassays [
25] and cancer therapy applications [
26]. However, recent theoretical work by Sikora et al
. showed that bis-isothiocyanide molecules can also be used as alternative to dithiols as conjugation molecules for bridging two or more nanocrystals together [
27], whereby the lowest reactivity of isothiocyanide groups compared to thiols for citrate stabilized gold nanoparticles was shown useful to achieve good control in the growth rate of nanoparticle clusters.
In this paper we have used a bis-isocyanide Rhenium complex to synthesize gold nanocrystal clusters of defined stochiometry and geometry, also known as plasmonic molecules. Increasing aliquots of linker molecule were mixed with nanocrystal solutions and the evolution of cluster formation was monitored by UV-vis spectroscopy. Correlated scanning electron microscopy (SEM)—dark-field spectroscopy was used to characterize discrete monomer, dimer and trimer plasmonic molecules. Compared to the scattering spectra of single nanocrystals, additional features appeared in the assembled structures, due to plasmonic coupling between nanocrystals. Polarization-dependent scattering spectra of dimer structures showed highly polarized scattering response, due to their highly asymmetric geometry (D∞h). In contrast, trimer structures displayed symmetric geometry (D3h), which showed small polarization-dependent response. The experimental data from both dimer and trimers matched well with theoretically calculated data, which were used to further understand and attribute the origin of plasmonic bands arising during linker-induced formation of plasmonic molecules.
2. Results
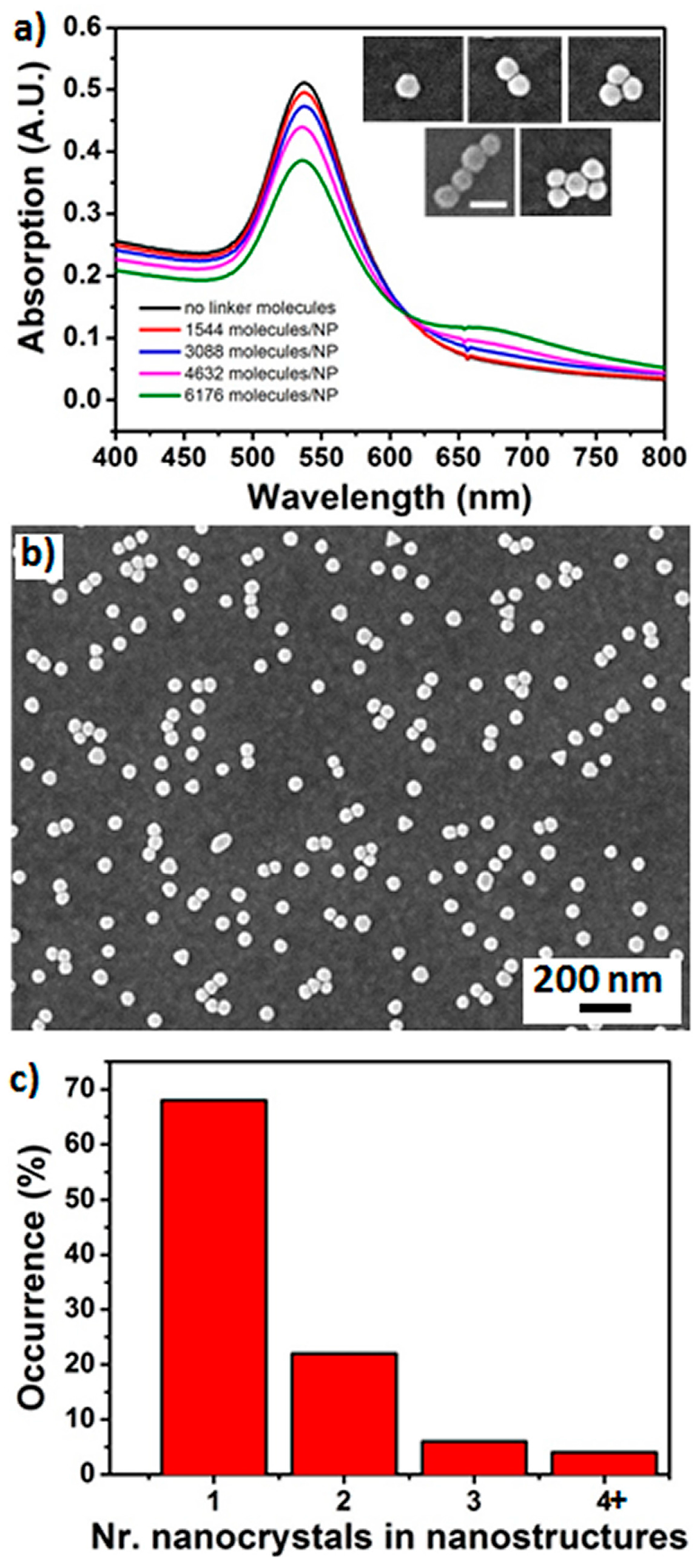
In assembled nanostructures the geometry and monodispersity of the nanocrystal building blocks is critical to control the self-assembly process and for accurate comparison between experimental and theoretical data. Used gold nanocrystals had an average size of 60 nm and a maximum absorption band centered at 540 nm (see
Figure S1). Therefore, prior to self-assembly experiments statistical correlated scanning electron microscopy (SEM) and darkfield microscopy were used to assess the size and shape distribution of nanocrystals in the used solution (see
Figures S2 and S3). The majority of the particles (88% occurrence) were spherical or near-spherical, with a measured plasmon resonance peak wavelength between 526 nm and 536 nm. Hexagons (9% occurrence) displayed scattering peaks in the range 554–572 nm and triangles (2% occurrence) in the range 583–606 nm. Small percentage of tetrahedrons (1%) were also found exhibiting the same resonance wavelength (534 nm) as spherical particles with, however, a slightly broader full width half maximum (FWHM).
Scheme 1 shows a graphical representation of the gold nanocrystals and the Re
