Effect of Chitin Nanocrystals on Crystallization and Properties of Poly(lactic acid)-Based Nanocomposites

 , ,
, , 

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of ChNCs
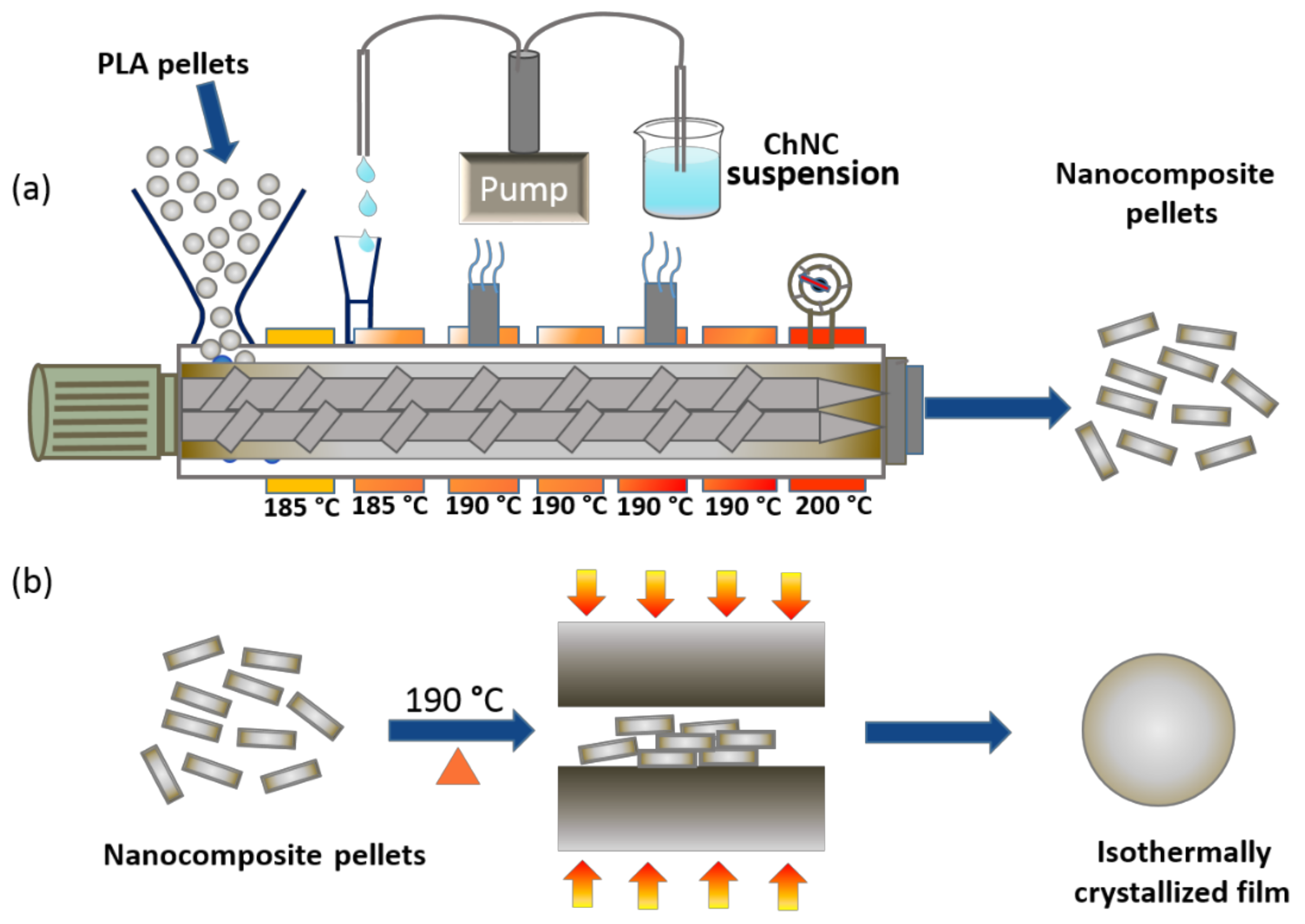
2.3. Preparation of Nanocomposite Pellets via Liquid-Assisted Extrusion
2.4. Preparation of Isothermal-Crystallized Films
2.5. Characterizations
3. Results
3.1. Morphology of ChNCs and Visual Appearance of Neat Films
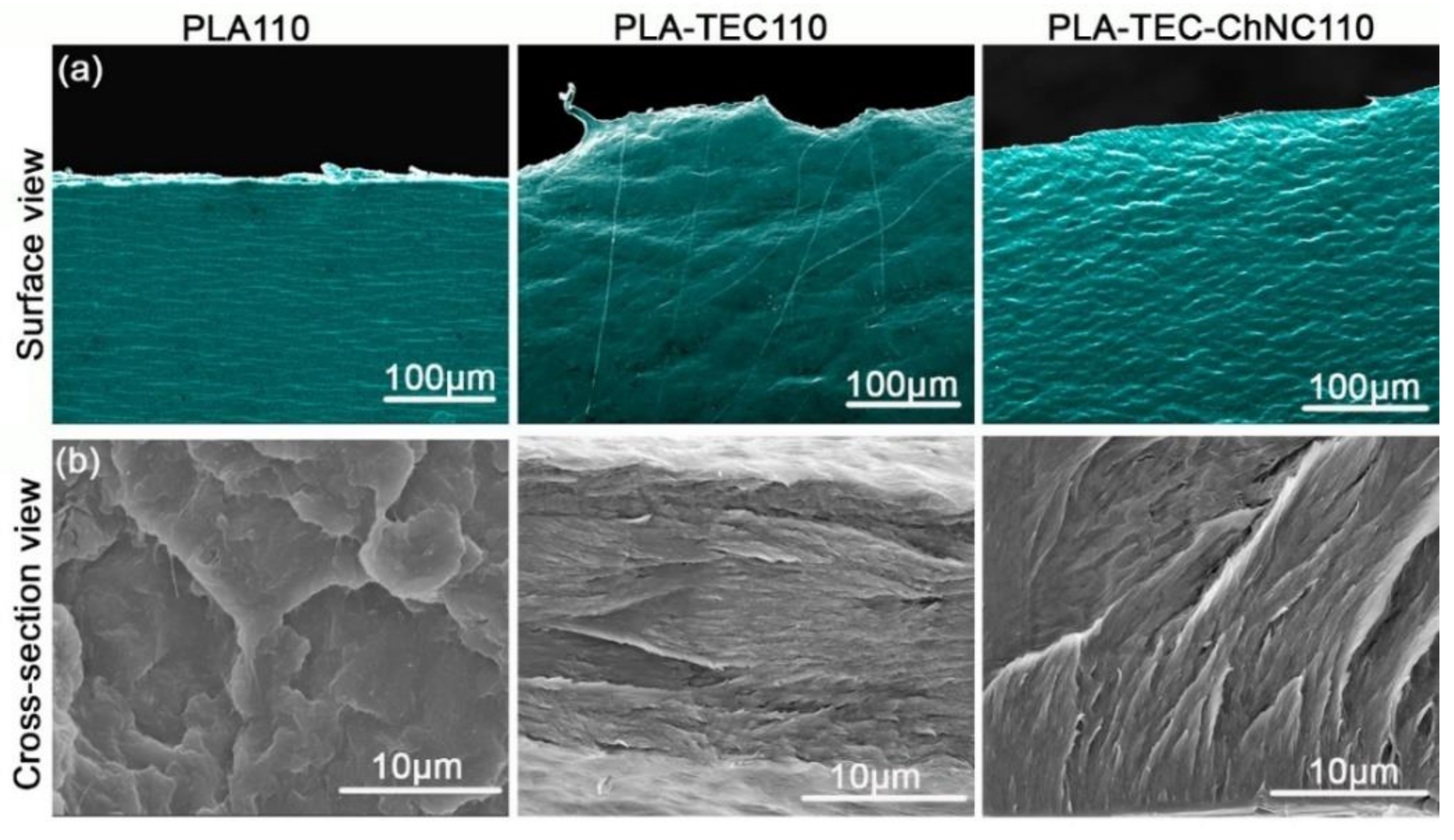
3.1.1. Surface Morphology
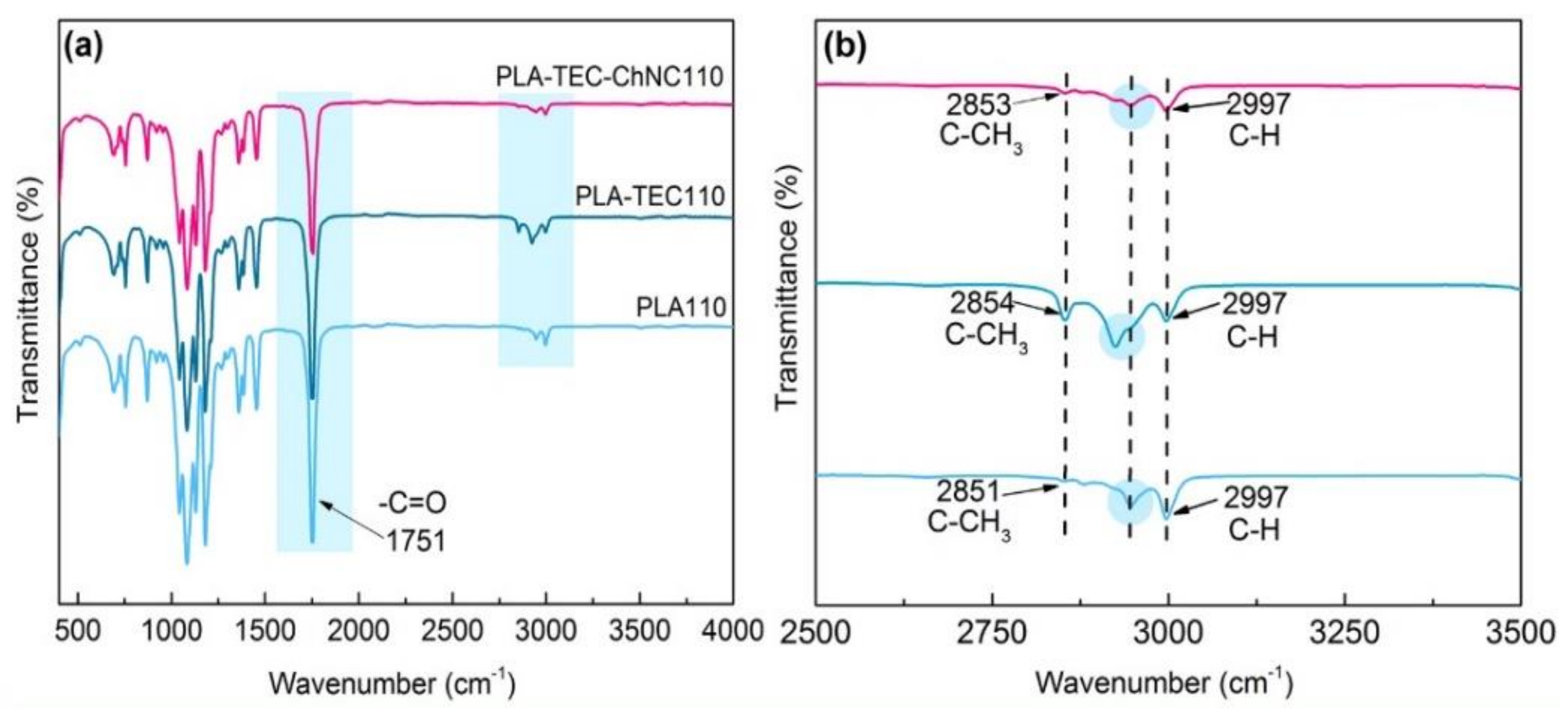
3.1.2. Surface Interaction between PLA, TEC, and ChNC
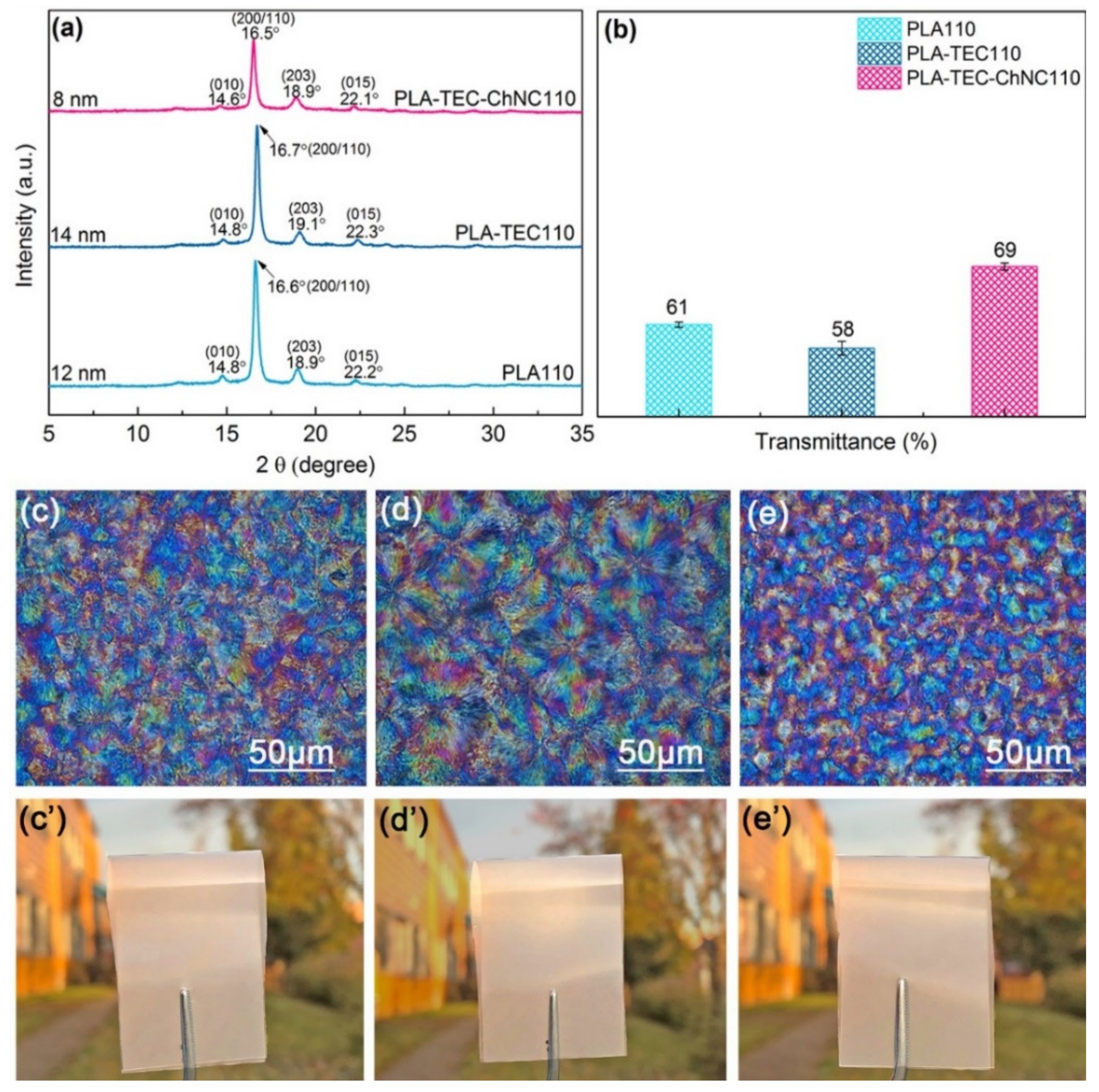
3.2. Thermal Properties, Crystallinity and Crystal Strucutre
3.3. Optical Properties
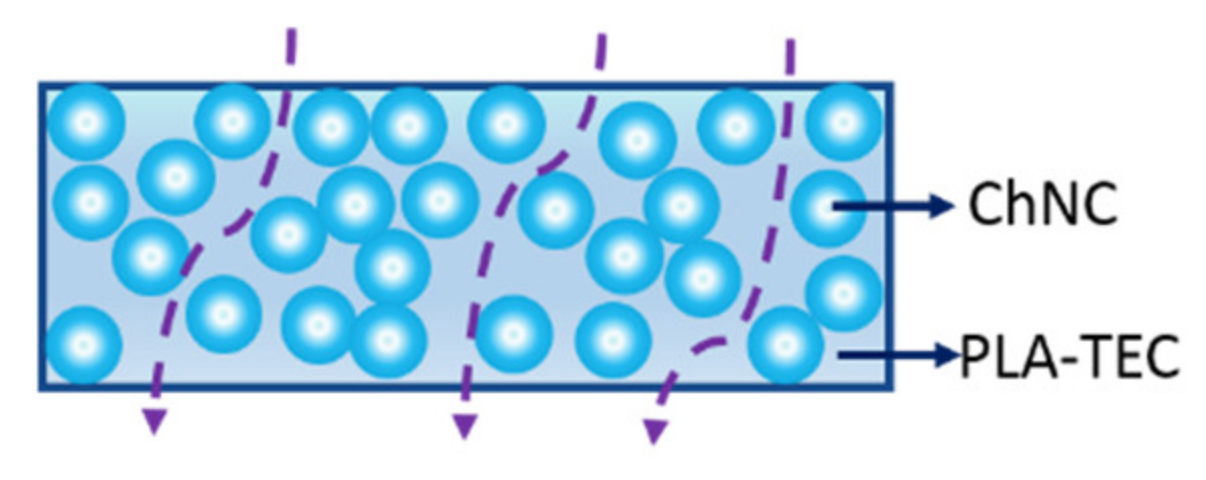
3.4. Barrier Properties
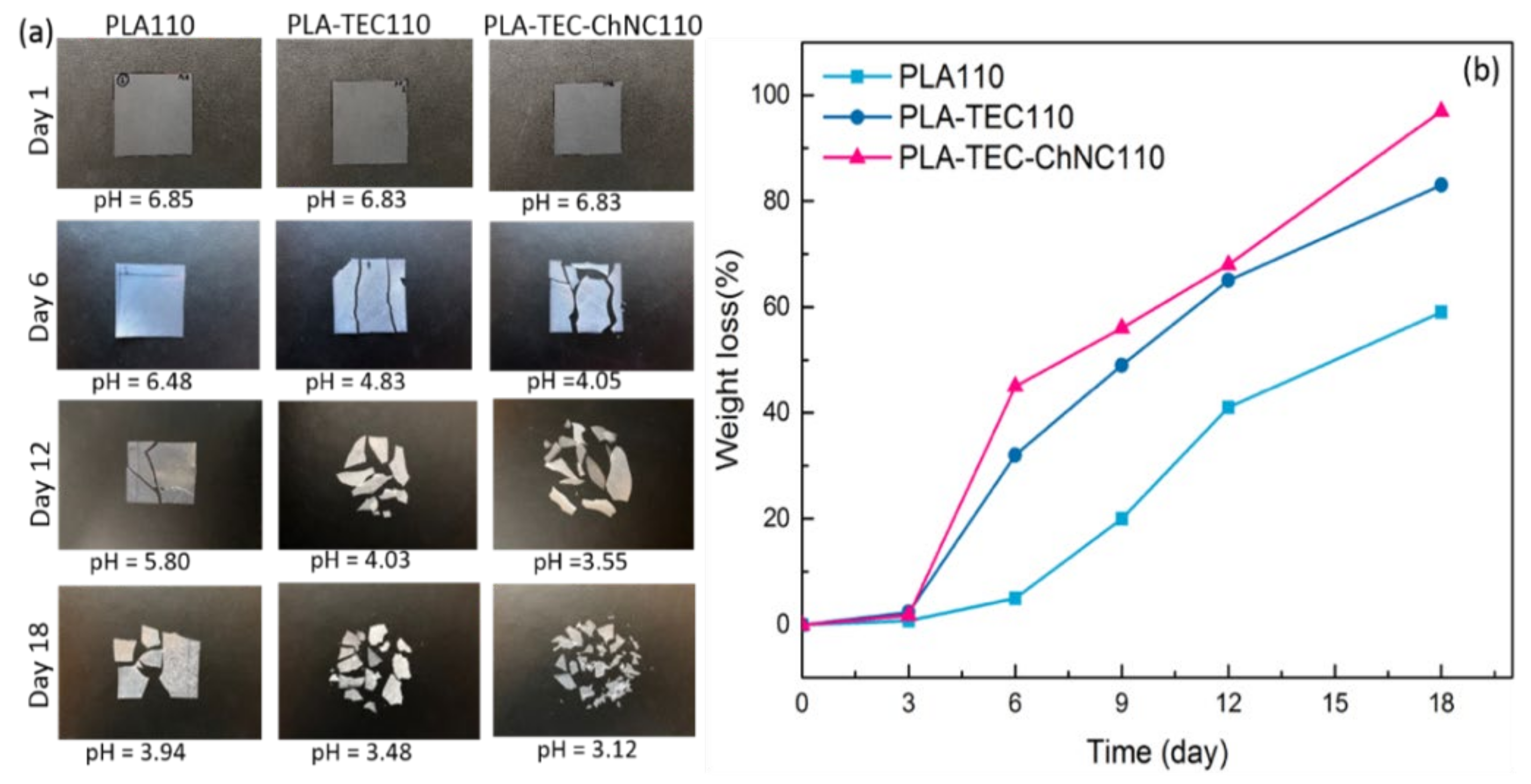
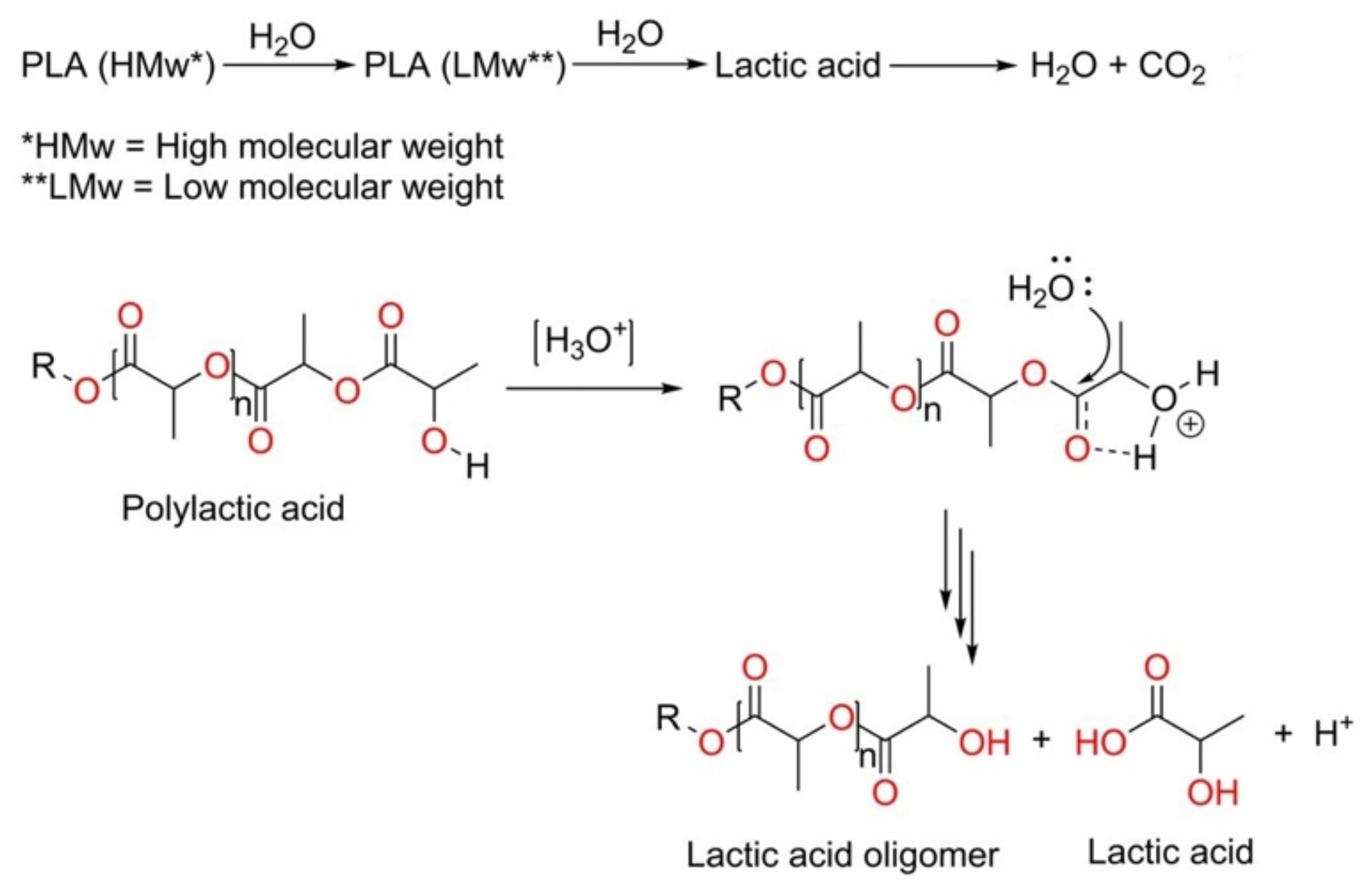
3.5. Hydrolytic Degradation
Effect of Hydrolytic Degradation on Thermal Properties
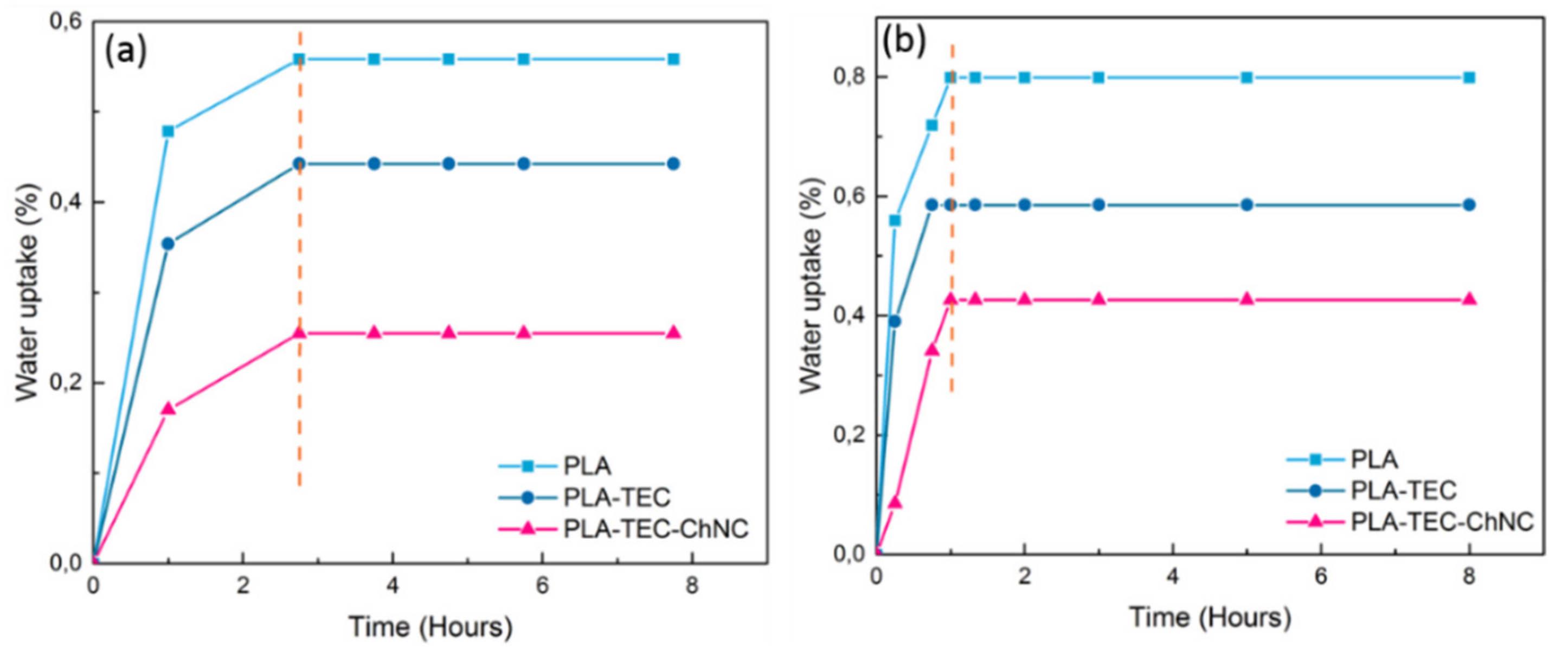
3.6. Water Uptake Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lim, L.T.; Auras, R.; Rubino, M. Processing technologies for poly(lactic acid). Prog. Polym. Sci. 2008, 33, 820–852. [Google Scholar] [CrossRef]
- Cailloux, J.; Hakim, R.N.; Santana, O.O.; Bou, J.; Abt, T.; Carrasco, F.; Maspoch, M.L. Reactive extrusion: A useful process to manufacture structurally modified PLA/o-MMT composites. Compos. Part A 2016, 88, 106–115. [Google Scholar] [CrossRef]
- Gamez-Perez, J.; Nascimento, L.; Bou, J.J.; Franco-Urquiza, E.; Santana, O.O.; Carrasco, F.; Maspoch, M.L. Influence of crystallinity on the fracture toughness of poly(lactic acid)/montmorillonite nanocomposites prepared by twin-screw extrusion. J. Appl. Polym. Sci. 2011, 120, 896–905. [Google Scholar] [CrossRef]
