Synthesis of Poly(Dimethylmalic Acid) Homo- and Copolymers to Produce Biodegradable Nanoparticles for Drug Delivery: Cell Uptake and Biocompatibility Evaluation in Human Heparg Hepatoma Cells

 , , and
, , and 
Abstract
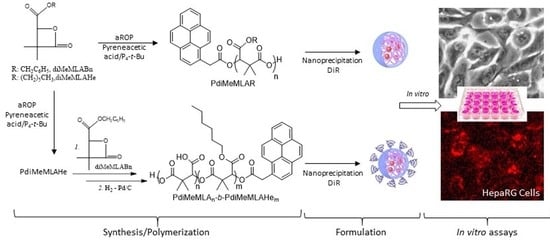
:
1. Introduction
2. Materials and Methods
2.1. Materials and Apparatus
2.2. Synthesis of Monomers and (co)Polymers
2.3. Formulation of Nanoparticles
2.4. In Vitro Assays
3. Results and Discussion
3.1. Synthesis and Characterization of diMeMLABn and diMeMLAHe
3.2. Synthesis and Characterization of Hydrophobic Homopolymers and Amphiphilic Block Copolymers Based on Pdimemla Derivatives
3.3. Formulation and Characterization of Nanoparticles
3.4. In Vitro Cell Viability Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticles design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef] [PubMed]
- Moghini, S.M.; Hunter, A.C.; Murray, J.C. Long-circulating and target-specific nanoparticles: Theory to practice. Pharmacol. Rev. 2001, 53, 283–318. [Google Scholar]
- Arvizo, R.R.; Miranda, O.R.; Moyano, D.F.; Walden, C.A.; Giri, K.; Bhattacharya, R.; Robertson, J.D.; Rotello, V.M.; Reid, J.M.; Mukherjee, P. Modulating pharmacokinetics, tumor uptake and biodistribution by engineered nanoparticles. PLoS ONE 2011, 6, e24374. [Google Scholar] [CrossRef] [Green Version]
- Mahon, E.; Salvati, A.; Baldelli Bombelli, F.; Lynch, I.; Dawson, K.A. Designing the nanoparticles-biomolecule interface for “targeting and therapeutic delivery”. J. Control. Release 2012, 161, 164–174. [Google Scholar] [CrossRef]
- Pearce, A.K.; O’Reilly, R.K. Insights into active targeting of nanoparticles in drug delivery: Advances in clinical studies and design considerations for cancer nanomedecine. Bioconjug. Chem. 2019, 30, 2300–2311. [Google Scholar] [CrossRef]
- Englert, C.; Brendel, J.C.; Majdanski, T.C.; Yildirim, T.; Schubert, S.; Gottschaldt, M.; Windhab, N.; Schubert, U.S. Pharmacopolymers in the 21st century: Synthetic polymers in drug delivery applications. Prog. Polym. Sci. 2018, 87, 107–164. [Google Scholar] [CrossRef]
- Venditto, V.J.; Szoka, F.C., Jr. Cancer nanomedicines: So many papers and so few drugs! Adv. Drug Deliv. Rev. 2013, 65, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Khan, H.A.; Sakharkar, M.K.; Nayak, A.; Kishore, U.; Khan, A. Nanoparticles for biomedical applications: An overview. In Nanomaterials: Nanostructured Materials for Biomedical Applications; Narayan, R., Ed.; Woodhead publishing Series in Biomaterials; Elsevier Ltd.: Duxfort, UK, 2018; pp. 357–377. [Google Scholar]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef]
- Copi, S.; Amalraj, A.; Sukumaran, N.P.; Haponiuk, J.T.; Thomas, S. Biopolymers and their composites for drug delivery: A brief overview. Macromol. Symp. 2018, 380, 1800114. [Google Scholar]