2(DMAA)
4(NCS)
2 linker molecule used in this work as well as a schematic of the coupled structures formed during the assembly process comprising one-nanocrystal (monomer), two nanocrystals (dimers), three nanocrystals (trimers) and higher order
n-mer structures. The attachment of the linker molecule to the gold nanocrystals was promoted by the high affinity of the sulphur atom of the isothiocyanate group for gold. The distance between the end thiol groups was calculated from X-ray crystallography data to be 1.2 nm [
28].
Figure 1a shows the formation of gold nanocrystal structures monitored by UV-vis spectroscopy. The spectrum of isolated nanocrystals in water solution (60 nm diameter) was characterized by a plasmon resonance peak at 540 nm. Progressive addition of linker molecule resulted in formation of an additional band at 675 nm, representative of the coupling between nanocrystals and formation of nanocrystal clusters. The near-infrared band reached its maximum after addition of 40 μmol of linker molecule, corresponding to a linker molecule/nanocrystal ratio of 6179:1. Theoretical calculations performed on smaller nanocrystals showed that the maximum coverage of linker molecule for 40 nm diameter nanocrystals was equal to 4273, which could be in agreement with the experimentally added amount of 6179 for a 60 nm diameter particle [
27]. This value corresponded to a nanocrystal coverage of 4.9% calculated approximating the nanocrystal to a sphere of radius 30 nm and the linker molecule to a sphere of 0.6 nm radius and assuming that all added linker molecule was bound to the nanocrystal surface. The range of cluster size and shapes obtained was analyzed by SEM. As shown in the inset of
Figure 1a, a number of structures formed, including dimers, trimers, quadrumers and small clusters. Formed n-mer structures are also shown in a representative image in
Figure 1b and in the additional images reported in
Figure S4: Trimers were usually arranged in triangular shape, some of them showing high D
3h symmetry, and quadrumers usually arranged in D
∞h symmetry, although more geometrical arrangements would be possible for such structures. Statistical analysis reported in
Figure 1c, performed on 100 structures, showed that the majority of structures in solution were single nanocrystals (70%), which explains the small change of the plasmon band at 540 nm upon addition of linker molecule. Dimers constituted ca. 20% of the formed structures, whereas the remaining 10% was constituted by trimers, quadrumers and larger clusters. Control experiments on unmodified nanocrystals prepared without addition of the linker molecules yielded over 95% of monomers with a low incidence of dimers and negligible incidence of higher order
n-mers (data not shown).