- Marchessault, R.H.; Morehead, F.F.; Walter, N.M. Liquid crystal systems from fibrillar polysaccharides. Nature 1959, 184, 632–633. [Google Scholar] [CrossRef]
- Ravi Kumar, M.N.V. A review of chitin and chitosan applications. React. Funct. Polym. 2000, 46, 1–27. [Google Scholar] [CrossRef]
- Paillet, M.; Dufresne, A. Chitin whisker reinforced thermoplastic nanocomposites. Macromolecules 2001, 34, 6527–6530. [Google Scholar] [CrossRef]
- Zeng, J.-B.; He, Y.-S.; Li, S.-L.; Wang, Y.-Z. Chitin whiskers: An overview. Biomacromolecules 2012, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Liu, J.; Qiang, J.; Yang, L.; Wan, Y.; Wang, H.; Gao, W.; Ko, F. Antibacterial performance of chitin nanowhisker reinforced Poly(lactic acid) composite nanofiber membrane. Adv. Sci. Lett. 2012, 10, 649–651. [Google Scholar] [CrossRef]
- Salaberria, A.; Diaz, R.H.; Andrés, M.; Fernandes, S.; Labidi, J. The antifungal activity of functionalized chitin nanocrystals in poly (Lactid Acid) films. Materials 2017, 10, 546. [Google Scholar] [CrossRef] [Green Version]
- Morin, A.; Dufresne, A. Nanocomposites of chitin whiskers from riftia tubes and poly(caprolactone). Macromolecules 2002, 35, 2190–2199. [Google Scholar] [CrossRef]
- Lu, Y.; Weng, L.; Zhang, L. Morphology and properties of soy protein isolate thermoplastics reinforced with chitin whiskers. Biomacromolecules 2004, 5, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Uddin, A.J.; Fujie, M.; Sembo, S.; Gotoh, Y. Outstanding reinforcing effect of highly oriented chitin whiskers in PVA nanocomposites. Carbohydr. Polym. 2012, 87, 799–805. [Google Scholar] [CrossRef]