- Chen, W.; Zhou, S.; Ge, L.; Wu, W.; Jiang, X. Translatable high drug loading drug delivery systems based on biocompatible polymers nanocarriers. Biomacromolecules 2018, 19, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.R. Delivering combination chemotherapies and targeting oncogenic pathways via polymeric drug delivery systems. Polymers 2019, 11, 630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loyer, P.; Cammas-Marion, S. Natural and synthetic poly(malic acid)-based derivates: A family of versatile biopolymers for the design of drug nanocarriers. J. Drug Target. 2014, 22, 556–575. [Google Scholar] [CrossRef]
- Casajus, H.; Saba, S.; Vlach, M.; Vène, E.; Ribault, C.; Tranchimand, S.; Nugier-Chauvin, C.; Dubreucq, E.; Loyer, P.; Cammas-Marion, S.; et al. Cell Uptake and Biocompatibility of Nanoparticles Prepared from Poly(benzyl malate) (Co)polymers Obtained through Chemical and Enzymatic Polymerization in Human HepaRG Cells and Primary Macrophages. Polymers (Basel) 2018, 10, 1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, D.E.; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [Google Scholar]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. 2003, 2, 214–221. [Google Scholar] [CrossRef]
- Veronese, F.M. Peptide and protein PEGylation: A review of problems and solutions. Biomaterials 2001, 22, 405–417. [Google Scholar] [CrossRef]
- Kolate, A.; Baradia, D.; Patil, S.; Vhora, I.; Kore, G.; Misra, A. PEG–A versatile conjugating ligand for drugs and drug delivery systems. J. Control. Release 2014, 192, 67–81. [Google Scholar] [CrossRef]
- Liu, S.; Jiang, S. Chemical conjugation of zwitterionic polymers protects immunogenic enzyme and preserves bioactivity without polymer-specific antibody response. Nano Today 2016, 11, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.K.; Hempel, G.; Koling, S.; Chan, L.S.; Fisher, T.; Meiselman, H.J.; Garratty, G. Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer 2007, 110, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Dams, E.T.; Laverman, P.; Oyen, W.J.; Storm, G.; Scherphof, G.L.; van Der Meer, J.W.; Corstens, F.H.; Boerman, O.C. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J. Pharmacol. Exp. Ther. 2000, 292, 1071–1079. [Google Scholar] [PubMed]
- Kawai, F. Microbial degradation of polyethers. Appl. Microbiol. Biotechnol. 2002, 58, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Huh, M.S.; Lee, E.J.; Koo, H.; Yhee, J.Y.; Oh, K.S.; Son, S.; Lee, S.; Kim, S.H.; Kwon, I.C.; Kim, K. Polysaccharide-Based Nanoparticles for Gene Delivery. Top. Curr. Chem. 2017, 375, 1–19. [Google Scholar]
- Cammas, S.; Béar, M.M.; Harada, A.; Guérin, P.; Kataoka, K. New macromolecular micelles based on degradable amphiphilic block copolymers. Macromol. Chem. Phys. 2000, 201, 355–364. [Google Scholar] [CrossRef]
- Martinez Barbosa, M.E.; Cammas, S.; Appel, M.; Ponchel, G. Investigation of the degradation mechanisms of poly(malic acid) esters in vitro and their related cytotoxicities on J774 macrophages. Biomacromolecules 2004, 5, 137–143. [Google Scholar] [CrossRef]