Bringing nanocrystals in close proximity of each other, within 2.5 times the particle diameter to each other, led to coupling of their plasmon modes and resulted in more complex optical signatures. In order to analyse such features at discrete level correlated darkfield spectroscopy and SEM imaging of formed plasmonic molecules was performed. Representative scattering experimental and calculated spectra of a monomer, dimer and trimer formed by 60 nm gold nanocrystals are shown in
Figure 2 the peak positions are noted in
Table 1. For all three structures the scattering curve was calculated using a boundary element method to solve Maxwell’s equations [
29]. The nanocrystals were discretized by spheres with 1444 vertices, a diameter of 60 nm and an inter-particle separation of 1 nm, based on the linker molecule length. The dielectric function for the gold nanoparticles was taken from Johnson and Christy, the average dielectric constant of the environment was assumed as 1.33 [
30].
The experimental scattering spectrum of a representative nanocrystal monomer (
Figure 2a, black curve) showed one peak at a resonance wavelength of 540 nm. This value correlated well with the calculated resonance wavelength of 60 nm spherical particles, centered at 541 nm (
Figure 2a, red curve). Two scattering peaks were experimentally observed in dimer structures
Figure 2b centered at 541 nm and 693 nm, respectively. The correlated SEM image showed that the measured dimer structure was constituted by spherical particles of very close diameter size. For this reason the experimental peak wavelength positions correlated well with the calculated peak positions of 544 nm and 691 nm. The experimental spectrum of a representative trimer of D
3h symmetry displayed two peaks centered at 542 nm and 551 nm. Compared to the calculated values of 669 nm and 678 nm, experimental data were red-shifted by 9 nm. This could be related to small deviation from perfect equilateral triangle geometry or some breaking of the symmetry caused by slightly different size nanocrystals constituting the cluster.
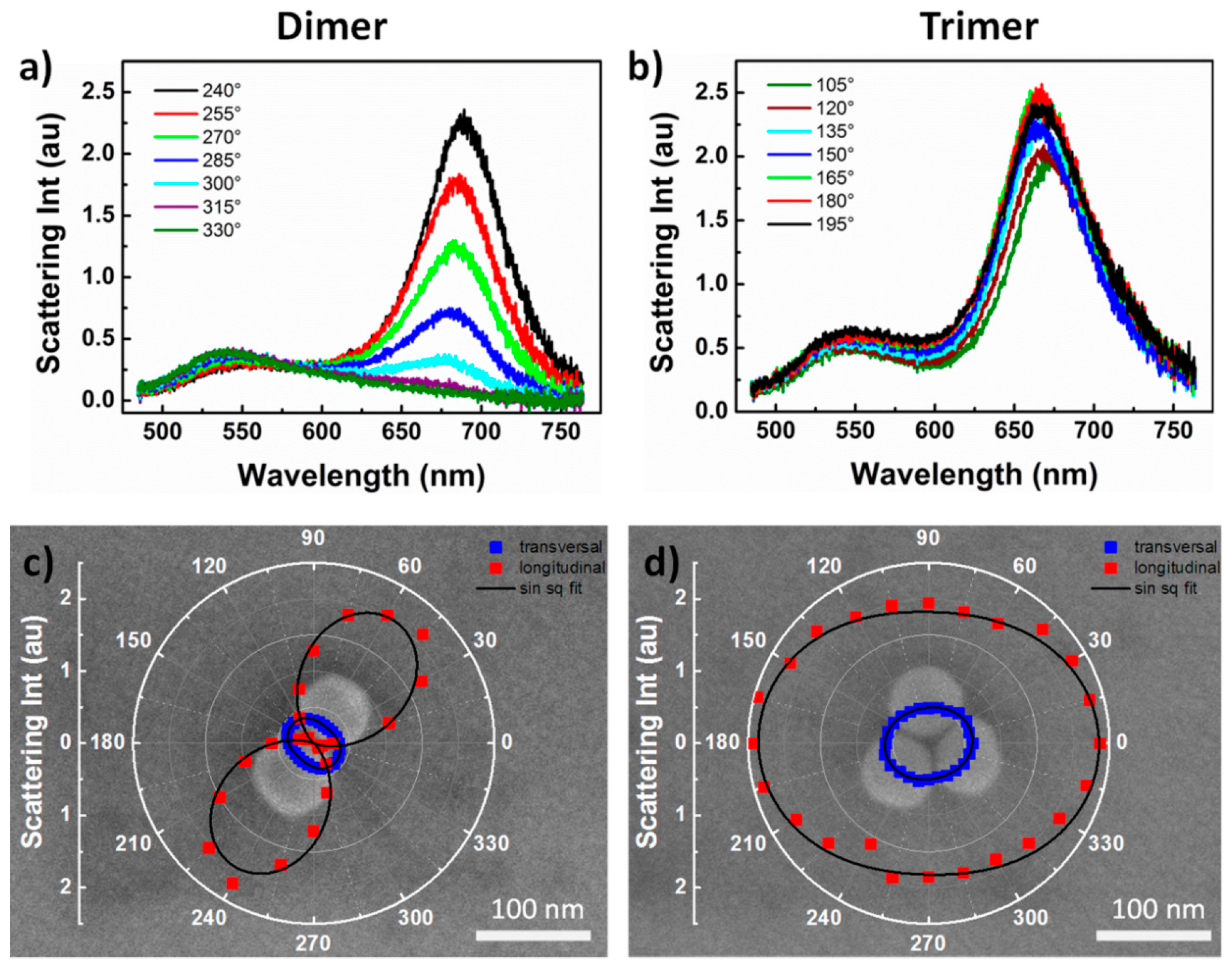