- Wang, B.; Li, J.; Zhang, J.; Li, H.; Chen, P.; Gu, Q.; Wang, Z. Thermo-mechanical properties of the composite made of poly (3-hydroxybutyrate-co-3-hydroxyvalerate) and acetylated chitin nanocrystals. Carbohydr. Polym. 2013, 95, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Scaffaro, R.; Botta, L.; Lopresti, F.; Maio, A.; Sutera, F. Polysaccharide nanocrystals as fillers for PLA based nanocomposites. Cellulose 2017, 24, 447–478. [Google Scholar] [CrossRef]
- Coltelli, M.B.; Cinelli, P.; Gigante, V.; Aliotta, L.; Morganti, P.; Panariello, L.; Lazzeri, A. Chitin nanofibrils in poly(Lactic acid) (PLA) nanocomposites: Dispersion and thermo-mechanical properties. Int. J. Mol. Sci. 2019, 20, 504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagaito, A.; Kanzawa, S.; Takagi, H. Polylactic Acid Reinforced with Mixed Cellulose and Chitin Nanofibers—Effect of Mixture Ratio on the Mechanical Properties of Composites. J. Compos. Sci. 2018, 2, 36. [Google Scholar] [CrossRef] [Green Version]
- Oksman, K.; Mathew, A.P.; Bondeson, D.; Kvien, I. Manufacturing process of cellulose whiskers/polylactic acid nanocomposites. Compos. Sci. Technol. 2006, 66, 2776–2784. [Google Scholar] [CrossRef]
- Herrera, N.; Salaberria, A.M.; Mathew, A.P.; Oksman, K. Plasticized polylactic acid nanocomposite films with cellulose and chitin nanocrystals prepared using extrusion and compression molding with two cooling rates: Effects on mechanical, thermal and optical properties. Compos. Part A Appl. Sci. Manuf. 2016, 83, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Herrera, N.; Roch, H.; Salaberria, A.M.; Pino-Orellana, M.A.; Labidi, J.; Fernandes, S.C.M.; Radic, D.; Leiva, A.; Oksman, K. Functionalized blown films of plasticized polylactic acid/chitin nanocomposite: Preparation and characterization. Mater. Des. 2016, 92, 846–852. [Google Scholar] [CrossRef]
