Dihydrogen Bonds in Salts of Boron Cluster Anions [BnHn]2− with Protonated Heterocyclic Organic Bases



Abstract
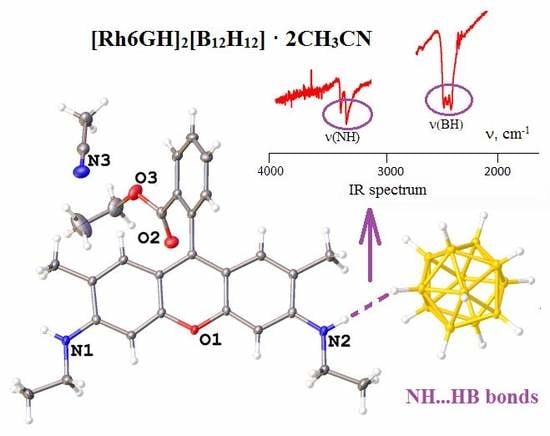
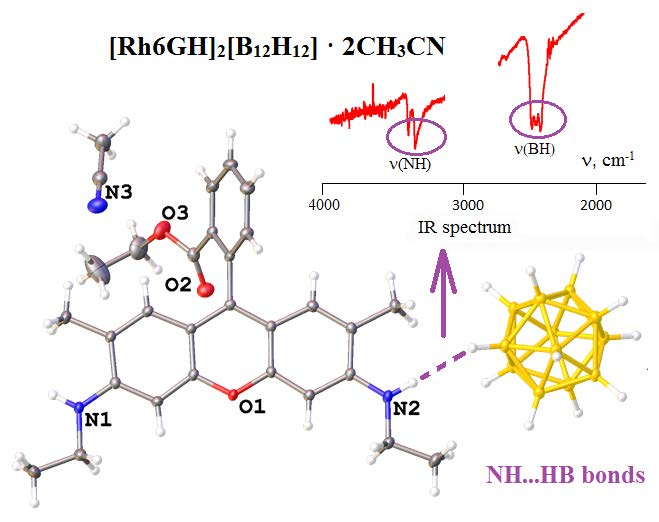
:
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. X-ray Diffraction
3. Results and Discussion
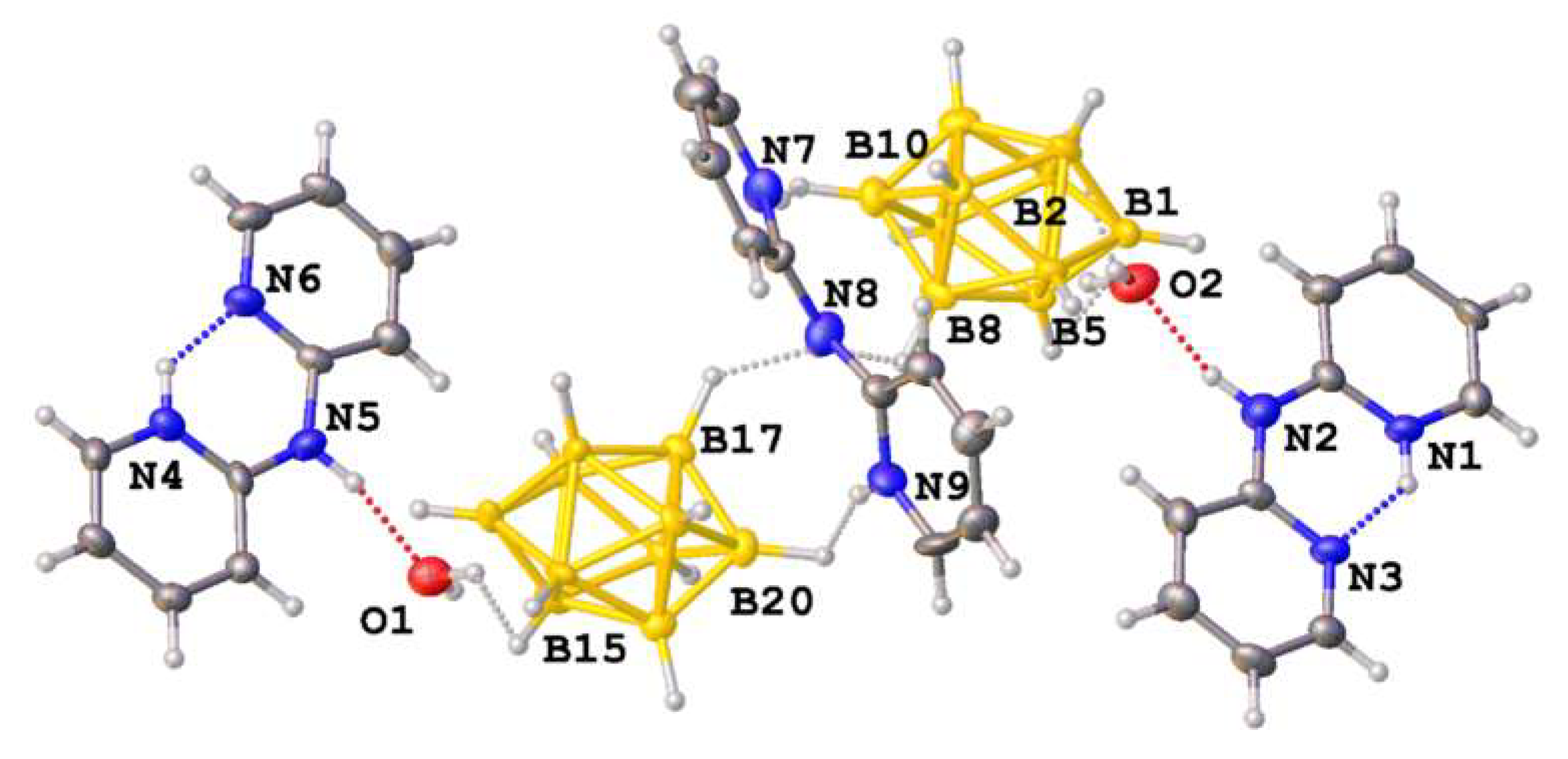
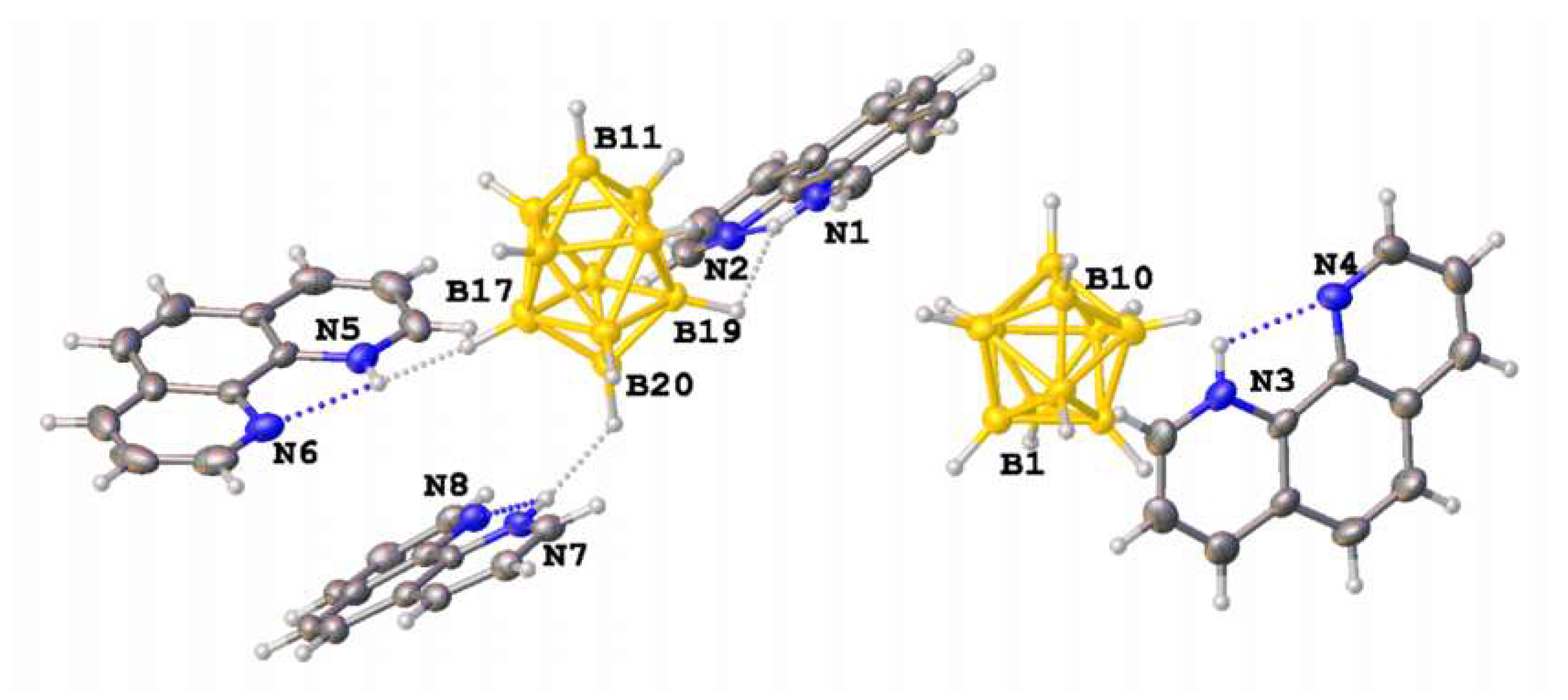
3.1. Synthesis and Structures of Salts of the Boron Clusters with Monoprotonated and Diprotonated Organic Bases
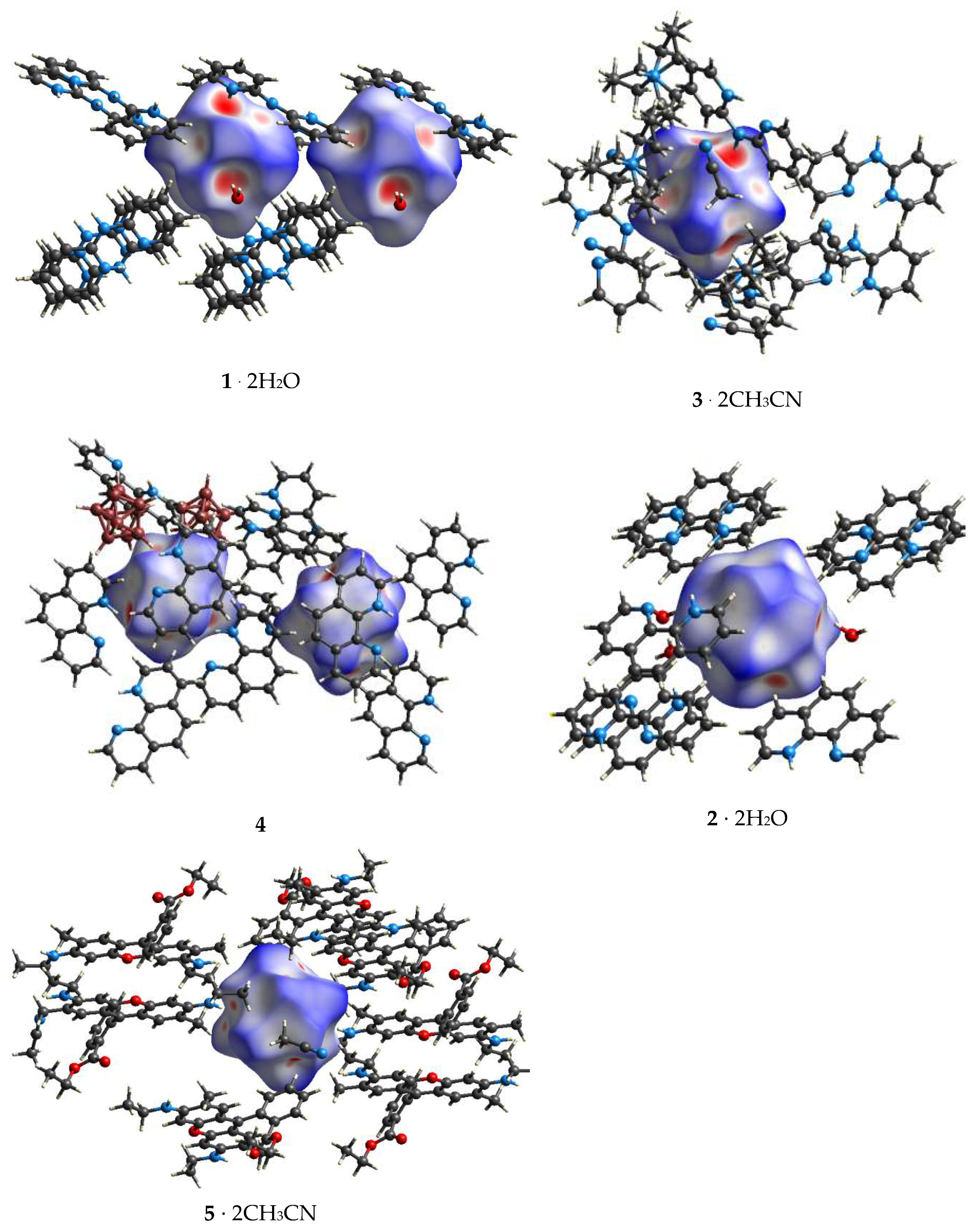
3.2. Hirshfeld Surface Analysis
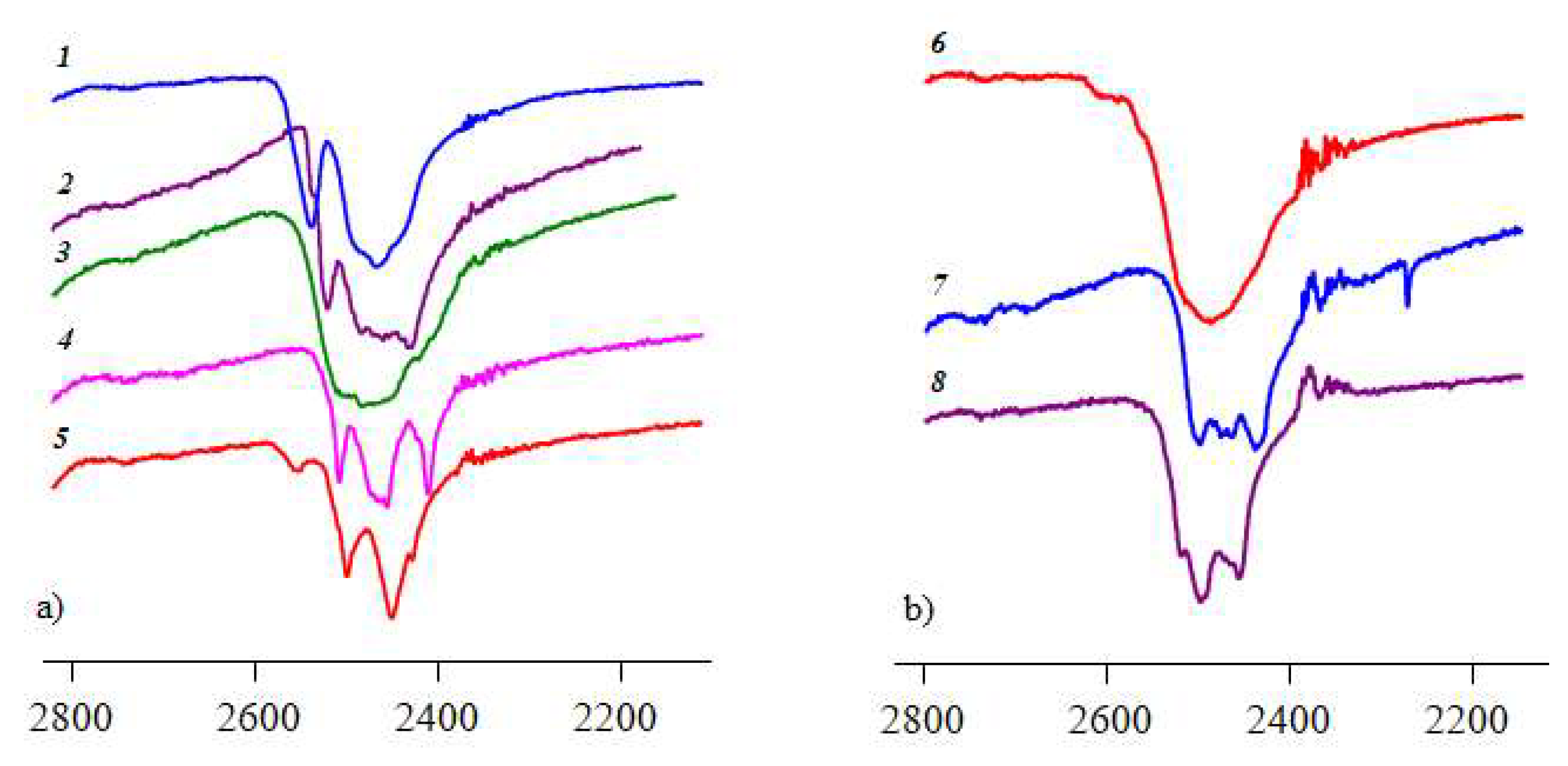
3.3. IR Spectroscopy Data of Compounds Synthesized
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alcock, N.W. Secondary Bonding to Nonmetallic Elements. In Advances in Inorganic Chemistry and Radiochemistry; Emeléus, H.J., Sharpe, A.G., Eds.; Elsevier: Amsterdam, Netherlands, 1972; Volume 15, pp. 1–444. [Google Scholar]
- Glidewell, C. Some chemical and structural consequences of non-bonded interactions. Inorg. Chim. Acta 1975, 12, 219–227. [Google Scholar] [CrossRef]
- Kuz'mina, L.G.; Struchkov, Y.T. Structural chemistry of organomercury compounds – role of secondary interactions. Croat. Chim. Acta 1984, 57, 701–724. [Google Scholar]