In order to better analyze the origin of peak formation, scattering spectra of nanocrystal clusters were taken at different polarization angles, as shown in
Figure 3. For the dimer structure
Figure 3a the peak at 541 nm showed a small change in intensity with the change of the polariser angle, equivalent to a factor 1.7 difference in intensity between scattering maxima at crossed polarized positions. In contrast, the peak at 693 nm showed a dramatic change in intensity with the rotation of the polarizer angle. The peak intensity decreased as the incident light polarization was rotated from parallel to perpendicular to the long axis of the dimer. Specifically, the alignment of the polarization angle parallel to the long dimer axis resulted in the maximum intensity of the peak at 693 nm whose intensity was 25 times higher than the intensity displayed for an alignment of the polarization angle perpendicular to the long dimer axis. This suggested that the resonance wavelength at 693 nm generated as a result of the formation of a strongly coupled plasmon mode along the longitudinal axis of the dimer.
Figure 3c shows a SEM image of the analyzed dimer with an overlaid plot of the plasmon peak intensity variation in dependence of the polarization angle. The plot clearly showed the generation of a strong dipole coupling parallel to the dimer long axis. In contrast, the lack of polarization dependence of the 540 nm plasmon mode was attributed to a weak dipole coupling perpendicular to the dimer long axis.
The polarization dependent spectra of the D
3h symmetry trimer (
Figure 3b) showed a very small intensity change with the variation of the polarizer angle for both 542 nm and 669 nm peaks. This was expected, due to the increased anisotropy of D
3h trimer structures compared to D
∞h dimer structures. Specifically, a perfectly symmetrical trimer would be expected to give rise to overlapping spectra in all polarization directions [
31]. Instead a slight anisotropy was recorded for this structure with a factor 1.3 decrease in intensity between plasmonic modes recorded at 90 degrees shifted polarization positions.