- Singh, S.; Maspoch, M.L.; Oksman, K. Crystallization of triethyl-citrate-plasticized poly(lactic acid) induced by chitin nanocrystals. J. Appl. Polym. Sci. 2019, 136, 47936. [Google Scholar] [CrossRef]
- Herrera, N.; Singh, A.A.; Salaberria, A.M.; Labidi, J.; Mathew, A.P.; Oksman, K. Triethyl citrate (TEC) as a dispersing aid in polylactic acid/chitin nanocomposites prepared via liquid-assisted extrusion. Polymers 2017, 9, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nečas, D.; Klapetek, P. Gwyddion: An open-source software for SPM data analysis. Cent. Eur. J. Phys. 2012, 10, 181–188. [Google Scholar] [CrossRef]
- Mathew, A.P.; Oksman, K.; Sain, M. The effect of morphology and chemical characteristics of cellulose reinforcements on the crystallinity of polylactic acid. J. Appl. Polym. Sci. 2006, 101, 300–310. [Google Scholar] [CrossRef]
- Sullivan, E.M.; Moon, R.J.; Kalaitzidou, K. Processing and characterization of cellulose nanocrystals/polylactic acid nanocomposite films. Materials 2015, 8, 8106–8116. [Google Scholar] [CrossRef] [Green Version]
- Somord, K.; Somord, K.; Suwantong, O.; Thanomsilp, C.; Peijs, T.; Soykeabkaew, N. Self-reinforced poly(lactic acid) nanocomposites with integrated bacterial cellulose and its surface modification. Nanocomposites 2018, 4, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Garlotta, D. A literature review of poly(lactic acid). J. Polym. Environ. 2001, 9, 63–84. [Google Scholar] [CrossRef]
- Maiza, M.; Benaniba, M.T.; Massardier-Nageotte, V. Plasticizing effects of citrate esters on properties of poly(lactic acid). J. Polym. Eng. 2016, 36, 371–380. [Google Scholar] [CrossRef]
- Fortunati, E.; Peltzer, M.; Armentano, I.; Torre, L.; Jiménez, A.; Kenny, J.M. Effects of modified cellulose nanocrystals on the barrier and migration properties of PLA nano-biocomposites. Carbohydr. Polym. 2012, 90, 948–956. [Google Scholar] [CrossRef]