- Kuz'mina, L.G. Secondary bonds and their role in chemistry. Russ. J. Coord. Chem. 1999, 25, 599–617. [Google Scholar]
- Virovets, A.V.; Podberezskaya, N.V. Specific nonbonded interactions in the structures of M3X74+ and M3X44+ (M = Mo, W; X = O, S, Se) clusters. J. Struct. Chem. 1993, 34, 306–322. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- George, A.J. An Introduction to Hydrogen Bonding (Topics in Physical Chemistry); Oxford University Press: New York, NY, USA, 1997; pp. 4–320. [Google Scholar]
- Muetterties, E.L.; Knoth, W.H. Polyhedral Boranes; Marcel Dekker: New York, NY, USA, 1968; pp. 104–168. [Google Scholar]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements, 2nd ed.; Butterworth–Heinemann: Oxford, UK, 1997; pp. 4–1600. [Google Scholar]
- Malinina, E.A.; Avdeeva, V.V.; Goeva, L.V.; Kuznetsov, N.T. Coordination compounds of electron-deficient boron cluster anions BnHn2− (n = 6, 10, 12). Russ. J. Inorg. Chem. 2010, 55, 2148–2202. [Google Scholar] [CrossRef]
- Avdeeva, V.V.; Malinina, E.A.; Sivaev, I.B.; Bregadze, V.I.; Kuznetsov, N.T. Silver and Copper Complexes with closo-Polyhedral Borane, Carborane and Metallacarborane Anions: Synthesis and X-ray Structure. Crystals 2016, 6, 60. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Prikaznov, A.V.; Naoufal, D. Fifty years of the closo-decaborate anion chemistry. Collect. Czech. Chem. Commun. 2010, 75, 1149–1199. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.I.; Sjoberg, S. Chemistry of closo-Dodecaborate Anion [B12H12]2-: A Review. Collect. Czech. Chem. Commun. 2002, 67, 679–727. [Google Scholar] [CrossRef]
- Zhizhin, K.Y.; Zhdanov, A.P.; Kuznetsov, N.T. Derivatives of closo-decaborate anion [B10H10]2− with exo-polyhedral substituents. Russ. J. Inorg. Chem. 2010, 55, 2089–2127. [Google Scholar] [CrossRef]
- Grimes, R. Carboranes, 2nd ed.; Academic Press: Cambridge, MA, USA, 2011; 1139p. [Google Scholar]
- Klanberg, F.; Muetterties, E.L.; Guggenberger, L.J. Metalloboranes. I. Metal complexes of B3, B9, B9S, B10, and B11 borane anions. Inorg. Chem. 1968, 7, 2272–2278. [Google Scholar] [CrossRef]
- Housecroft, C.E.; Fehlner, T.P. Metalloboranes: Their Relationships to Metal-Hydrocarbon Complexes and Clusters. Adv. Organomet. Chem. 1982, 21, 57–112. [Google Scholar]