- Barouti, G.; Khalil, A.; Orione, C.; Jarnouen, K.; Cammas-Marion, S.; Loyer, P.; Guillaume, S.M. Poly(trimethylene carbonate)/poly(malic acid) amphiphilic diblock copolymers as original biocompatible nanoparticles. Chem. Eur. J. 2016, 22, 2819–2830. [Google Scholar] [CrossRef]
- Zia, K.M.; Noreen, A.; Zuber, M.; Tabasum, S.; Mujahid, M. Recent Developments and Future Prospects on Bio-Based Polyesters Derived from Renewable Resources: A Review. Int. J. Biol. Macromol. 2016, 82, 1028–1040. [Google Scholar] [CrossRef]
- Ikada, Y.; Tsuji, H. Biodegradable Polyesters for Medical and Ecological Applications. Macromol. Rapid Commun. 2000, 21, 117–132. [Google Scholar] [CrossRef]
- Xu, M.; Yang, R.; Huang, Q.; Zhao, X.; Ma, C.; Li, W.; Li, J.; Liu, S. Preparation and characterization of acetylated Nanocrystalline cellulose-reinforced polylactide highly regular porous films. Bioresources 2018, 13, 8432–8443. [Google Scholar] [CrossRef]
- Manitchotpisit, P.; Skory, C.D.; Peterson, S.W.; Price, N.P.J.; Vermillion, K.E.; Leathers, T.D. Poly(β-L-malic acid) production by diverse phylogenetic clades of Aureobasidium Pullulans. J. Ind. Microbiol. Biotechnol. 2012, 39, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Hall, H.K., Jr.; Scheneider, A.K. Polymerization of cyclic esters, urethans, ureas and imides. J. Am. Chem. Soc. 1958, 80, 6409–6412. [Google Scholar] [CrossRef]
- Khalil, A.; Cammas-Marion, S.; Coulembier, O. Organocatalysis applied to the ring-opening polymerization of β-lactones: A brief overview. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 657–672. [Google Scholar] [CrossRef]
- Barbaud, C.; Guerrouache, M.; Guérin, P. Synthesis of novel α,α′,β-trisubstituted β-lactones. Tetrahedron Lett. 2002, 43, 9513–9515. [Google Scholar] [CrossRef]
- Barbaud, C.; Faÿ, F.; Abdillah, F.; Randriamahefa, S.; Guérin, P. Synthesis of new homopolyester and copolyesters by anionic ring-opening polymerization of α,α′,β-trisubstituted β-lactones. Macromol. Chem. Phys. 2004, 205, 199–207. [Google Scholar] [CrossRef]
- Ouhib, F.; Randriamahefa, S.; Guérin, P.; Barbaud, C. Synthesis of new statistical and block co-polyesters by ROP of α,α,β-trisubstituted β-lactones and their characterizations. Des. Monomers Polym. 2005, 8, 25–35. [Google Scholar] [CrossRef]
- Schott, M.A.; Domurado, M.; Leclercq, L.; Barbaud, C.; Domurado, D. Solubilization of water-insoluble drugs due to random amphiphilic and degradable poly(dimethylmalic acid) derivatives. Biomacromolecules 2013, 14, 1936–1944. [Google Scholar] [CrossRef]
- Belibel, R.; Barbaud, C. Synthesis and characterizations of hemiditactic homopolymers derived of poly(3-allyl-3-methylmalic acid): An example of a new class of polymer’s ditacticity. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2408–2418. [Google Scholar] [CrossRef]
- Belibel, R.; Barbaud, C. Synthesis of new optically active α,α′,β-trisubstituted-β-lactones as monomers for stereoregular biopolyesters. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 2586–2597. [Google Scholar] [CrossRef]
- Tetraethylammonium. Available online: https://www.drugbank.ca/drugs/DB08837 (accessed on 2 November 2018).