- Sanchez-Garcia, M.D.; Gimenez, E.; Lagaron, J.M. Morphology and barrier properties of solvent cast composites of thermoplastic biopolymers and purified cellulose fibers. Carbohydr. Polym. 2008, 71, 235–244. [Google Scholar] [CrossRef]
- Martino, V.P.; Ruseckaite, R.A.; Jiménez, A.; Averous, L. Correlation between composition, structure and properties of poly(lactic acid)/polyadipate-based nano-biocomposites. Macromol. Mater. Eng. 2010, 295, 551–558. [Google Scholar] [CrossRef]
- Trifol, J.; Plackett, D.; Sillard, C.; Hassager, O.; Daugaard, A.E.; Bras, J.; Szabo, P. A comparison of partially acetylated nanocellulose, nanocrystalline cellulose, and nanoclay as fillers for high-performance polylactide nanocomposites. J. Appl. Polym. Sci. 2016, 133, 43257. [Google Scholar] [CrossRef]
- Martínez-Sanz, M.; Lopez-Rubio, A.; Lagaron, J.M. Optimization of the dispersion of unmodified bacterial cellulose nanowhiskers into polylactide via melt compounding to significantly enhance barrier and mechanical properties. Biomacromolecules 2012, 13, 3887–3899. [Google Scholar] [CrossRef] [PubMed]
- Fukuzumi, H.; Saito, T.; Iwata, T.; Kumamoto, Y.; Isogai, A. Transparent and high gas barrier films of cellulose nanofibers prepared by TEMPO-mediated oxidation. Biomacromolecules 2009, 10, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.A.; Delcourt, C.; Alexandre, M.; Degée, P.; Monteverde, F.; Dubois, P. Polylactide/montmorillonite nanocomposites: Study of the hydrolytic degradation. Polym. Degrad. Stab. 2005, 87, 535–542. [Google Scholar] [CrossRef]