- Shubina, E.S.; Bakhmutova, E.V.; Filin, A.M.; Sivaev, I.B.; Teplitskaya, L.N.; Chistyakov, A.L.; Stankevich, I.V.; Bakhmutov, V.I.; Bregadze, V.I.; Epstein, L.M. Dihydrogen bonding of decahydro-closo-decaborate(2-) and dodecahydro-closo-dodecaborate(2-) anions with proton donors: Experimental and theoretical investigation. J. Organomet. Chem. 2002, 657, 155–162. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, J.-C.; Shore, S.G. The Roles of Dihydrogen Bonds in Amine Borane Chemistry. Acc. Chem. Res. 2013, 46, 2666–2675. [Google Scholar] [CrossRef] [PubMed]
- Mebs, S.; Kalinowski, R.; Grabowsky, S.; Förster, D.; Kickbusch, R.; Justus, E.; Morgenroth, W.; Paulmann, C.; Luger, P.; Gabel, D.; et al. Charge transfer via the dative N-B bond and dihydrogen contacts. Experimental and theoretical electron density studies of four deltahedral boranes. J. Phys. Chem. A. 2011, 115, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, R.H.; Siegbahn, P.E.M.; Eisenstein, O.; Rheingold, A.L.; Koetzle, T.F. A new intermolecular interaction: unconventional hydrogen bonds with element-hydride bonds as proton acceptor. Acc. Chem. Res. 1996, 29, 348–354. [Google Scholar] [CrossRef]