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [Green Version]
- Cerec, V.; Glaise, D.; Garnier, D.; Morosan, S.; Turlin, B.; Drenou, B.; Gripon, P.; Kremsdorf, D.; Guguen-Guillouzo, C.; Corlu, A. Transdifferentiation of hepatocyte-like cells from the human hepatoma HepaRG cell line through bipotent progenitor. Hepatology 2007, 45, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Corlu, A.; Loyer, P. Culture Conditions Promoting Hepatocyte Proliferation and Cell Cycle Synchronization. Methods Mol. Biol. 2015, 1250, 27–51. [Google Scholar] [PubMed]
- Coulembier, O.; Degée, P.; Cammas-Marion, S.; Guérin, P.; Dubois, P. New amphiphilic poly[(R,S)-β-malic acid-b-ε-caprolactone] diblock copolymers by combining anionic and coordination-insertion ring-opening polymerization. Macromolecules 2002, 35, 9896–9903. [Google Scholar] [CrossRef]
- Zhang, L.; Nederberg, F.; Messman, J.M.; Pratt, R.C.; Hedrick, J.L.; Wade, C.G. Organocatalytic stereoselective ring-opening polymerization of lactide with dimeric phosphazene bases. J. Am. Chem. Soc. 2007, 129, 12610–12611. [Google Scholar] [CrossRef]
- Zhang, L.; Nederberg, F.; Pratt, R.C.; Waymouth, R.M.; Hedrick, J.L.; Wade, C.G. Phopsphazene bases: A new category of organocatalysts for the living ring-opening polymerization of cyclic esters. Macromolecules 2007, 40, 4154–4158. [Google Scholar] [CrossRef]
- De Winter, J.; Coulembier, O.; Gerbaux, P.; Dubois, P. High molecular weight poly(α,α′,β-trisubstituted β-lactones) as generated by metal-free phosphazene catalysts. Macromolecules 2010, 43, 10291–10296. [Google Scholar] [CrossRef]
- Yang, H.; Xu, J.; Pispas, S.; Zhang, G. Hybrid copolymerization of ε-caprolactone and methyl methacrylate. Macromolecules 2012, 45, 3312–3317. [Google Scholar] [CrossRef]
- Kawalec, M.; Coulembier, O.; Gerbaux, P.; Sobata, M.; De Winter, J.; Dubois, P.; Kowalczuk, M.; Kurcok, P. Trace do matter-Purity of 4-methyl-2-oxetanone and its effect on anionic ring-opening polymerization as evidence by phosphazene superbase catalysis. Reac. Funct. Polym. 2012, 72, 509–520. [Google Scholar] [CrossRef]
- Xu, J.; Yang, H.; Zhang, G. Synthesis of poly(ε-caprolactone-co-methacrylic acid) copolymer via phosphazene-catalyzed hybrid copolymerization. Macromol. Chem. Phys. 2013, 214, 378–385. [Google Scholar] [CrossRef]
- Giammona, G.; Craparo, E.F. Biomedical applications of polylactide (PLA) and its copolymers. Molecules 2018, 23, 980. [Google Scholar] [CrossRef] [Green Version]
- Vène, E.; Barouti, G.; Jarnouen, K.; Gicquel, T.; Rauch, C.; Ribault, C.; Guillaume, S.M.; Cammas-Marion, S.; Loyer, P. Opsonisation of nanoparticles prepared from poly(β-hydroxybutyrate) and poly(trimethylene carbonate)-b-poly(malic acid) amphiphilic diblock copolymers: Impact on the in vitro cell uptake by primary human macrophages and HepaRG hepatoma cells. Int. J. Pharm. 2016, 513, 38–452. [Google Scholar] [CrossRef] [PubMed]
- Barouti, G.; Jarnouen, K.; Cammas-Marion, S.; Loyer, P.; Guillaume, S.M. Polyhydroxyalkanoate-based amphiphilic diblock copolymers as original biocompatible nanovectors. Polym. Chem. 2015, 6, 5414–5429. [Google Scholar] [CrossRef]