- De Jong, S.J.; Arias, E.R.; Rijkers, D.T.S.; Van Nostrum, C.F.; Kettenes-Van Den Bosch, J.J.; Hennink, W.E. New insights into the hydrolytic degradation of poly(lactic acid): Participation of the alcohol terminus. Polymer 2001, 42, 2795–2802. [Google Scholar] [CrossRef]
- Maharana, T.; Mohanty, B.; Negi, Y.S. Melt-solid polycondensation of lactic acid and its biodegradability. Prog. Polym. Sci. 2009, 34, 99–124. [Google Scholar] [CrossRef]
- Schliecker, G.; Schmidt, C.; Fuchs, S.; Kissel, T. Characterization of a homologous series of D,L-lactic acid oligomers; a mechanistic study on the degradation kinetics in vitro. Biomaterials 2003, 24, 3835–3844. [Google Scholar] [CrossRef]
- Proikakis, C.S.; Mamouzelos, N.J.; Tarantili, P.A.; Andreopoulos, A.G. Swelling and hydrolytic degradation of poly(d,l-lactic acid) in aqueous solutions. Polym. Degrad. Stab. 2006, 91, 614–619. [Google Scholar] [CrossRef]
- Elsawy, M.A.; Kim, K.H.; Park, J.W.; Deep, A. Hydrolytic degradation of polylactic acid (PLA) and its composites. Renew. Sustain. Energy Rev. 2017, 79, 1346–1352. [Google Scholar] [CrossRef]
- Zhou, Q.; Xanthos, M. Nanoclay and crystallinity effects on the hydrolytic degradation of polylactides. Polym. Degrad. Stab. 2008, 93, 1450–1459. [Google Scholar] [CrossRef]
- Chow, W.S.; Leu, Y.Y.; Mohd Ishak, Z.A. Water Absorption of Poly(lactic acid) Nanocomposites: Effects of Nanofillers and Maleated Rubbers. Polym. Plast. Technol. Eng. 2014, 53, 858–863. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Feeding Rate (kg/h) | Composition of Materials (wt %) | |||
|---|---|---|---|---|---|
| PLA | Suspension | PLA | TEC | ChNCs | |
| PLA | 2.00 | 0.00 | 100 | 0 | 0 |
| PLA-TEC | 1.80 | 0.75 * | 90 | 10 | 0 |
| PLA-TEC-ChNC | 1.78 | 0.77 * | 89 | 10 | 1 |
| Sample Codes | ICTEMP (°C) | ICTIME (min) |
|---|---|---|
| PLA | N/A | 0 |
| PLA–TEC | N/A | 0 |