- Richardson, T.B.; de Gala, S.; Siegbahn, P.E.M.; Crabtree, R.H. Unconventional Hydrogen Bonds: Intermolecular B-H...H-N Interactions. J. Am. Chem. Soc. 1995, 117, 12875–12876. [Google Scholar] [CrossRef]
- Hawthorne, M.F.; Beno, C.L.; Harwell, D.E.; Jalisatgi, S.S.; Knobler, C.B. Intra- and inter-molecular hydrogen bonding in some cobaltacarboranes. J. Mol. Struct. (Theochem) 2003, 656, 239–247. [Google Scholar] [CrossRef]
- Virovets, A.V.; Vakulenko, N.N.; Volkov, V.V.; Podberezskaya, N.V. Crystal structure of di(1-n-dodecylpyridine) decahydro-closo-decaborate(2-) (C5H5N-C12H25)2[B10H10]. J. Struct. Chem 1994, 35, 339–344. [Google Scholar] [CrossRef]
- Custelcean, R.; Jackson, J.E. Dihydrogen Bonding: Structures, Energetics, and Dynamics. Chem. Rev. 2001, 101, 1963–1980. [Google Scholar] [CrossRef]
- Belkova, N.V.; Epstein, L.M.; Filippov, O.A.; Shubina, E.S. Hydrogen and dihydrogen bonds in the reactions of metal hydrides. Chem. Rev. 2016, 116, 8545–8587. [Google Scholar] [CrossRef] [PubMed]
- Bakhmutov, V.I. Dihydrogen Bonds: Principles, Experiments and Applications; Wiley-Interscience: Hoboken, NJ, USA, 2008. [Google Scholar]