Figure 3d shows a SEM image of the analyzed trimer with an overlaid plot of the plasmon peak intensity variation in dependence of the polarization angle. The recorded anisotropy was attributed to slight imperfections in the symmetry of the studied trimer as close observation revealed that one particle was slightly elongated compared to the others.
Our results were in agreement with results recently reported by Tian et al
. for silver nanoparticle assemblies where solvent evaporation-induced aggregation was used as process to create random and discrete nanoparticle assemblies which were subsequently analyzed by correlated electron microscopy/darkfield spectroscopy [
32]. In spite of the high monodispersity of used nanocrystals the random nature of the assembly process lowered the degree of symmetry reached by formed structures. For this reason, Tian et al
. found a mismatch between experimental and theoretical optical signatures of obtained trimers, of which geometry significantly deviated from the perfect D3
h symmetry. In our work, the yield of formed structures was significantly lower than what found by Tian et al. However, some examples of high symmetry clusters were found among formed structures. We argued that the formation of such clusters was facilitated by the use of a linker molecule with a bi-anchoring moiety and structural constraints (triple bond between Re atoms, short length and double bonds in the NCS group).
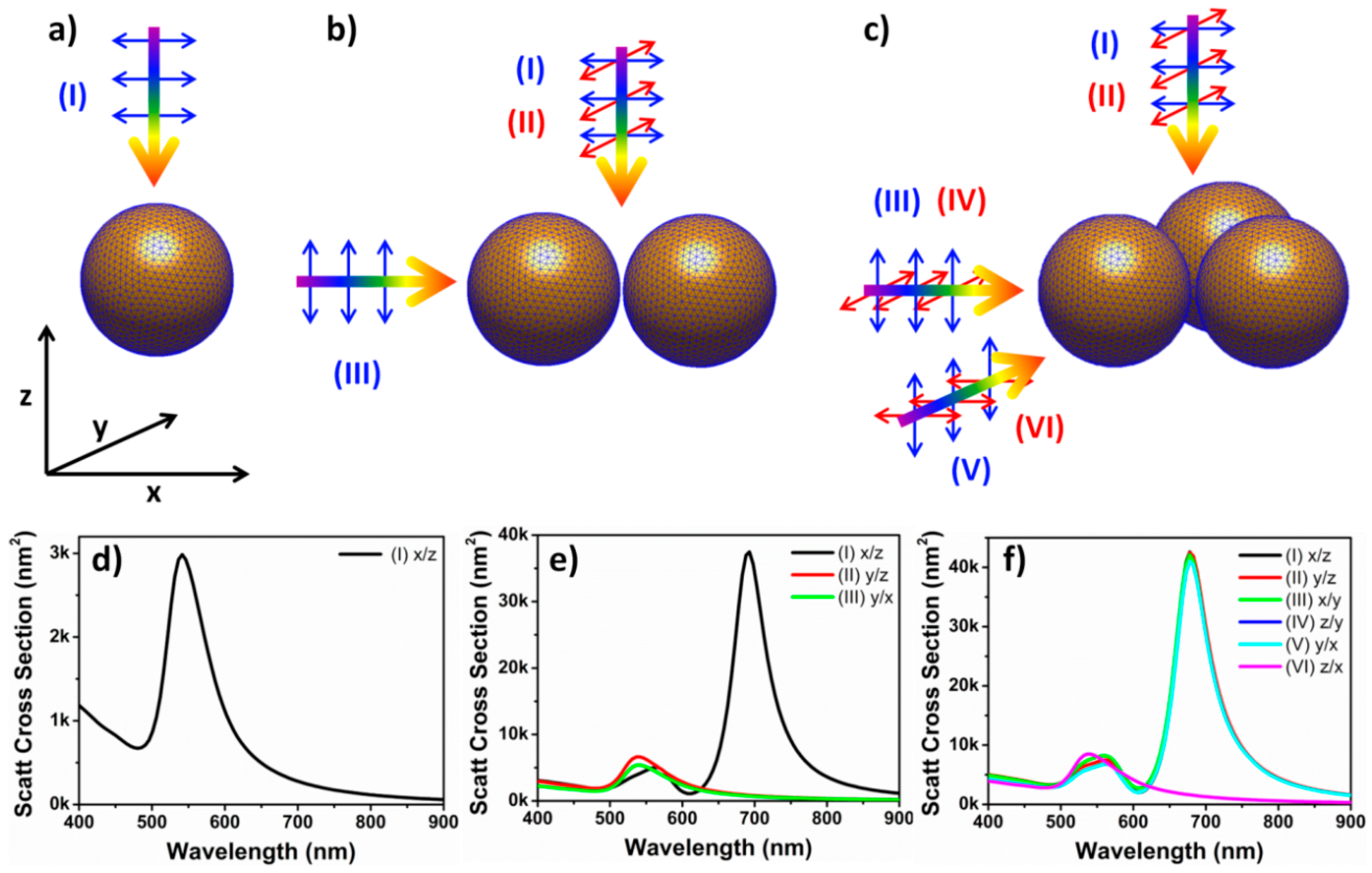
In order to further analyze dimer and trimer polarization behavior we calculated polarization resolved scattering spectra of the analyzed nanoparticle structures, shown in
Figure 4.
For perfectly symmetrical monomers only one light propagation and polarization direction (I)
Figure 4a had to be considered and the calculated scattering spectrum showed one plasmon peak at 541 nm. For a dimer with D
∞h symmetry three combinations of polarizations and propagation directions of incident light were considered: Light incident perpendicular to the dimer long axis with polarization direction parallel (I) and perpendicular (II) to the long axis of the dimer and light propagation direction parallel and light polarization perpendicular (III) to the dimer long axis. Additionally, due to the illumination geometry of the darkfield condenser a contribution of out-of-plane plasmon excitation was expected and the scattering spectrum was calculated for light incident parallel to the long dimer axis (III). At condition (I), the spectrum was dominated by a plasmon peak at 691 nm, indicating strongly interacting dipoles arranged head to tail and excited with the electric field component parallel to the dimer long axis. In contrast, when the plasmon modes were excited perpendicular to the dimer long axis or