| PLA–TEC–ChNC | N/A | 0 |
| PLA135–5 | 135 | 5 * |
| PLA–TEC135–5 | 135 | 5 * |
| PLA–TEC–ChNC135–5 | 135 | 5 * |
| PLA135–15 | 135 | 15 * |
| PLA–TEC135–15 | 135 | 15 * |
| PLA–TEC–ChNC135–15 | 135 | 15 * |
| PLA130–5 | 130 | 5 * |
| PLA–TEC130–5 | 130 | 5 * |
| PLA–TEC–ChNC130–5 | 130 | 5 * |
| PLA130–15 | 130 | 15 * |
| PLA–TEC130–15 | 130 | 15 * |
| PLA–TEC–ChNC130–15 | 130 | 15 * |
| PLA125 | 125 | 5 * |
| PLA–TEC125 | 125 | 5 * |
| PLA–TEC–ChNC125 | 125 | 5 * |
| PLA125 | 125 | 15 * |
| PLA–TEC125 | 125 | 15 * |
| PLA–TEC–ChNC125 | 125 | 15 * |
| PLA110 | 110 | 40 ** |
| PLA–TEC110 | 110 | 40 ** |
| PLA–TEC–ChNC110 | 110 | 5 ** |
| Materials | Tg (°C) | Tcc (°C) | Tm (°C) | Crystallinity (%) |
|---|---|---|---|---|
| PLA | 61.7 | 110.4 | 170.4 | 4.0 |
| PLA–TEC | 48.8 | 98.0 | 164.8 | 6.6 |
| PLA–TEC–ChNC | 48.1 | 95.4 | 164.4 | 6.7 |
| PLA135-5 | 58.9 | 108.6 | 169.6 | 7.5 |
| PLA–TEC135–5 | 46.0 | 97.4 | 164.6 | 7.8 |
| PLA–TEC–ChNC135–5 | 46.3 | 94.7 | 163.2 | 8.6 |
| PLA135–15 | 60.6 | 110.6 | 170.0 | 1.3 |
| PLA–TEC135–15 | 47.5 | 97.4 | 164.4 | 4.5 |
| PLA–TEC–ChNC135–15 | 44.8 | 93.4 | 163.4 | 14.2 |
| PLA130–5 | 59.4 | 108.4 | 169.4 | 2.6 |
| PLA–TEC130–5 | 46.8 | 96.2 | 164.5 | 7.5 |
| PLA–TEC–ChNC130–5 | 46.3 | 94.5 | 163.6 | 8.2 |
| PLA130–15 | 58.6 | 108.5 | 168.6 | 7.0 |
| PLA–TEC130–15 | 45.7 | 96.0 | 163.3 | 7.2 |
| PLA–TEC–ChNC130–15 | 39.1 | 87.1 | 163.3 | 34.5 |
| PLA125–5 | 59.5 | 109.7 | 169.5 | 6.0 |
| PLA–TEC125–5 | 46.9 | 97.0 | 164.8 | 7.2 |
| PLA–TEC–ChNC125–5 | 46.5 | 95.8 | 164.7 | 7.3 |
| PLA125–15 | 59.8 | 110.4 | 169.6 | 5.8 |
| PLA–TEC125–15 | 47.3 | 96.9 | 163.7 | 6.0 |
| PLA–TEC–ChNC125–15 | 37.7 | 86.9 | 162.7 | 36.5 |
| PLA110–40 | 59.4 | 111.8 | 164.9 | 45.5 |
| PLA–TEC110–40 | 44.7 | 113.7 | 164.8 | 49.9 |