- Malinina, E.A.; Avdeeva, V.V.; Goeva, L.V.; Polyakova, I.N.; Kuznetsov, N.T. Specific interactions in metal salts and complexes with cluster boron anions BnHn2− (n = 6, 10, 12). Russ. J. Inorg. Chem. 2011, 56, 687–697. [Google Scholar] [CrossRef]
- Avdeeva, V.V.; Kravchenko, E.A.; Gippius, A.A.; Vologzhanina, A.V.; Ugolkova, E.A.; Minin, V.V.; Malinina, E.A.; Kuznetsov, N.T. Synthesis, structure, and physicochemical properties of triply-bridged binuclear copper(II) complex [Cu2Phen2(μ-CH3CO2)2(μ-OH)]2[B10Cl10]. Inorg. Chim. Acta 2019, 487, 208–213. [Google Scholar] [CrossRef]
- Kravchenko, E.A.; Gippius, A.A.; Vologzhanina, A.V.; Avdeeva, V.V.; Malinina, E.A.; Ulitin, E.O.; Kuznetsov, N.T. Secondary interactions in decachloro-closo-decaborates of alkali metals M2[B10Cl10] (M = K+ and Cs+): 35Cl NQR and X-ray studies. Polyhedron 2016, 117, 561–568. [Google Scholar] [CrossRef]
- Kravchenko, E.A.; Gippius, A.A.; Vologzhanina, A.V.; Avdeeva, V.V.; Malinina, E.A.; Demikhov, E.I.; Kuznetsov, N.T. Secondary interactions as defined by 35Cl NQR spectra in cesium decachloro-closo-decaborates prepared in non-aqueous solutions. Polyhedron 2017, 138, 140–144. [Google Scholar] [CrossRef]