- Caron, A.; Braud, C.; Bunel, C.; Vert, M. Blocky structure of copolymers obtained by Pd/C catalyzed hydrogenolysis of benzyl protecting groups as shown by sequence-selective hydrolytic degradation in poly(β-malic acid) derivatives. Polymer 1990, 31, 1797–1802. [Google Scholar] [CrossRef]
- Kataoka, K.; Kwon, G.S.; Yokoyama, M.; Okano, T.; Sakurai, Y. Block copolymer micelles as vehicles for drug delivery. J. Control. Release 1993, 24, 119–132. [Google Scholar]
- Thioune, O.; Fessi, H.; Devissaguet, J.P.; Puisieux, F. Preparation of pseudolatex by nanoprecipitation: Influence of the solvent nature on intrinsic viscosity and interaction constant. Int. J. Pharm. 1997, 146, 233–238. [Google Scholar] [CrossRef]
- Martínez Rivas, C.J.; Tarhini, M.; Badri, W.; Miladi, K.; Greige-Gerges, H.; Nazari, Q.A.; Galindo Rodríguez, S.A.; Román, R.Á.; Fessi, H.; Elaissari, A. Nanoprecipitation process: From encapsulation to drug delivery. Int. J. Pharm. 2017, 532, 66–81. [Google Scholar] [CrossRef]
- Msolli, I.; Belibel, R.; Chauvet, F.; Maaroufi, M.R.; Barbaud, C. Synthesis of nanoparticles based on PDMMLA derivative copolymers and study of warfarin encapsulation and controlled release. RCS Adv. 2017, 7, 6704–6711. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S. DLS and zeta potential-What they are and what they are not? J. Control. Release 2016, 235, 337–351. [Google Scholar] [CrossRef]
- Limayem Blouza, I.; Charcosset, C.; Sfar, S.; Fessi, H. Preparation and characterization of spironolactone-loaded nanocapsules for paediatric use. Int. J. Pharm. 2006, 325, 124–131. [Google Scholar] [CrossRef]
- Aninat, C.; Piton, A.; Glaise, D.; Le Charpentier, T.; Langouet, S.; Morel, F.; Guguen-Guillouzo, C.; Guillouzo, A. Expression of cytochrome P450, conjugating enzymes and nuclear receptors in human hepatoma HepaRG cells. Drug Metab. Dispos. 2006, 34, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Quesnot, N.; Bucher, S.; Gade, C.; Vlach, M.; Vène, E.; Valenca, S.; Gicquel, T.; Holst, H.; Robin, M.A.; Loyer, P. Production of chlorzoxazone glucuronides via cytochrome P4502E1 dependent and independent pathways in human hepatocytes. Arch. Tox. 2018, 92, 3077–3091. [Google Scholar] [CrossRef] [PubMed]
- Vlach, M.; Quesnot, N.; Dubois-Pot-Schneider, H.; Ribault, C.; Verres, Y.; Petitjean, K.; Rauch, C.; Morel, F.; Robin, M.A.; Corlu, A.; et al. Cytochrome P450 1A1/2, 2B6 and 3A4 HepaRG Cell-Based Biosensors to Monitor Hepatocyte Differentiation, Drug Metabolism and Toxicity. Sensors (Basel) 2019, 19, 2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.H.; Laurent, V.; Chetouani, G.; Ljubimova, J.Y.; Holler, E.; Benvegnu, T.; Loyer, P.; Cammas-Marion, S. New functional degradable and bio-compatible nanoparticles based on poly(malic acid) derivatives for site-specific anti-cancer drug delivery. Int. J. Pharm. 2012, 423, 84–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Homopolymer | [Monomer]:[1-pyreneacetic acid]:[P4-t-Bu] | Mn, theo (g/mol) | Mn, NMR a (g/mol) | Mn, SEC b (g/mol) | Đ b |
|---|---|---|---|---|---|
| PdiMeMLABn30 | 32:1:1 | 7500 | 7000 | 2700 | 1.20 |
| PdiMeMLAHe30 | 33:1:1 | 7500 | 6800 | 5500 | 1.16 |