| PLA–TEC–ChNC110–5 | 36.8 | - | 164.6 | 47.5 |
| Materials | WVTR [g/m2 day] | OTR [ml/(m2 24h)] | OP [(ml μm)/(m2 24h kPa)] | |||
|---|---|---|---|---|---|---|
| Ave | std | Ave | std | Ave | std | |
| PLA | 200 [25] | - | 746 [33] | - | - | - |
| PLA–TEC110 | 44 | (3) | 165 | (21) | 19,425 | (3076) |
| PLA–TEC–ChNC110 | 28 | (2) | 113 | (18) | 11,563 | (2210) |
| Materials | Diffusional Exponent (n) | Kinetic Constant (K) | Diffusion Coefficient (D) (m2 s−1) |
|---|---|---|---|
| PLA110 | 0.248 | 0.909 | 1.62 × 10−6 |
| PLA–TEC110 | 0.315 | 1.095 | 1.09 × 10−6 |
| PLA–TEC–ChNC110 | 1.230 | 0.979 | 0.10 × 10−6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.; Patel, M.; Schwendemann, D.; Zaccone, M.; Geng, S.; Maspoch, M.L.; Oksman, K. Effect of Chitin Nanocrystals on Crystallization and Properties of Poly(lactic acid)-Based Nanocomposites. Polymers 2020, 12, 726. https://doi.org/10.3390/polym12030726
Singh S, Patel M, Schwendemann D, Zaccone M, Geng S, Maspoch ML, Oksman K. Effect of Chitin Nanocrystals on Crystallization and Properties of Poly(lactic acid)-Based Nanocomposites. Polymers. 2020; 12(3):726. https://doi.org/10.3390/polym12030726
Chicago/Turabian StyleSingh, Shikha, Mitul Patel, Daniel Schwendemann, Marta Zaccone, Shiyu Geng, Maria Lluisa Maspoch, and Kristiina Oksman. 2020. "Effect of Chitin Nanocrystals on Crystallization and Properties of Poly(lactic acid)-Based Nanocomposites" Polymers 12, no. 3: 726. https://doi.org/10.3390/polym12030726
APA StyleSingh, S., Patel, M., Schwendemann, D., Zaccone, M., Geng, S., Maspoch, M. L., & Oksman, K. (2020). Effect of Chitin Nanocrystals on Crystallization and Properties of Poly(lactic acid)-Based Nanocomposites. Polymers, 12(3), 726. https://doi.org/10.3390/polym12030726