- Chantler, C.T.; Maslen, E.N. Charge transfer and three-centre bonding in monoprotonated and diprotonated 2,2'-bipyridylium decahydro-closo-decaborate(2-). Acta Crystallogr., Sect. B 1989, 45, 290–297. [Google Scholar] [CrossRef]
- Fuller, D.J.; Kepert, D.L.; Skelton, B.W.; White, A.H. Structure, Stereochemistry and Novel 'Hydrogen Bonding' in Two Bipyridinium Salts of the B10H102−Anion. Aust. J. Chem. 1987, 40, 2097–2105. [Google Scholar] [CrossRef]
- Miller, H.C.; Miller, N.E.; Muetterties, E.L. Synthesis of polyhedral boranes. J. Am. Chem. Soc. 1963, 85, 3885–3886. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Morris, J.H. Novel Synthesis of the B12H122− Anion. Proc. Chem. Soc. 1963, 11, 338. [Google Scholar]
- Avdeeva, V.V.; Vologzhanina, A.V.; Goeva, L.V.; Malinina, E.A.; Kuznetsov, N.T. Reactivity of boron cluster anions [B10H10]2−, [B10Cl10]2– and [B12H12]2– in cobalt(II)/cobalt(III) complexation with 1,10-phenanthroline. Inorg. Chim. Acta 2015, 428, 154–162. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17 (2017). University of Western Australia. Available online: http://hirshfeldsurface.net (accessed on 27 June 2019).