| Entry | [diMeMLAHe]0/[diMeMLABn]0 Initial Ratio | Theoretical Values | Experimental Values | Mn b (g/mol) | Đ b | ||
|---|---|---|---|---|---|---|---|
| MPdiMeMLAHe (g/mol) | MPdiMeMLABn (g/mol) | MPdiMeMLAHe a (g/mol) | MPdiMeMLABn a (g/mol) | ||||
| 1 | 32:49 | 7300 | 11,500 | 6600 | 12,600 | 12,000 | 1.25 |
| 2 | 49:24 | 11,100 | 5700 | 11,800 | 6400 | 7400 | 1.31 |
| Block Copolymers | MPdiMeMLA a (g/mol) | MPdiMeMLAHe a (g/mol) | Mglobal (g/mol) | diMeMLA/ diMeMLAHe |
|---|---|---|---|---|
| PdiMeMLA54-b-PdiMeMLAHe29 | 7720 (n = 54) | 6600 (m = 29) | 14,320 | 50/50 |
| PdiMeMLA27-b-PdiMeMLAHe50 | 3850 (n = 27) | 11,800 (n = 50) | 15,650 | 25/75 |
| Polymers | Dh (nm) | PDI | Zeta Potential (mV) |
|---|---|---|---|
| PdiMeMLABn30 | 100 | 0.19 | −37 |
| PdiMeMLAHe30 | 130 | 0.12 | −53 |
| Copolymers | Dh (nm) | PDI | Zeta Potential (mV) |
|---|---|---|---|
| PdiMeMLA54-b-PdiMeMLAHe29 | 50 | 0.08 | −36 |
| PdiMeMLA27-b-PdiMeMLAHe50 | 150 | 0.17 | −32 |
| Polymers | DiR (wt%) | Dh (nm) | PDI |
|---|---|---|---|
| PdiMeMLABn30 | 0.5 | 110 | 0.15 |
| PdiMeMLAHe30 | 0.5 | 120 | 0.13 |
| PdiMeMLA54-b-PdiMeMLAHe29 | 1 | 50 | 0.07 |
| PdiMeMLA27-b-PdiMeMLAHe50 | 1 | 120 | 0.20 |
| NPs | PdiMeMLA54-b-PdiMeMLAHe29 | PdiMeMLA27-b-PdiMeMLAHe50 | PdiMeMLABn30 | PdiMeMLAHe30 |
|---|---|---|---|---|
| IC50 (μg/mL) | 4.65 ± 0.49 | 22.11 ± 0.84 | 6.95 ± 2.2 | 23.56 ± 2.12 |
| NPs | PdiMeMLA54-b-PdiMeMLAHe29 | PdiMeMLA27-b-PdiMeMLAHe50 | PdiMeMLABn30 | PdiMeMLAHe30 | |
|---|---|---|---|---|---|
| IC50 (μg/mL) | 4.65 ± 0.49 | 22.11 ± 0.84 | 6.95 ± 2.2 | 23.56 ± 2.12 | |
| P4-t-Bu (% mol) | 1.3 | 0.94 | 2.94 | 2.87 | |
| Positive cells (%) | At 6 μg/mL | 66.6 | 96.9 | 18.75 | 11.75 |
| At 25 μg/mL | 24.8 | 99.5 | 78.4 | 97.3 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalil, A.; Saba, S.; Ribault, C.; Vlach, M.; Loyer, P.; Coulembier, O.; Cammas-Marion, S. Synthesis of Poly(Dimethylmalic Acid) Homo- and Copolymers to Produce Biodegradable Nanoparticles for Drug Delivery: Cell Uptake and Biocompatibility Evaluation in Human Heparg Hepatoma Cells. Polymers 2020, 12, 1705. https://doi.org/10.3390/polym12081705
Khalil A, Saba S, Ribault C, Vlach M, Loyer P, Coulembier O, Cammas-Marion S. Synthesis of Poly(Dimethylmalic Acid) Homo- and Copolymers to Produce Biodegradable Nanoparticles for Drug Delivery: Cell Uptake and Biocompatibility Evaluation in Human Heparg Hepatoma Cells. Polymers. 2020; 12(8):1705. https://doi.org/10.3390/polym12081705
Chicago/Turabian StyleKhalil, Ali, Saad Saba, Catherine Ribault, Manuel Vlach, Pascal Loyer, Olivier Coulembier, and Sandrine Cammas-Marion. 2020. "Synthesis of Poly(Dimethylmalic Acid) Homo- and Copolymers to Produce Biodegradable Nanoparticles for Drug Delivery: Cell Uptake and Biocompatibility Evaluation in Human Heparg Hepatoma Cells" Polymers 12, no. 8: 1705. https://doi.org/10.3390/polym12081705
APA StyleKhalil, A., Saba, S., Ribault, C., Vlach, M., Loyer, P., Coulembier, O., & Cammas-Marion, S. (2020). Synthesis of Poly(Dimethylmalic Acid) Homo- and Copolymers to Produce Biodegradable Nanoparticles for Drug Delivery: Cell Uptake and Biocompatibility Evaluation in Human Heparg Hepatoma Cells. Polymers, 12(8), 1705. https://doi.org/10.3390/polym12081705