out-of-plane, the spectra showed only one plasmon peak, centered at 539 nm. This peak position was only slightly blue shifted compared to the plasmon peak of a single nanoparticle at 541 nm and indicated a very weak coupling of dipoles arranged shoulder to shoulder and excited with the electric field component perpendicular to the long axis of the dimer. The comparison between these calculated spectra and the experimentally recorded spectra of
Figure 3 showed good agreement and indicated that the recorded spectra at any polarization angle are linear combinations of the calculated plasmon modes [
5]. Moreover, our data are in agreement with theoretical data reported in literature showing that the plasmonic modes of a dimer are formed by hybridization of single particle modes and that, indeed, such structures can be treated as plasmonic molecules [
33,
34].
For the more complex structure of the nanoparticle trimer (
Figure 4c) all possible incident light propagation and polarization directions were considered and the corresponding scattering spectra were calculated. For perpendicular incident light (I) and (II) identical scattering spectra were calculated with peaks at 678 nm and 562 nm, indicating strong plasmon coupling irrespective of polarization direction. For incident light conditions (III) and (V) nearly identical scattering spectra to (I) and (II) were calculated, also originating from strong plasmon coupling. Only out-of-plane excitation (IV) and (VI) resulted in single peak scattering spectra (539 nm), showing very weak plasmon coupling. Again, as for the nanoparticle dimer, the calculated scattering spectra agreed well with the experimental data obtained for the trimer in
Figure 3, which can be considered as linear combinations of the calculated plasmon modes. While for the nanoparticle dimer the two different plasmon modes can be understood as longitudinal and transversal plasmon modes, the case of a trimer is more complex. The origin of plasmon modes was analyzed by a model developed by Nordlander for the most symmetrical case of individual particles arranged in the form of an equilateral triangle [
30]. According the group symmetry theory this plasmonic molecule trimer sustains nine dipolar plasmon modes. Six of the modes are in-plane, three can be classified as bonding and three are non-bonding, with energies lower and higher than that of an individual particle, respectively. Two bonding and two non-bonding modes are degenerate and can be excited by linearly polarized light so that the corresponding plasmon bands are located at a longer and shorter wavelength compared the individual particle, respectively. The state of lowest energy is dark for excitation with linearly polarized light, but can be excited with radially polarized light.