- Qi, G.; Wang, K.; Yang, K.; Zou, B. Pressure-Induced Phase Transition of Hydrogen Storage Material Hydrazine Bisborane: Evolution of Dihydrogen Bonds. J. Phys. Chem. C. 2016, 120, 21293–21298. [Google Scholar] [CrossRef]
- Qi, G.; Wang, K.; Xiao, G.; Zou, B. High pressure, a protocol to identify the weak dihydrogen bonds: experimental evidence of C–H···H–B interaction. Sci. Chin. Chem. 2018, 61, 276–280. [Google Scholar] [CrossRef]
- Smol'yakov, A.F.; Korlyukov, A.A.; Dolgushin, F.M.; Balagurova, E.V.; Chizhevsky, I.T.; Vologzhanina, A.V. Studies of Multicenter and Intermolecular Dihydrogen B–H···H–C Bonding in [4,8,8′-exo-{PPh3Cu}-4,8,8′-(μ-H)3-commo-3,3′-Co(1,2-C2B9H9)(1′,2′-C2B9H10)]. Eur. J. Inorg. Chem. 2015, 5847–5855. [Google Scholar] [CrossRef]
- Bennour, I.; Haukka, M.; Teixidor, F.; Vinas, C.; Kabadou, A. Crystal structure and Hirshfeld surface analysis of [N(CH3)4][2,2′-Fe(1,7-closo-C2B9H11)2]. J. Organomet. Chem. 2017, 846, 74–80. [Google Scholar] [CrossRef]
- Sagan, F.; Filas, R.; Mitoraj, M.P. Non-Covalent Interactions in Hydrogen Storage Materials LiN(CH3)2BH3 and KN(CH3)2BH3. Crystals 2016, 6. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ν(NH) | ν(BH) |
|---|---|---|
| K2[B10H10] | — | 2529, 2461 |
| Phen | — | — |
| 4 | 3095 | 2510, 2477, 2451, 2421 |
| 6·0.5H2O | 3164, 3152 | 2495, 2476, 2414 |
| BPA | 3255, 3181, 3102 | — |
| 1·2H2O | 3286, 3236, 3204, 3136 | 2543, 2492, 2444, 2420 |
| 3·2CH3CN | 3467, 3316, 3255, 3212, 3145, 3111 | 2499, 2466, 2448, 2404 |
| Cs2[B12H12] | — | 2465 |
| 5·CH3CN | 3410, 3361 | 2484, 2461, 2447, 2422 |
| 2·2H2O | 3098, 3063 | 2487, 2443 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avdeeva, V.V.; Vologzhanina, A.V.; Malinina, E.A.; Kuznetsov, N.T. Dihydrogen Bonds in Salts of Boron Cluster Anions [BnHn]2− with Protonated Heterocyclic Organic Bases. Crystals 2019, 9, 330. https://doi.org/10.3390/cryst9070330
Avdeeva VV, Vologzhanina AV, Malinina EA, Kuznetsov NT. Dihydrogen Bonds in Salts of Boron Cluster Anions [BnHn]2− with Protonated Heterocyclic Organic Bases. Crystals. 2019; 9(7):330. https://doi.org/10.3390/cryst9070330
Chicago/Turabian StyleAvdeeva, Varvara V., Anna V. Vologzhanina, Elena A. Malinina, and Nikolai T. Kuznetsov. 2019. "Dihydrogen Bonds in Salts of Boron Cluster Anions [BnHn]2− with Protonated Heterocyclic Organic Bases" Crystals 9, no. 7: 330. https://doi.org/10.3390/cryst9070330
APA StyleAvdeeva, V. V., Vologzhanina, A. V., Malinina, E. A., & Kuznetsov, N. T. (2019). Dihydrogen Bonds in Salts of Boron Cluster Anions [BnHn]2− with Protonated Heterocyclic Organic Bases. Crystals, 9(7), 330. https://doi.org/10.3390/cryst9070330