4. Materials and Methods
Materials: Citrate-stabilized gold nanocrystals with core diameter
d = 60 ± 6 nm were purchased from BBInternational (Cardiff, UK). Ultrapure deionized water with resistivity ~18.2 MΩ cm (ELGA PURELAB Ultra, Celbridge, Ireland) was used in all experiments. All glassware and cuvettes were cleaned using aqua regia and Piranha solutions prior use in order to prevent spurious aggregation of the nanocrystals. To improve the stability of the nanocrystals, 3 mM aqueous tri-sodium citrate was added prior to use. Linker molecule Re
2(DMAA)
4(NCS)
2 was synthesized according to literature [
28] and was provided by Prof. Gallagher group at University College Cork.
Synthesis of gold nanocrystal clusters: The bi-functional linker molecule, Re2(DMAA)4(NCS)2 was dissolved in acetone (ACS grade). 3 mL of a 1:1 nanocrystal: tri-sodium citrate solution (1.5 mL of 1 × 10–10 M nanocrystal, mixed with 1.5 mL of 3 mM aqueous tri-sodium citrate) was decanted into a clean cuvette. Aliquots of 10 μL Re2(DMAA)4(NCS)2 linker solution (10 μM) were added at 10 minute intervals (typically four aliquots). The solution was mixed after each addition to ensure complete mixing of the nanocrystal/linker components.
Optical characterization: UV-Vis spectra of nanocrystal clusters were acquired with an Agilent/HP 8453 UV-Vis Spectrophotometer (200nm < λ < 1100nm, Dublin, Ireland). The evolution of cluster formation was monitored by taking UV-vis spectra after each aliquot of linker addition.
Scattering spectra of individual nanocrystals and nanocrystal clusters were acquired with an inverted IX-71 Olympus microscope with an oil immersion darkfield condenser (Olympus U-DCW, 1.4 NA, Berlin, Germany) and 100× objective (Olympus MPlanApo 100×/0.95 NA). Samples were illuminated with a 100 W halogen lamp. The light collected by the objective was directed either onto a color CMOS camera (DCC1645C, ThorLabs, Exeter, UK) for image acquisition or onto the entrance slit of a monochromator (SP-2356, Acton Research) equipped with a thermoelectrically cooled, back illuminated CCD (Spec10:100B, Princeton Instruments, Berlin, Germany) for spectra acquisition. The sample polarization rate was determined by a polarizer placed in collection between the sample and the monochromator. The experiments were performed by first recording an image of the sample with a mirror in the light path of the spectrometer. Once a suitable nanostructure was selected, the mirror was switched to a grating (300 groves mm–1) to disperse the scattered light and a spectrum was recorded using an acquisition time of 60 s. Normalized scattering spectra from individual nanocrystals were obtained by subtracting and dividing by a background scattering spectrum taken from a nearby clean area on the sample.
Electron Microscopy: Scanning electron microscopy (SEM) images of gold clusters were acquired using a field emission SEM (JSM-6700F, JEOL UK Ltd., Welwyn Garden City, UK) operating at beam voltages of 10 kV. For navigation on the substrate, lithographically etched alignment marks in the Indium Tin Oxide (ITO) coating of glass cover slips were used.
Calculation of Nanostructure Scattering Spectra: To calculate the scattering spectra of gold nanoparticle monomers, dimers and trimers a boundary element method based toolbox for MATLAB was utilized.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}